ImmunoPET in Multiple Myeloma—What? So What? Now What?

 , , , , ,
, , , , , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. What?
3. So What?
4. Now What?
5. What’s More?
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B
References
- Röllig, C.; Knop, S.; Bornhäuser, M. Multiple myeloma. Lancet 2015, 385, 2197–2208. [Google Scholar] [CrossRef]
- Kazandjian, D. Multiple myeloma epidemiology and survival: A unique malignancy. Semin. Oncol. 2016, 43, 676–681. [Google Scholar] [CrossRef] [Green Version]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Bladé, J.; Merlini, G.; Mateos, M.-V.; Rajkumar, S.V.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Nandakumar, B.; Binder, M.; Dispenzieri, A.; Kapoor, P.; Buadi, F.; Gertz, M.A.; Lacy, M.; Dingli, D.; Hwa, L.; Leung, N.; et al. Continued improvement in survival in multiple myeloma (MM) including high-risk patients. J. Clin. Oncol. 2019, 37, 8039. [Google Scholar] [CrossRef]
- Blimark, C.H.; Turesson, I.; Genell, A.; Ahlberg, L.; Björkstrand, B.; Carlson, K.; Forsberg, K.; Juliusson, G.; Linder, O.; Mellqvist, U.-H.; et al. Outcome and survival of myeloma patients diagnosed 2008–2015. Real-world data on 4904 patients from the Swedish Myeloma Registry. Haematology 2017, 103, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Branagan, A.; Lei, M.; Lou, U.; Raje, N. Current Treatment Strategies for Multiple Myeloma. JCO Oncol. Pr. 2020, 16, 5–14. [Google Scholar] [CrossRef]
- Bailly, C.; Cléry, P.-F.; Faivre-Chauvet, A.; Bourgeois, M.; Guérard, F.; Haddad, F.; Barbet, J.; Chérel, M.; Kraeber-Bodere, F.; Carlier, T.; et al. Immuno-PET for Clinical Theranostic Approaches. Int. J. Mol. Sci. 2016, 18, 57. [Google Scholar] [CrossRef] [Green Version]
- Rolfe, G.; Freshwater, D.; Jasper, M. Critical Reflection for Nursing and the Helping Professions: A User’s Guide; Palgrave: London, UK, 2001; ISBN 978-0-333-77795-4. [Google Scholar]
- Giuliani, N.; Malavasi, F. Editorial: Immunotherapy in Multiple Myeloma. Front. Immunol. 2019, 10, 1945. [Google Scholar] [CrossRef] [Green Version]
- Touzeau, C.; Moreau, P.; Dumontet, C. Monoclonal antibody therapy in multiple myeloma. Leukemia 2017, 31, 1039–1047. [Google Scholar] [CrossRef]
- Mateos, M.-V.; Dimopoulos, M.A.; Cavo, M.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lucio, P.; Nagy, Z.; Kaplan, P.; et al. Daratumumab plus Bortezomib, Melphalan, and Prednisone for Untreated Myeloma. N. Engl. J. Med. 2018, 378, 518–528. [Google Scholar] [CrossRef]
- Moreau, P.; Attal, M.; Hulin, C.; Arnulf, B.; Belhadj, K.; Benboubker, L.; Béné, M.C.; Broijl, A.; Caillon, H.; Caillot, D.; et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): A randomised, open-label, phase 3 study. Lancet 2019, 394, 29–38. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. New Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Mun, Y.C.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef]
- Zamagni, E.; Tacchetti, P.; Pantani, L.; Cavo, M. Anti-CD38 and anti-SLAMF7: The future of myeloma immunotherapy. Expert Rev. Hematol. 2018, 11, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Durie, B.; Palumbo, A.; San-Miguel, J. Monoclonal antibodies in the treatment of multiple myeloma: Current status and future perspectives. Leukemia 2015, 30, 526–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plesner, T.; Krejcik, J. Daratumumab for the Treatment of Multiple Myeloma. Front. Immunol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Afifi, S.; Michael, A.; Lesokhin, A. Immunotherapy. Ann. Pharmacother. 2016, 50, 555–568. [Google Scholar] [CrossRef] [Green Version]
- Van De Donk, N.W.C.J.; Richardson, P.G.; Malavasi, F. CD38 antibodies in multiple myeloma: Back to the future. Blood 2018, 131, 13–29. [Google Scholar] [CrossRef]
- Dostalek, M.; Gardner, I.; Gurbaxani, B.M.; Rose, R.H.; Chetty, M. Pharmacokinetics, Pharmacodynamics and Physiologically-Based Pharmacokinetic Modelling of Monoclonal Antibodies. Clin. Pharm. 2013, 52, 83–124. [Google Scholar] [CrossRef]
- Keizer, R.J.; Huitema, A.D.R.; Schellens, J.H.M.; Beijnen, J.H. Clinical Pharmacokinetics of Therapeutic Monoclonal Antibodies. Clin. Pharm. 2010, 49, 493–507. [Google Scholar] [CrossRef]
- Ahamadi, M.; Freshwater, T.; Prohn, M.; Li, C.H.; De Alwis, D.P.; De Greef, R.; Elassaiss-Schaap, J.; Kondic, A.; Stone, J.A. Model-Based Characterization of the Pharmacokinetics of Pembrolizumab: A Humanized Anti–PD-1 Monoclonal Antibody in Advanced Solid Tumors. CPT Pharmacomet. Syst. Pharmacol. 2016, 6, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, G.; Wang, X.; Agrawal, S.; Gupta, M.; Roy, A.; Feng, Y. Model-Based Population Pharmacokinetic Analysis of Nivolumab in Patients With Solid Tumors. CPT Pharmacomet. Syst. Pharmacol. 2016, 6, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Ogungbenro, K.; Patel, A.; Duncombe, R.; Nuttall, R.; Clark, J.; Lorigan, P.C. Dose Rationalization of Pembrolizumab and Nivolumab Using Pharmacokinetic Modeling and Simulation and Cost Analysis. Clin. Pharmacol. Ther. 2017, 103, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Clemens, P.L.; Yan, X.; Lokhorst, H.M.; Lonial, S.; Losic, N.; Khan, I.; Jansson, R.; Ahmadi, T.; Lantz, K.; Zhou, H.; et al. Pharmacokinetics of Daratumumab Following Intravenous Infusion in Relapsed or Refractory Multiple Myeloma After Prior Proteasome Inhibitor and Immunomodulatory Drug Treatment. Clin. Pharm. 2017, 56, 915–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.S.; Yan, X.; Puchalski, T.; Lonial, S.; Lokhorst, H.M.; Voorhees, P.M.; Plesner, T.; Liu, K.; Khan, I.; Jansson, R.; et al. Clinical Implications of Complex Pharmacokinetics for Daratumumab Dose Regimen in Patients With Relapsed/Refractory Multiple Myeloma. Clin. Pharmacol. Ther. 2017, 101, 721–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van De Donk, N.W.; Usmani, S.Z. CD38 Antibodies in Multiple Myeloma: Mechanisms of Action and Modes of Resistance. Front. Immunol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Mogollón, P.; Díaz-Tejedor, A.; Algarín, E.; Paíno, T.; Garayoa, M.; Ocio, E.M. Biological Background of Resistance to Current Standards of Care in Multiple Myeloma. Cells 2019, 8, 1432. [Google Scholar] [CrossRef] [Green Version]
- Oliva, S.; Troia, R.; D’Agostino, M.; Boccadoro, M.; Gay, F. Promises and Pitfalls in the Use of PD-1/PD-L1 Inhibitors in Multiple Myeloma. Front. Immunol. 2018, 9, 2749. [Google Scholar] [CrossRef]
- Rosenblatt, J.; Avigan, D. Targeting the PD-1/PD-L1 axis in multiple myeloma: A dream or a reality? Blood 2017, 129, 275–279. [Google Scholar] [CrossRef] [Green Version]
- Tamura, H.; Ishibashi, M.; Yamashita, T.; Tanosaki, S.; Okuyama, N.; Kondo, A.; Hyodo, H.; Shinya, E.; Takahashi, H.; Dong, H.; et al. Marrow stromal cells induce B7-H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia 2012, 27, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Jelinek, T.; Paiva, B.; Hajek, R. Update on PD-1/PD-L1 Inhibitors in Multiple Myeloma. Front. Immunol. 2018, 9, 2431. [Google Scholar] [CrossRef] [Green Version]
- Costa, F.; Das, R.; Bailur, J.K.; Dhodapkar, K.; Dhodapkar, M.V. Checkpoint Inhibition in Myeloma: Opportunities and Challenges. Front. Immunol. 2018, 9, 2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paiva, B.; Azpilikueta, A.; Puig, N.; Ocio, E.M.; Sharma, R.; Oyajobi, B.O.; Labiano, S.; San-Segundo, L.; Rodriguez, A.; Aires-Mejía, I.; et al. PD-L1/PD-1 presence in the tumor microenvironment and activity of PD-1 blockade in multiple myeloma. Leukemia 2015, 29, 2110–2113. [Google Scholar] [CrossRef] [PubMed]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribrag, V.; Avigan, D.E.; Green, D.J.; Wise-Draper, T.; Posada, J.G.; Vij, R.; Zhu, Y.; Farooqui, M.Z.H.; Marinello, P.; Siegel, D.S. Phase 1b trial of pembrolizumab monotherapy for relapsed/refractory multiple myeloma: KEYNOTE-013. Br. J. Haematol. 2019, 186, e41–e44. [Google Scholar] [CrossRef] [Green Version]
- Mateos, M.-V.; Orlowski, R.Z.; Ocio, E.M.; Rodríguez-Otero, P.; Reece, N.; Moreau, P.; Munshi, N.; Avigan, D.E.; Siegel, D.S.; Ghori, R.; et al. Pembrolizumab combined with lenalidomide and low-dose dexamethasone for relapsed or refractory multiple myeloma: Phase I KEYNOTE -023 study. Br. J. Haematol. 2019, 186, e117–e121. [Google Scholar] [CrossRef] [Green Version]
- Usmani, S.Z.; Schjesvold, F.; Oriol, A.; Karlin, L.; Cavo, M.; Rifkin, R.M.; Yimer, H.A.; Leblanc, R.; Takezako, N.; McCroskey, R.D.; et al. Pembrolizumab plus lenalidomide and dexamethasone for patients with treatment-naive multiple myeloma (KEYNOTE-185): A randomised, open-label, phase 3 trial. Lancet Haematol. 2019, 6, e448–e458. [Google Scholar] [CrossRef]
- Mateos, M.-V.; Blacklock, H.; Schjesvold, F.; Oriol, A.; Simpson, D.; George, A.; Goldschmidt, H.; LaRocca, A.; Chanan-Khan, A.; Sherbenou, D.; et al. Pembrolizumab plus pomalidomide and dexamethasone for patients with relapsed or refractory multiple myeloma (KEYNOTE-183): A randomised, open-label, phase 3 trial. Lancet Haematol. 2019, 6, e459–e469. [Google Scholar] [CrossRef]
- Costello, C. The future of checkpoint inhibition in multiple myeloma? Lancet Haematol. 2019, 6, e439–e440. [Google Scholar] [CrossRef]
- Zelle-Rieser, C.; Thangavadivel, S.; Biedermann, R.; Brunner, A.; Stoitzner, P.; Willenbacher, E.; Greil, R.; Jöhrer, K. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J. Hematol. Oncol. 2016, 9, 116. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danhof, S.; Strifler, S.; Hose, D.; Kortüm, M.; Bittrich, M.; Hefner, J.; Einsele, H.; Knop, S.; Schreder, M. Clinical and biological characteristics of myeloma patients influence response to elotuzumab combination therapy. J. Cancer Res. Clin. Oncol. 2018, 145, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Jakubowiak, A.; Offidani, M.; Pegourie, B.; De La Rubia, J.; Garderet, L.; Laribi, K.; Bosi, A.; Marasca, R.; Laubach, J.; Mohrbacher, A.; et al. Randomized phase 2 study: Elotuzumab plus bortezomib/dexamethasone vs bortezomib/dexamethasone for relapsed/refractory MM. Blood 2016, 127, 2833–2840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijhof, I.S.; Casneuf, T.; Van Velzen, J.; Van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.; Van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef] [Green Version]
- Pick, M.; Vainstein, V.; Goldschmidt, N.; Lavie, D.; Libster, D.; Gural, A.; Grisariu, S.; Avni, B.; Ben-Yehuda, D.; Gatt, M. Daratumumab resistance is frequent in advanced-stage multiple myeloma patients irrespective of CD38 expression and is related to dismal prognosis. Eur. J. Haematol. 2018, 100, 494–501. [Google Scholar] [CrossRef]
- Kitadate, A.; Kobayashi, H.; Abe, Y.; Narita, K.; Miura, D.; Takeuchi, M.; Matsue, K. CD38 Expression Levels on Myeloma Cells and the Frequency of Circulating CD38-Positive Treg Cells Are Associated with the Response to Daratumumab in Multiple Myeloma. Blood 2018, 132, 1883. [Google Scholar] [CrossRef]
- Viola, D.; Dona, A.; Gunes, E.G.; Troadec, E.; Wu, X.; Branciamore, S.; McDonald, T.; Ghoda, L.; Streatfield, A.; Sanchez, J.F.; et al. Immune Mediated Mechanisms of Resistance to Daratumumab. Blood 2018, 132, 3201. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, E.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Yan, X.; Clemens, P.L.; Puchalski, T.; Lonial, S.; Lokhorst, H.M.; Orlowski, R.Z.; Losic, N.; Khan, I.; Jansson, R.; Ahmadi, T.; et al. Target-Mediated Drug Disposition of Daratumumab Following Intravenous Infusion in Relapsed or Refractory Multiple Myeloma after Prior Proteasome Inhibitors and Immunomodulatory Drugs: A Population Pharmacokinetic Analysis. Blood 2015, 126, 4222. [Google Scholar] [CrossRef]
- Centanni, M.; Moes, D.J.A.R.; Trocóniz, I.F.; Ciccolini, J.; Van Hasselt, J.G.C. Clinical Pharmacokinetics and Pharmacodynamics of Immune Checkpoint Inhibitors. Clin. Pharm. 2019, 58, 835–857. [Google Scholar] [CrossRef] [Green Version]
- Deng, R.; Jin, F.; Prabhu, S.; Iyer, S. Monoclonal antibodies: What are the pharmacokinetic and pharmacodynamic considerations for drug development? Expert Opin. Drug Metab. Toxicol. 2012, 8, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Deslandes, A. Comparative clinical pharmacokinetics of antibody-drug conjugates in first-in-human Phase 1 studies. mAbs 2014, 6, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Bartelink, I.H.; Jones, E.F.; Shahidi-Latham, S.K.; Lee, P.R.E.; Zheng, Y.; Vicini, P.; van ‘t Veer, L.; Wolf, D.; Iagaru, A.; Kroetz, D.L.; et al. Tumor Drug Penetration Measurements Could Be the Neglected Piece of the Personalized Cancer Treatment Puzzle. Clin. Pharmacol. Ther. 2018, 106, 148–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Cerdá, L.; Asín-Prieto, E.; Parra-Guillen, Z.P.; Trocóniz, I.F. The Long Neglected Player: Modeling Tumor Uptake to Guide Optimal Dosing. Clin. Cancer Res. 2018, 24, 3236–3238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasche, L.; Chavan, S.S.; Stephens, O.W.; Patel, P.H.; Tytarenko, R.; Ashby, C.; Bauer, M.A.; Stein, C.; Deshpande, S.; Wardell, C.; et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat. Commun. 2017, 8, 268. [Google Scholar] [CrossRef]
- Rasche, L.; Kortüm, K.M.; Raab, M.S.; Weinhold, N. The Impact of Tumor Heterogeneity on Diagnostics and Novel Therapeutic Strategies in Multiple Myeloma. Int. J. Mol. Sci. 2019, 20, 1248. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.S.; Dimopoulos, M.A.; Sonneveld, P.; Ho, P.J.; Belch, A.; Leiba, M.; Capra, M.; Gomez, D.; Medvedova, E.; Iida, S.; et al. Pharmacokinetics and Exposure–Response Analyses of Daratumumab in Combination Therapy Regimens for Patients with Multiple Myeloma. Adv. Ther. 2018, 35, 1859–1872. [Google Scholar] [CrossRef] [Green Version]
- Chillemi, A.; Quarona, V.; Zito, A.; Morandi, F.; Marimpietri, D.; Cuccioloni, M.; Robert, O.J.; Mark, C.S.; Bolzoni, M.; Toscani, D.; et al. Generation and Characterization of Microvesicles after Daratumumab Interaction with Myeloma Cells. Blood 2015, 126, 1849. [Google Scholar] [CrossRef]
- Krejcik, J.; Frerichs, K.A.; Nijhof, I.S.; Van Kessel, B.; Van Velzen, J.F.; Bloem, A.C.; Broekmans, M.E.; Zweegman, S.; Van Meerloo, J.; Musters, R.J.; et al. Monocytes and Granulocytes Reduce CD38 Expression Levels on Myeloma Cells in Patients Treated with Daratumumab. Clin. Cancer Res. 2017, 23, 7498–7511. [Google Scholar] [CrossRef] [Green Version]
- Zonder, J.A.; Mohrbacher, A.F.; Singhal, S.; Van Rhee, F.; Bensinger, W.I.; Ding, H.; Fry, J.; Afar, D.E.H.; Singhal, A.K. A phase 1, multicenter, open-label, dose escalation study of elotuzumab in patients with advanced multiple myeloma. Blood 2012, 120, 552–559. [Google Scholar] [CrossRef]
- Beckman, R.A.; Von Roemeling, R.; Scott, A.M. Monoclonal antibody dose determination and biodistribution into solid tumors. Ther. Deliv. 2011, 2, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Strik, A.S.; Wang, Y.-M.C.; Ruff, L.E.; Yashar, W.; Messmer, B.T.; Mould, D.R. Individualized Dosing of Therapeutic Monoclonal Antibodies—A Changing Treatment Paradigm? AAPS J. 2018, 20, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, V.A.; Balthasar, J.P. Understanding Inter-Individual Variability in Monoclonal Antibody Disposition. Antibodies 2019, 8, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gormley, N.J.; Pazdur, R. Immunotherapy Combinations in Multiple Myeloma—Known Unknowns. N. Engl. J. Med. 2018, 379, 1791–1795. [Google Scholar] [CrossRef]
- Day, D.; Siu, L.L. Approaches to modernize the combination drug development paradigm. Genome Med. 2016, 8, 115. [Google Scholar] [CrossRef] [Green Version]
- Hofmarcher, T.; Lindgren, P.; Wilking, N.; Jonsson, B. The cost of cancer in Europe 2018. Eur. J. Cancer 2020, 129, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Romero, D. To all involved—we have a problem. Nat. Rev. Clin. Oncol. 2018, 15, 397. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.K.K.; Danta, M.; Day, R.; Ma, D.D.F. Dealing with the spiralling price of medicines: Issues and solutions. Intern. Med. J. 2018, 48, 16–24. [Google Scholar] [CrossRef] [Green Version]
- Salas-Vega, S.; Iliopoulos, O.; Mossialos, E. Assessment of Overall Survival, Quality of Life, and Safety Benefits Associated With New Cancer Medicines. JAMA Oncol. 2017, 3, 382. [Google Scholar] [CrossRef] [Green Version]
- Turner, J.H. Theranostic Outcomes in Clinical Practice of Oncology: What, So What, Now What? What’s More. Cancer Biother. Radiopharm 2019, 34, 135–140. [Google Scholar] [CrossRef]
- Sotelo-Rodríguez, D.C.; Ruíz-Patiño, A.; Ricaurte, L.; Arrieta, O.; Zatarain-Barrón, Z.L.; Cardona, A.F.; Trinca, F.; Infante, P.; Dinis, R.; Inácio, M.; et al. Challenges and shifting paradigms in clinical trials in oncology: The case for immunological and targeted therapies. Ecancermedicalscience 2019, 13, 936. [Google Scholar] [CrossRef] [PubMed]
- Hoering, A.; Durie, B.; Wang, H.; Crowley, J. End points and statistical considerations in immuno-oncology trials: Impact on multiple myeloma. Future Oncol. 2017, 13, 1181–1193. [Google Scholar] [CrossRef] [PubMed]
- Gerwing, M.; Herrmann, K.; Helfen, A.; Schliemann, C.; Berdel, W.E.; Eisenblätter, M.; Wildgruber, M. The beginning of the end for conventional RECIST—novel therapies require novel imaging approaches. Nat. Rev. Clin. Oncol. 2019, 16, 442–458. [Google Scholar] [CrossRef] [PubMed]
- Nishino, M.; Ramaiya, N.H.; Hatabu, H.; Hodi, F.S. Monitoring immune-checkpoint blockade: Response evaluation and biomarker development. Nat. Rev. Clin. Oncol. 2017, 14, 655–668. [Google Scholar] [CrossRef]
- Romano, A.; Palumbo, G.A.; Parrinello, N.L.; Conticello, C.; Martello, M.; Terragna, C. Minimal Residual Disease Assessment Within the Bone Marrow of Multiple Myeloma: A Review of Caveats, Clinical Significance and Future Perspectives. Front. Oncol. 2019, 9, 699. [Google Scholar] [CrossRef]
- Moreau, P.; Zamagni, E. MRD in multiple myeloma: More questions than answers? Blood Cancer J. 2017, 7, 639. [Google Scholar] [CrossRef]
- Kumar, S.K.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.C.; Lonial, S.; Bladé, J.; Mateos, M.-V.; et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016, 17, e328–e346. [Google Scholar] [CrossRef]
- Van Dongen, G.; Visser, G.W.; Hooge, M.N.L.-D.; De Vries, E.G.; Perk, L.R. Immuno-PET: A Navigator in Monoclonal Antibody Development and Applications. Oncology 2007, 12, 1379–1389. [Google Scholar] [CrossRef] [Green Version]
- Guang, M.H.Z.; McCann, A.; Bianchi, G.; Zhang, L.; Dowling, P.; Bazou, D.; O’Gorman, P.; Anderson, K.C. Overcoming multiple myeloma drug resistance in the era of cancer ‘omics’. Leuk. Lymphoma 2017, 59, 542–561. [Google Scholar] [CrossRef]
- Kraeber-Bodere, F.; Bailly, C.; Chérel, M.; Chatal, J.-F. ImmunoPET to help stratify patients for targeted therapies and to improve drug development. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 2166–2168. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Rosenkrans, Z.T.; Liu, J.; Huang, G.; Luo, Q.-Y.; Cai, W. ImmunoPET: Concept, Design, and Applications. Chem. Rev. 2020, 120, 3787–3851. [Google Scholar] [CrossRef] [PubMed]
- McKnight, B.N.; Viola, N. 89Zr-ImmunoPET companion diagnostics and their impact in clinical drug development. J. Label. Compd. Radiopharm. 2018, 61, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Ghai, A.; Maji, D.; Cho, N.; Chanswangphuwana, C.; Rettig, M.; Shen, D.; DiPersio, J.; Akers, W.; Dehdashti, F.; Achilefu, S.; et al. Preclinical Development of CD38-Targeted [89Zr]Zr-DFO-Daratumumab for Imaging Multiple Myeloma. J. Nucl. Med. 2017, 59, 216–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caserta, E.; Chea, J.; Minnix, M.; Poku, E.K.; Viola, D.; Vonderfecht, S.; Yazaki, P.; Crow, D.; Khalife, J.; Sanchez, J.F.; et al. Copper 64–labeled daratumumab as a PET/CT imaging tracer for multiple myeloma. Blood 2018, 131, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Ulaner, G.; Sobol, N.; O’Donoghue, J.; Burnazi, E.; Staton, K.; Weber, W.; Lyashchenko, S.; Lewis, J.; Landgren, C.O. Preclinical development and First-in-human imaging of 89Zr-Daratumumab for CD38 targeted imaging of myeloma. J. Nucl. Med. 2019, 60, 203. [Google Scholar]
- Pandit-Taskar, N. Functional Imaging Methods for Assessment of Minimal Residual Disease in Multiple Myeloma: Current Status and Novel ImmunoPET Based Methods. Semin. Hematol. 2018, 55, 22–32. [Google Scholar] [CrossRef]
- Ulaner, G.A.; Sobol, N.B.; O’Donoghue, J.A.; Kirov, A.S.; Riedl, C.C.; Min, R.; Smith, E.; Carter, L.M.; Lyashchenko, S.K.; Lewis, J.S.; et al. CD38-targeted Immuno-PET of Multiple Myeloma: From Xenograft Models to First-in-Human Imaging. Radiology 2020, 295, 606–615. [Google Scholar] [CrossRef]
- Bailly, C.; Bodet-Milin, C.; Bourgeois, M.; Gouard, S.; Ansquer, C.; Barbaud, M.; Sébille, J.-C.; Chérel, M.; Kraeber-Bodere, F.; Carlier, T. Exploring Tumor Heterogeneity Using PET Imaging: The Big Picture. Cancers 2019, 11, 1282. [Google Scholar] [CrossRef] [Green Version]
- Bensch, F.; Van Der Veen, E.L.; Hooge, M.N.L.-D.; Jorritsma-Smit, A.; Boellaard, R.; Kok, I.C.; Oosting, S.F.; Schröder, C.P.; Hiltermann, T.J.N.; Van Der Wekken, A.J.; et al. 89Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat. Med. 2018, 24, 1852–1858. [Google Scholar] [CrossRef]
- Niemeijer, A.N.; Leung, D.; Huisman, M.C.; Bahce, I.; Hoekstra, O.S.; Van Dongen, G.A.M.S.; Boellaard, R.; Du, S.; Hayes, W.; Smith, R.; et al. Whole body PD-1 and PD-L1 positron emission tomography in patients with non-small-cell lung cancer. Nat. Commun. 2018, 9, 4664. [Google Scholar] [CrossRef]
- Dijkers, E.C.; Munnink, T.H.O.; Kosterink, J.G.; Brouwers, A.H.; Jager, P.L.; De Jong, J.R.; Van Dongen, G.A.; Schröder, C.P.; Hooge, M.N.L.-D.; De Vries, E.G. Biodistribution of 89Zr-trastuzumab and PET Imaging of HER2-Positive Lesions in Patients With Metastatic Breast Cancer. Clin. Pharmacol. Ther. 2010, 87, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Muylle, K.; Flamen, P.; Vugts, D.J.; Guiot, T.; Ghanem, G.; Meuleman, N.; Bourgeois, P.; Vanderlinden, B.; Van Dongen, G.A.M.S.; Everaert, H.; et al. Tumour targeting and radiation dose of radioimmunotherapy with (90)Y-rituximab in CD20+ B-cell lymphoma as predicted by (89)Zr-rituximab immuno-PET: Impact of preloading with unlabelled rituximab. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1304–1314. [Google Scholar] [CrossRef] [PubMed]
- Oordt, C.W.M.-V.D.H.V.; McGeoch, A.; Bergstrom, M.; McSherry, I.; Smith, D.A.; Cleveland, M.; Al-Azzam, W.; Chen, L.; Verheul, H.; Hoekstra, O.S.; et al. Immuno-PET Imaging to Assess Target Engagement: Experience from 89Zr-Anti-HER3 mAb (GSK2849330) in Patients with Solid Tumors. J. Nucl. Med. 2019, 60, 902–909. [Google Scholar] [CrossRef] [Green Version]
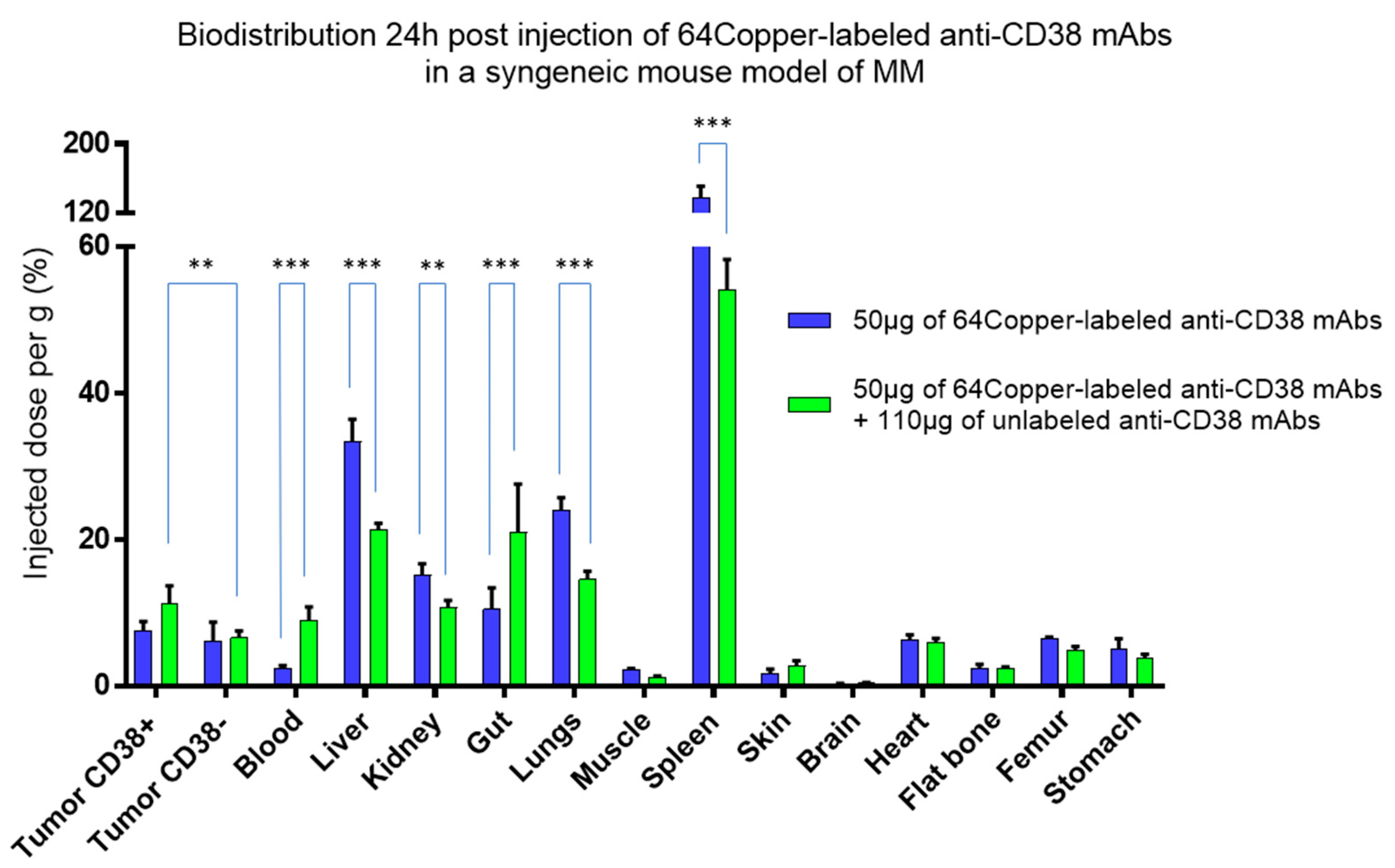
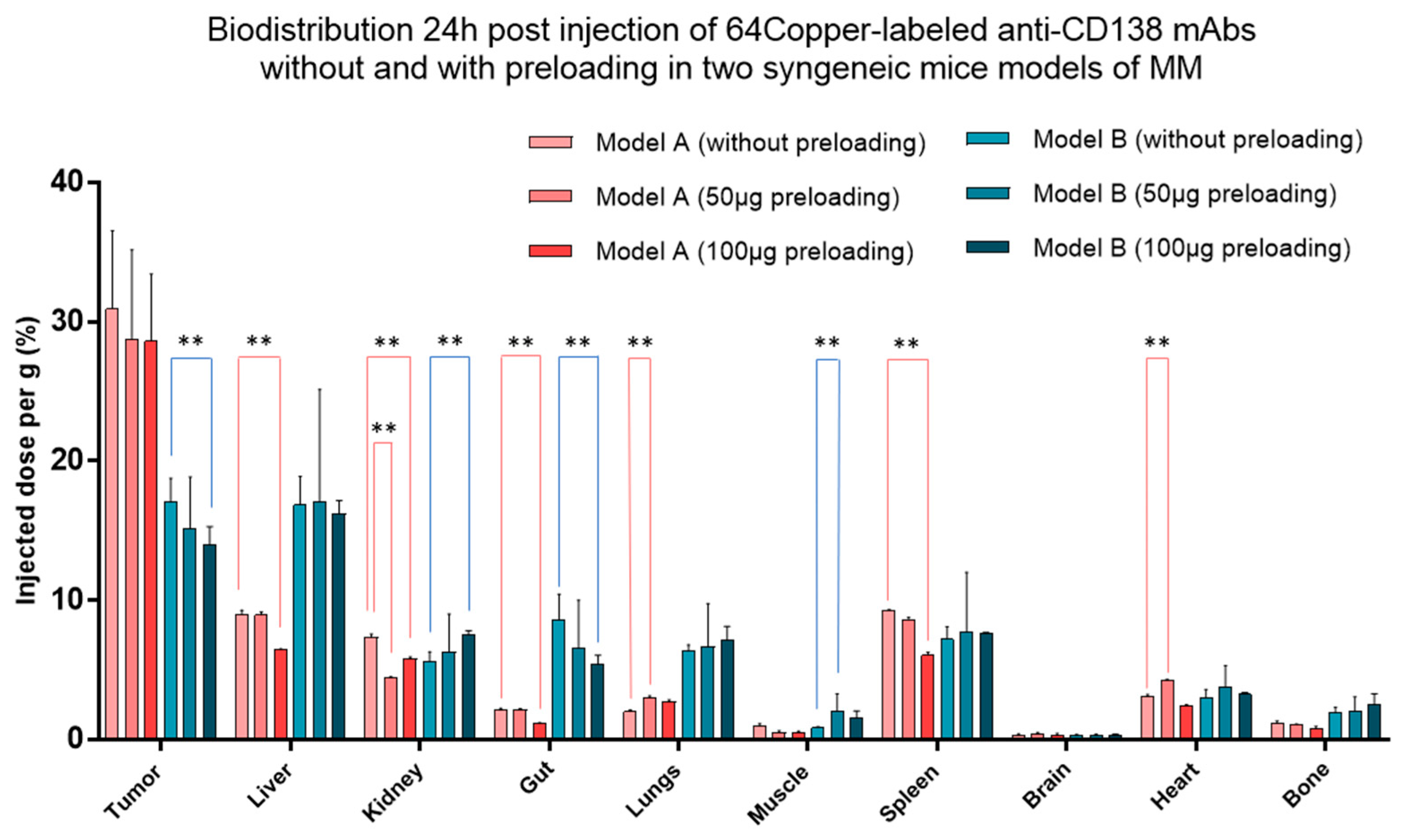
- Bailly, C.; Gouard, S.; Guérard, F.; Chalopin, B.; Carlier, T.; Faivre-Chauvet, A.; Saëc, P.R.-L.; Bourgeois, M.; Chouin, N.; Rbah-Vidal, L.; et al. What is the Best Radionuclide for Immuno-PET of Multiple Myeloma? A Comparison Study Between 89Zr- and 64Cu-Labeled Anti-CD138 in a Preclinical Syngeneic Model. Int. J. Mol. Sci. 2019, 20, 2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.K.; Chow, A.; Monette, S.; Vivier, D.; Pourat, J.; Edwards, K.J.; Dilling, T.R.; Abdel-Atti, D.; Zeglis, B.M.; Poirier, J.T.; et al. Fc-Mediated Anomalous Biodistribution of Therapeutic Antibodies in Immunodeficient Mouse Models. Cancer Res. 2018, 78, 1820–1832. [Google Scholar] [CrossRef] [Green Version]
- Paton-Hough, J.; Chantry, A.; Lawson, M.A. A review of current murine models of multiple myeloma used to assess the efficacy of therapeutic agents on tumour growth and bone disease. Bone 2015, 77, 57–68. [Google Scholar] [CrossRef]
- Sanderson, R.D.; Yang, Y. Syndecan-1: A dynamic regulator of the myeloma microenvironment. Clin. Exp. Metastasis 2007, 25, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Cavo, M.; Terpos, E.; Nanni, C.; Moreau, P.; Lentzsch, S.; Zweegman, S.; Hillengass, J.; Engelhardt, M.; Usmani, S.Z.; Vesole, D.H.; et al. Role of 18F-FDG PET/CT in the diagnosis and management of multiple myeloma and other plasma cell disorders: A consensus statement by the International Myeloma Working Group. Lancet Oncol. 2017, 18, e206–e217. [Google Scholar] [CrossRef]
- Green, D.J.; Press, O. Whither radioimmunotherapy: To be or not to be? Cancer Res. 2017, 77, 2191–2196. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, S.N.F.; Visser, O.J.; Vosjan, M.J.W.D.; Van Lingen, A.; Hoekstra, O.S.; Zijlstra, J.M.; Huijgens, P.C.; Van Dongen, G.A.M.S.; Lubberink, M. Biodistribution, radiation dosimetry and scouting of 90Y-ibritumomab tiuxetan therapy in patients with relapsed B-cell non-Hodgkin’s lymphoma using 89Zr-ibritumomab tiuxetan and PET. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 512–520. [Google Scholar] [CrossRef] [Green Version]
- Morschhauser, F.; Radford, J.; Van Hoof, A.; Vitolo, U.; Soubeyran, P.; Tilly, H.; Huijgens, P.C.; Kolstad, A.; D’Amore, F.; Diaz, M.G.; et al. Phase III Trial of Consolidation Therapy With Yttrium-90–Ibritumomab Tiuxetan Compared With No Additional Therapy After First Remission in Advanced Follicular Lymphoma. J. Clin. Oncol. 2008, 26, 5156–5164. [Google Scholar] [CrossRef] [PubMed]
- Witzig, T.E.; Flinn, I.W.; Gordon, L.I.; Emmanouilides, C.; Czuczman, M.S.; Saleh, M.N.; Cripe, L.; Wiseman, G.; Olejnik, T.; Multani, P.S.; et al. Treatment With Ibritumomab Tiuxetan Radioimmunotherapy in Patients With Rituximab-Refractory Follicular Non-Hodgkin’s Lymphoma. J. Clin. Oncol. 2002, 20, 3262–3269. [Google Scholar] [CrossRef] [PubMed]
- Martínez, A.; Martínez-Ramirez, M.; Martínez-Caballero, D.; Beneit, P.; Clavel, J.; Figueroa, G.; Verdú, J. Radioinmunoterapia en el linfoma no Hodgkin, posicionamiento, seguridad y eficacia de 90Y-ibritumomab. Experiencia y seguimiento a los 10 años. Revista Española de Medicina Nuclear e Imagen Molecular 2017, 36, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.W.; Pinto, A.; Linkesch, W.; Lindén, O.; Viardot, A.; Keller, U.; Hess, G.; Lastoria, S.; Lerch, K.; Frigeri, F.; et al. 90Yttrium-Ibritumomab-Tiuxetan as First-Line Treatment for Follicular Lymphoma: 30 Months of Follow-Up Data From an International Multicenter Phase II Clinical Trial. J. Clin. Oncol. 2013, 31, 308–313. [Google Scholar] [CrossRef]
- Hohloch, K. Radioimmunotherapy of lymphoma: An underestimated therapy option. Lancet Haematol. 2017, 4, e6–e7. [Google Scholar] [CrossRef]
- Illidge, T. Radioimmunotherapy of Lymphoma: A Treatment Approach Ahead of Its Time or Past Its Sell-By Date? J. Clin. Oncol. 2010, 28, 2944–2946. [Google Scholar] [CrossRef] [PubMed]
- Dadachova, E. Cancer Therapy with Alpha-Emitters Labeled Peptides. Semin. Nucl. Med. 2010, 40, 204–208. [Google Scholar] [CrossRef]
- Fichou, N.; Gouard, S.; Maurel, C.; Barbet, J.; Ferrer, L.; Morgenstern, A.; Bruchertseifer, F.; Faivre-Chauvet, A.; Bigot-Corbel, E.; Davodeau, F.; et al. Single-Dose Anti-CD138 Radioimmunotherapy: Bismuth-213 is More Efficient than Lutetium-177 for Treatment of Multiple Myeloma in a Preclinical Model. Front. Med. 2015, 2, 553. [Google Scholar] [CrossRef] [Green Version]
- Gouard, S.; Chalopin, B.; Saï-Maurel, C.; Guérard, F.; Navarro, L.; Gestin, J.-F.; Chouin, N.; Haddad, F.; Alliot, C.; Kraeber-Bodéré, F.; et al. Efficacy of Astatine-211 Radioimmunotherapy of Multiple Myeloma Using an Anti-mCD138 Monoclonal Antibody in a Syngeneic Murine Model. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, S166–S167. [Google Scholar]
- Gouard, S.; Pallardy, A.; Gaschet, J.; Faivre-Chauvet, A.; Bruchertseifer, F.; Morgenstern, A.; Maurel, C.; Matous, E.; Kraeber-Bodere, F.; Davodeau, F.; et al. Comparative analysis of multiple myeloma treatment by CD138 antigen targeting with bismuth-213 and Melphalan chemotherapy. Nucl. Med. Boil. 2014, 41, e30–e35. [Google Scholar] [CrossRef]
- Chérel, M.; Gouard, S.; Gaschet, J.; Saï-Maurel, C.; Bruchertseifer, F.; Morgenstern, A.; Bourgeois, M.; Gestin, J.-F.; Kraeber-Bodere, F.; Barbet, J.; et al. 213Bi Radioimmunotherapy with an Anti-mCD138 Monoclonal Antibody in a Murine Model of Multiple Myeloma. J. Nucl. Med. 2013, 54, 1597–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailly, C.; Gouard, S.; Lacombe, M.; Saec, P.R.-L.; Chalopin, B.; Bourgeois, M.; Chouin, N.; Tripier, R.; Halime, Z.; Haddad, F.; et al. Comparison of Immuno-PET of CD138 and PET imaging with 64CuCl2 and 18F-FDG in a preclinical syngeneic model of multiple myeloma. Oncotarget 2018, 9, 9061–9072. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bailly, C.; Chalopin, B.; Gouard, S.; Carlier, T.; Remaud-Le Saëc, P.; Marionneau-Lambot, S.; Moreau, P.; Touzeau, C.; Kraeber-Bodere, F.; Bodet-Milin, C.; et al. ImmunoPET in Multiple Myeloma—What? So What? Now What? Cancers 2020, 12, 1467. https://doi.org/10.3390/cancers12061467
Bailly C, Chalopin B, Gouard S, Carlier T, Remaud-Le Saëc P, Marionneau-Lambot S, Moreau P, Touzeau C, Kraeber-Bodere F, Bodet-Milin C, et al. ImmunoPET in Multiple Myeloma—What? So What? Now What? Cancers. 2020; 12(6):1467. https://doi.org/10.3390/cancers12061467
Chicago/Turabian StyleBailly, Clément, Benjamin Chalopin, Sébastien Gouard, Thomas Carlier, Patricia Remaud-Le Saëc, Séverine Marionneau-Lambot, Philippe Moreau, Cyrille Touzeau, Françoise Kraeber-Bodere, Caroline Bodet-Milin, and et al. 2020. "ImmunoPET in Multiple Myeloma—What? So What? Now What?" Cancers 12, no. 6: 1467. https://doi.org/10.3390/cancers12061467
APA StyleBailly, C., Chalopin, B., Gouard, S., Carlier, T., Remaud-Le Saëc, P., Marionneau-Lambot, S., Moreau, P., Touzeau, C., Kraeber-Bodere, F., Bodet-Milin, C., & Chérel, M. (2020). ImmunoPET in Multiple Myeloma—What? So What? Now What? Cancers, 12(6), 1467. https://doi.org/10.3390/cancers12061467