Detection of Colorectal Cancer and Advanced Adenoma by Liquid Biopsy (Decalib Study): The ddPCR Challenge

 ,
,
Abstract
:1. Introduction
2. Results
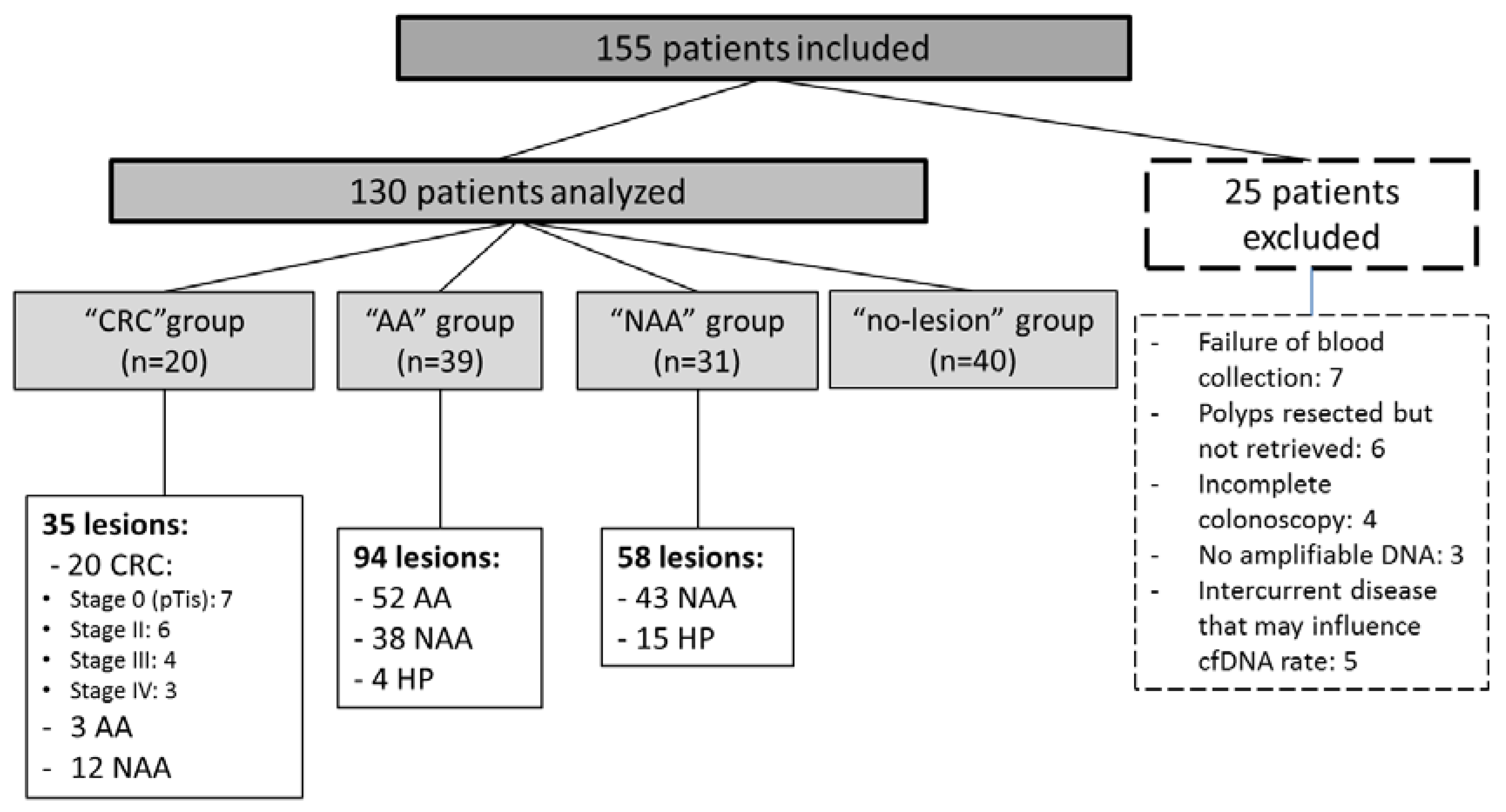
2.1. Study Population
2.2. Molecular Status of Colorectal Lesions
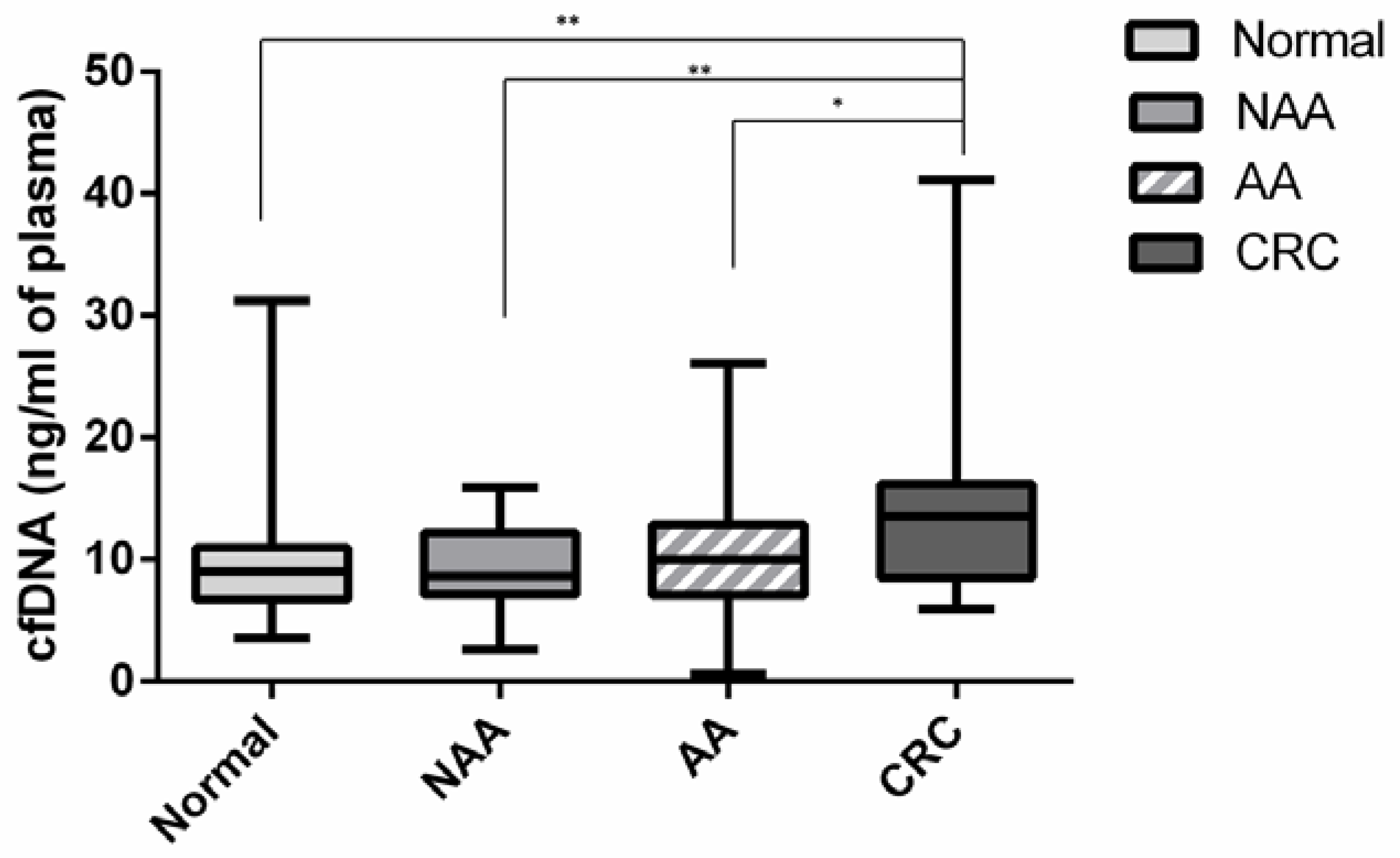
2.3. Circulating Cell Free DNA
2.4. Circulating Tumor DNA
3. Discussion
4. Materials and Methods
4.1. Patient Selection
4.2. Pathological Results
4.3. Blood Samples, Colorectal Lesions and DNA Extraction
4.4. Molecular Analyses
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lieberman, D.A.; Rex, D.K.; Winawer, S.J.; Giardiello, F.M.; Johnson, D.A.; Levin, T.R. Guidelines for Colonoscopy Surveillance After Screening and Polypectomy: A Consensus Update by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 2012, 143, 844–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denis, B.; Guittet, L. Dépistage du cancer colorectal par test immunologique quantitatif de recherche de sang occulte dans les selles: Une révolution? Hépato-Gastro. Oncol. Dig. 2015, 22, 119–129. [Google Scholar] [CrossRef]
- Park, D.I.; Ryu, S.; Kim, Y.-H.; Lee, S.-H.; Lee, C.K.; Eun, C.S.; Han, D.S. Comparison of Guaiac-Based and Quantitative Immunochemical Fecal Occult Blood Testing in a Population at Average Risk Undergoing Colorectal Cancer Screening. Am. J. Gastroenterol. 2010, 105, 2017–2025. [Google Scholar] [CrossRef] [PubMed]
- Altobelli, E.; Lattanzi, A.; Paduano, R.; Varassi, G.; di Orio, F. Colorectal cancer prevention in Europe: Burden of disease and status of screening programs. Prev. Med. 2014, 62, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Vitellius, C.; Laly, M.; Banaszuk, A.-S.; Deherce, I.; Cornet, N.; Bertrais, S.; Saulnier, P.; Caroli-Bosc, F.-X. Contribution of the OC Sensor® immunoassay in comparison to the Hemoccult II® guaiac-test in organized colorectal cancer screening. Eur. J. Epidemiol. 2018. [Google Scholar] [CrossRef]
- van Dam, L.; Korfage, I.J.; Kuipers, E.J.; Hol, L.; van Roon, A.H.C.; Reijerink, J.C.I.Y.; van Ballegooijen, M.; van Leerdam, M.E. What influences the decision to participate in colorectal cancer screening with faecal occult blood testing and sigmoidoscopy? Eur. J. Cancer Oxf. Engl. 1990 2013, 49, 2321–2330. [Google Scholar] [CrossRef]
- Piton, N.; Lonchamp, E.; Nowak, F.; Sabourin, J.-C. Real life distribution of KRAS and NRAS mutations in metastatic colorectal carcinoma from French routine genotyping. Cancer Epidemiol. Prev. Biomark. 2015, 24, 1416–1418. [Google Scholar] [CrossRef] [Green Version]
- Vaughn, C.P.; Zobell, S.D.; Furtado, L.V.; Baker, C.L.; Samowitz, W.S. Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes. Chromosomes Cancer 2011, 50, 307–312. [Google Scholar] [CrossRef]
- Zauber, P.; Marotta, S.; Sabbath-Solitare, M. KRAS gene mutations are more common in colorectal villous adenomas and in situ carcinomas than in carcinomas. Int. J. Mol. Epidemiol. Genet. 2013, 4, 1–10. [Google Scholar]
- Jass, J.R.; Baker, K.; Zlobec, I.; Higuchi, T.; Barker, M.; Buchanan, D.; Young, J. Advanced colorectal polyps with the molecular and morphological features of serrated polyps and adenomas: Concept of a “fusion” pathway to colorectal cancer. Histopathology 2006, 49, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.-M.; Mitchell, J.M.; Sepulveda, J.L.; Sepulveda, A.R. Molecular and histologic considerations in the assessment of serrated polyps. Arch. Pathol. Lab. Med. 2015, 139, 730–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Guleria, R.; Singh, V.; Bharti, A.C.; Mohan, A.; Das, B.C. Efficacy of circulating plasma DNA as a diagnostic tool for advanced non-small cell lung cancer and its predictive utility for survival and response to chemotherapy. Lung Cancer 2010, 70, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Hao, T.B.; Shi, W.; Shen, X.J.; Qi, J.; Wu, X.H.; Wu, Y.; Tang, Y.Y.; Ju, S.Q. Circulating cell-free DNA in serum as a biomarker for diagnosis and prognostic prediction of colorectal cancer. Br. J. Cancer 2014, 111, 1482–1489. [Google Scholar] [CrossRef] [Green Version]
- Lecomte, T.; Ceze, N.; Dorval, E.; Laurent-Puig, P. Circulating free tumor DNA and colorectal cancer. Gastroenterol. Clin. Biol. 2010, 34, 662–681. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrone, F.; Lampis, A.; Bertan, C.; Verderio, P.; Ciniselli, C.M.; Pizzamiglio, S.; Frattini, M.; Nucifora, M.; Molinari, F.; Gallino, G.; et al. Circulating free DNA in a screening program for early colorectal cancer detection. Tumori 2014, 100, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Gormally, E.; Caboux, E.; Vineis, P.; Hainaut, P. Circulating free DNA in plasma or serum as biomarker of carcinogenesis: Practical aspects and biological significance. Mutat. Res. 2007, 635, 105–117. [Google Scholar] [CrossRef]
- Hudecova, I. Digital PCR analysis of circulating nucleic acids. Clin. Biochem. 2015, 48, 948–956. [Google Scholar] [CrossRef]
- Pekin, D.; Skhiri, Y.; Baret, J.-C.; Le Corre, D.; Mazutis, L.; Salem, C.B.; Millot, F.; El Harrak, A.; Hutchison, J.B.; Larson, J.W.; et al. Quantitative and sensitive detection of rare mutations using droplet-based microfluidics. Lab. Chip 2011, 11, 2156–2166. [Google Scholar] [CrossRef]
- Youden, W.J. Index for rating diagnostic tests. Cancer 1950, 3, 32–35. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Stoehlmacher, J.; Pantel, K.; Goekkurt, E. Detection and monitoring of cell-free DNA in blood of patients with colorectal cancer. Ann. N. Y. Acad. Sci. 2008, 1137, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Frattini, M.; Gallino, G.; Signoroni, S.; Balestra, D.; Battaglia, L.; Sozzi, G.; Leo, E.; Pilotti, S.; Pierotti, M.A. Quantitative analysis of plasma DNA in colorectal cancer patients: A novel prognostic tool. Ann. N. Y. Acad. Sci. 2006, 1075, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Frattini, M.; Gallino, G.; Signoroni, S.; Balestra, D.; Lusa, L.; Battaglia, L.; Sozzi, G.; Bertario, L.; Leo, E.; Pilotti, S.; et al. Quantitative and qualitative characterization of plasma DNA identifies primary and recurrent colorectal cancer. Cancer Lett. 2008, 263, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Boni, L.; Cassinotti, E.; Canziani, M.; Dionigi, G.; Rovera, F.; Dionigi, R. Free circulating DNA as possible tumour marker in colorectal cancer. Surg. Oncol. 2007, 16 (Suppl. 1), S29–S31. [Google Scholar] [CrossRef]
- Lo, Y.M.; Rainer, T.H.; Chan, L.Y.; Hjelm, N.M.; Cocks, R.A. Plasma DNA as a prognostic marker in trauma patients. Clin. Chem. 2000, 46, 319–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehl, F.; Li, M.; Dressman, D.; He, Y.; Shen, D.; Szabo, S.; Diaz, L.A.; Goodman, S.N.; David, K.A.; Juhl, H.; et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 16368–16373. [Google Scholar] [CrossRef] [Green Version]
- Kopreski, M.S.; Benko, F.A.; Borys, D.J.; Khan, A.; McGarrity, T.J.; Gocke, C.D. Somatic mutation screening: Identification of individuals harboring K-ras mutations with the use of plasma DNA. J. Natl. Cancer Inst. 2000, 92, 918–923. [Google Scholar] [CrossRef] [Green Version]
- Lecomte, T.; Berger, A.; Zinzindohoué, F.; Micard, S.; Landi, B.; Blons, H.; Beaune, P.; Cugnenc, P.-H.; Laurent-Puig, P. Detection of free-circulating tumor-associated DNA in plasma of colorectal cancer patients and its association with prognosis. Int. J. Cancer 2002, 100, 542–548. [Google Scholar] [CrossRef]
- Lilleberg, S.L.; Durocher, J.; Sanders, C.; Walters, K.; Culver, K. High sensitivity scanning of colorectal tumors and matched plasma DNA for mutations in APC, TP53, K-RAS, and BRAF genes with a novel DHPLC fluorescence detection platform. Ann. N. Y. Acad. Sci. 2004, 1022, 250–256. [Google Scholar] [CrossRef]
- Church, T.R.; Wandell, M.; Lofton-Day, C.; Mongin, S.J.; Burger, M.; Payne, S.R.; Castaños-Vélez, E.; Blumenstein, B.A.; Rösch, T.; Osborn, N.; et al. Prospective evaluation of methylated SEPT9 in plasma for detection of asymptomatic colorectal cancer. Gut 2014, 63, 317–325. [Google Scholar] [CrossRef] [Green Version]
- Danese, E.; Minicozzi, A.M.; Benati, M.; Montagnana, M.; Paviati, E.; Salvagno, G.L.; Lima-Oliveira, G.; Gusella, M.; Pasini, F.; Lippi, G.; et al. Comparison of genetic and epigenetic alterations of primary tumors and matched plasma samples in patients with colorectal cancer. PLoS ONE 2015, 10, e0126417. [Google Scholar] [CrossRef] [PubMed]
- Mansukhani, S.; Barber, L.J.; Kleftogiannis, D.; Moorcraft, S.Y.; Davidson, M.; Woolston, A.; Proszek, P.Z.; Griffiths, B.; Fenwick, K.; Herman, B.; et al. Ultra-Sensitive Mutation Detection and Genome-Wide DNA Copy Number Reconstruction by Error-Corrected Circulating Tumor DNA Sequencing. Clin. Chem. 2018, 64, 1626–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547. [Google Scholar] [CrossRef] [PubMed]
- Navarro, M.; Nicolas, A.; Ferrandez, A.; Lanas, A. Colorectal cancer population screening programs worldwide in 2016: An update. World J. Gastroenterol. 2017, 23, 3632–3642. [Google Scholar] [CrossRef]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-Throughput Droplet Digital PCR System for Absolute Quantitation of DNA Copy Number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef]
- Cortes, U.; Guilloteau, K.; Rouvreau, M.; Archaimbault, C.; Villalva, C.; Karayan-Tapon, L. Development of pyrosequencing methods for the rapid detection of RAS mutations in clinical samples. Exp. Mol. Pathol. 2015, 99, 207–211. [Google Scholar] [CrossRef]
- Mas, L.; Bachet, J.-B.; Taly, V.; Bouché, O.; Taieb, J.; Cohen, R.; Meurisse, A.; Normand, C.; Gornet, J.-M.; Artru, P.; et al. BRAF Mutation Status in Circulating Tumor DNA from Patients with Metastatic Colorectal Cancer: Extended Mutation Analysis from the AGEO RASANC Study. Cancers 2019, 11, 988. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Molecular Status | Stage | |||||
|---|---|---|---|---|---|---|
| 0 | I | II | III | IV | Total (%) | |
| KRAS mutation | 4 | - | 3 | 1 | 1 | 9 (45) |
| BRAF mutation | - | - | 1 | 2 | - | 3 (15) |
| KRAS and BRAF WT | 3 | - | 2 | 1 | 2 | 8 (40) |
| Total (%) | 7 (35) | - | 6 (30) | 4 (20) | 3 (15) | 20 (100) |
| Molecular Status | Pathologic Subtype | |||||
|---|---|---|---|---|---|---|
| TALG ≥ 1 cm | TAHG | TVALG | TVAHG | SSA/p ≥ 1 cm or with dysplasia | Total (%) | |
| KRAS mutation | 5 | 4 | 8 | 1 | - | 18 (34.6) |
| BRAF mutation | - | - | - | - | 9 | 9 (17.3) |
| KRAS and BRAF WT | 13 | 1 | 9 | 1 | 1 | 25 (48.1) |
| Total (%) | 18 (34.6) | 5 (9.8) | 17 (32.6) | 2 (3.8) | 10 (19.2) | 52 (100) |
| Molecular Status | Pathologic Subtype | |||
|---|---|---|---|---|
| TALG < 1 cm | SSA/p < 1 cm with no dysplasia | HP | Total (%) | |
| KRAS mutation | 3 | - | 5 | 8 (13.8) |
| BRAF mutation | - | 2 | 10 | 12 (22.4) |
| KRAS and BRAF WT | 38 | - | - | 38 (67.2) |
| Total (%) | 41 (70.7) | 2 (3.4) | 15 (25.9) | 58 (100) |
| Tumor Stage | pTNM * | Vascular Emboli | Mutation Identified in Primary Tumor | cfDNA (ng/mL) | Fraction of KRAS-Mutated ctDNA (%) | Fraction of BRAF-Mutated ctDNA (%) |
|---|---|---|---|---|---|---|
| 0 | pTis | - | KRASG12C | 12.21 | 0 | 0 |
| pTis | - | WT | 12.61 | 0 | 0 | |
| pTis | - | KRASG12D | 14.30 | 0.09 | 0 | |
| pTis | - | KRASG13D | 5.92 | 0 | 0 | |
| pTis | - | WT | 8.69 | 0 | 0 | |
| pTis | - | WT | 16.02 | 0 | 0 | |
| pTis | - | KRASG12V | 8.48 | 0 | 0 | |
| II | pT3 | - | BRAFV600E | 15.09 | 0 | 0.58 |
| pT3 | + | KRASG13D | 9.87 | 0.25 | 0 | |
| pT3 | + | KRASG12D | 25.58 | 0.08 | 0 | |
| pT3 | - | KRASG12D | 16.17 | 0.15 | 0 | |
| pT3 | - | WT | 29.04 | 0 | 0 | |
| pT3 | + | WT | 8.33 | 0 | 0 | |
| III | pT4aN1b | - | BRAFV600E | 18.07 | 0 | 0.06 |
| pT3N1b | + | BRAFV600E | 16.17 | 0 | 0.16 | |
| cT3N1M0 | + | WT | 6.06 | 0 | 0 | |
| cT3N2M0 | + | KRASG13D | 12.76 | 0.59 | 0 | |
| IV | cTxNxM1 | + | KRASG12D | 14.66 | 8.6 | 0 |
| cTxNxM1 | + | WT | 41.14 | 0 | 0 | |
| pT2N0M1 | + | WT | 6.89 | 0 | 0 |
| Colorectal Lesion | Decalib All Colorectal Lesions Sensitivity/Specificity | Decalib KRAS- or BRAF- Mutated Colorectal Lesions Sensitivity/Specificity | Immunochemical Fecal Occult Blood Test (OC Sensor®) * Sensitivity/Specificity |
|---|---|---|---|
| CRC | 45.0%/100% | 75.0%/100% | 65–85%/90–95% |
| AA | 2.6%/100% | 4.3%/100% | 25–35%/90–95% |
| CRC plus AA | 16.9%/100% | 28.6%/100% | 33–48%/90–95% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Junca, A.; Tachon, G.; Evrard, C.; Villalva, C.; Frouin, E.; Karayan-Tapon, L.; Tougeron, D. Detection of Colorectal Cancer and Advanced Adenoma by Liquid Biopsy (Decalib Study): The ddPCR Challenge. Cancers 2020, 12, 1482. https://doi.org/10.3390/cancers12061482
Junca A, Tachon G, Evrard C, Villalva C, Frouin E, Karayan-Tapon L, Tougeron D. Detection of Colorectal Cancer and Advanced Adenoma by Liquid Biopsy (Decalib Study): The ddPCR Challenge. Cancers. 2020; 12(6):1482. https://doi.org/10.3390/cancers12061482
Chicago/Turabian StyleJunca, Audelaure, Gaëlle Tachon, Camille Evrard, Claire Villalva, Eric Frouin, Lucie Karayan-Tapon, and David Tougeron. 2020. "Detection of Colorectal Cancer and Advanced Adenoma by Liquid Biopsy (Decalib Study): The ddPCR Challenge" Cancers 12, no. 6: 1482. https://doi.org/10.3390/cancers12061482
APA StyleJunca, A., Tachon, G., Evrard, C., Villalva, C., Frouin, E., Karayan-Tapon, L., & Tougeron, D. (2020). Detection of Colorectal Cancer and Advanced Adenoma by Liquid Biopsy (Decalib Study): The ddPCR Challenge. Cancers, 12(6), 1482. https://doi.org/10.3390/cancers12061482