Only Hematopoietic Stem and Progenitor Cells from Cord Blood Are Susceptible to Malignant Transformation by MLL-AF4 Translocations

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
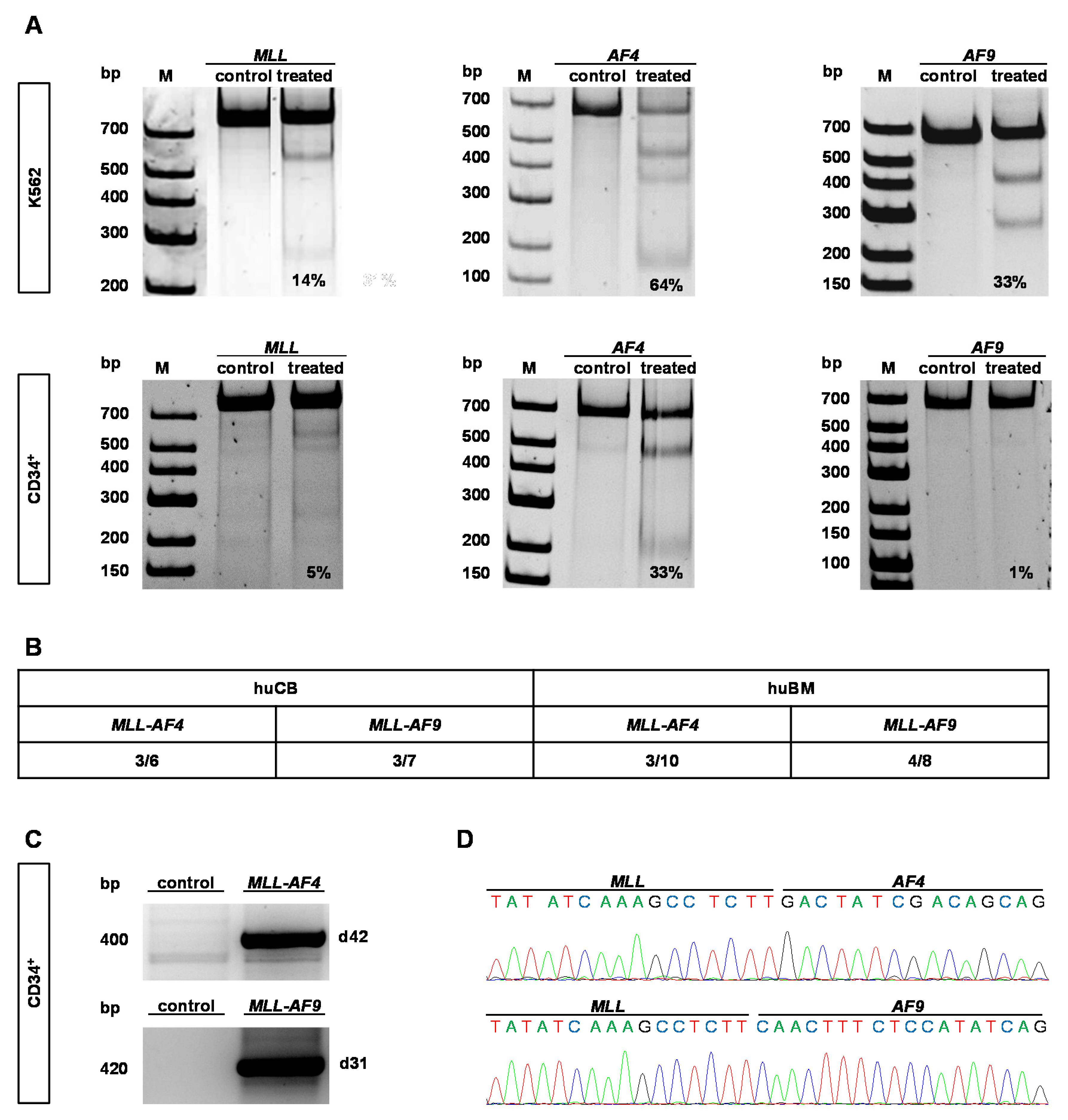
2.1. CRISPR/Cas9 Demonstrates High Cutting Efficiencies and Induces t(9;11) and t(4;11) Chromosomal Translocations in Human HSPCs Derived from huBM
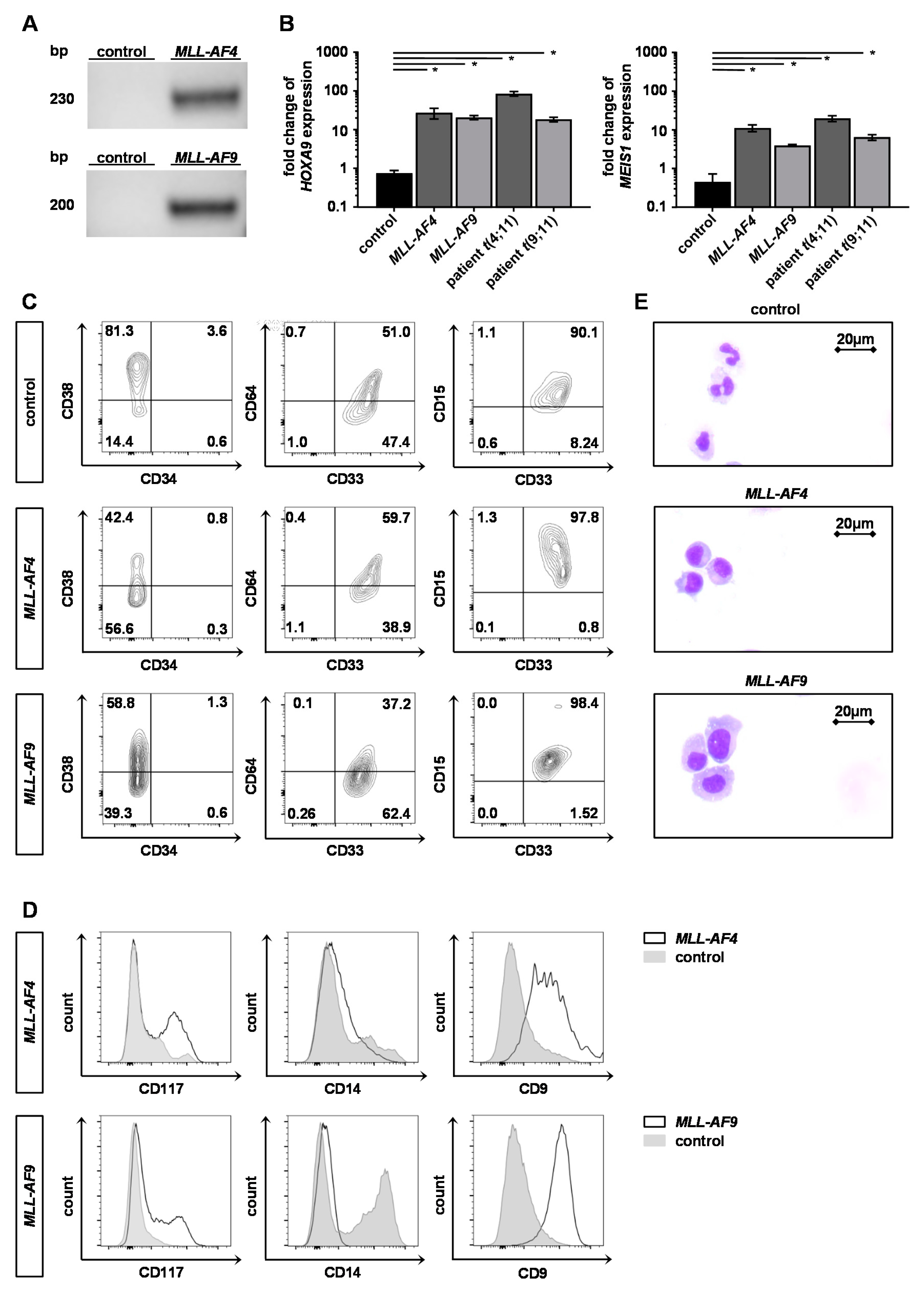
2.2. Engineered Adult KMT2Ar Cells Are Characterized by KMT2Ar-Typical Gene Expression, Phenotype and Morphology
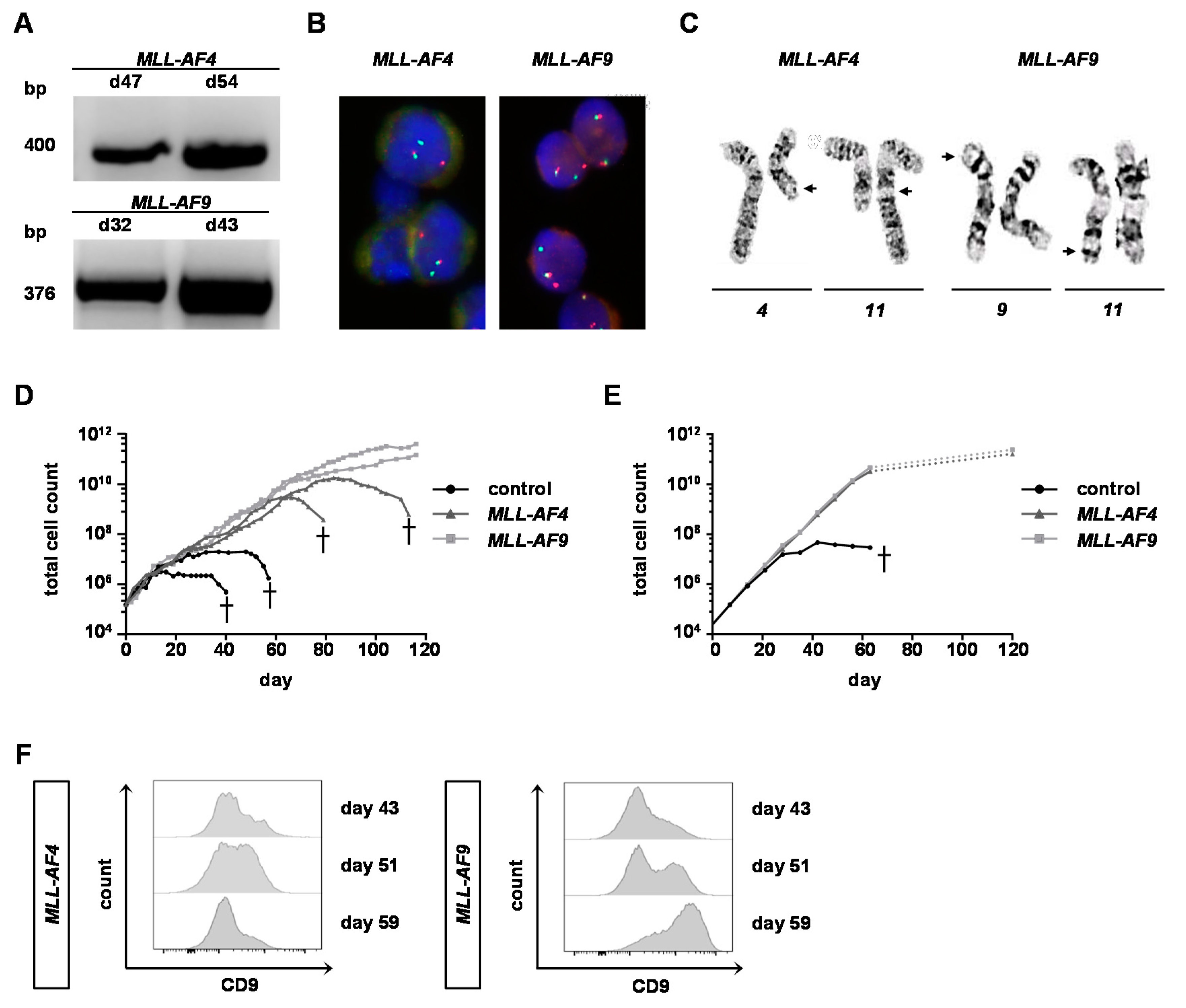
2.3. MLL-AF9 Can Immortalize Neonatal and Adult Cells, Whereas MLL-AF4 Only Immortalizes Neonatal Cells
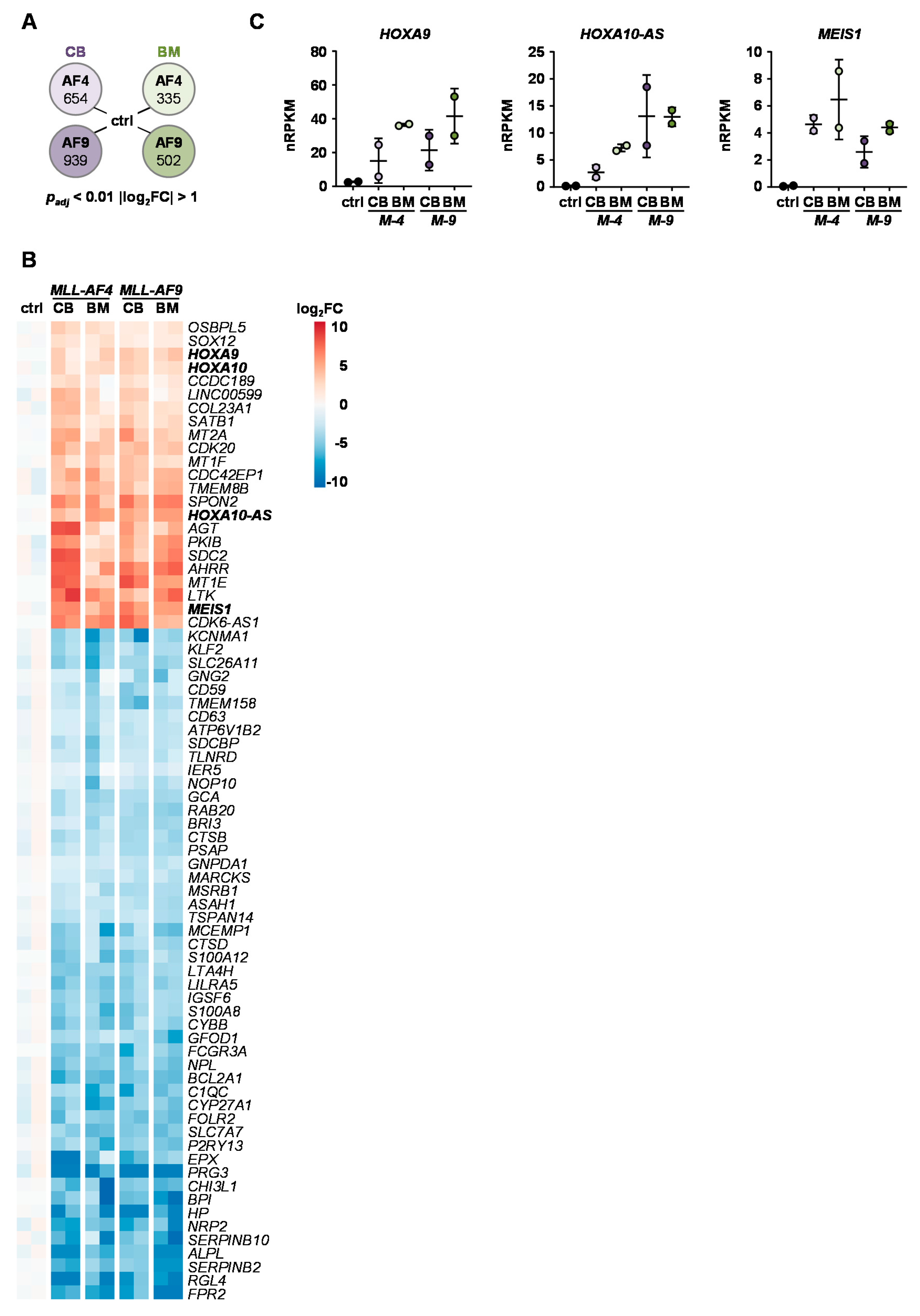
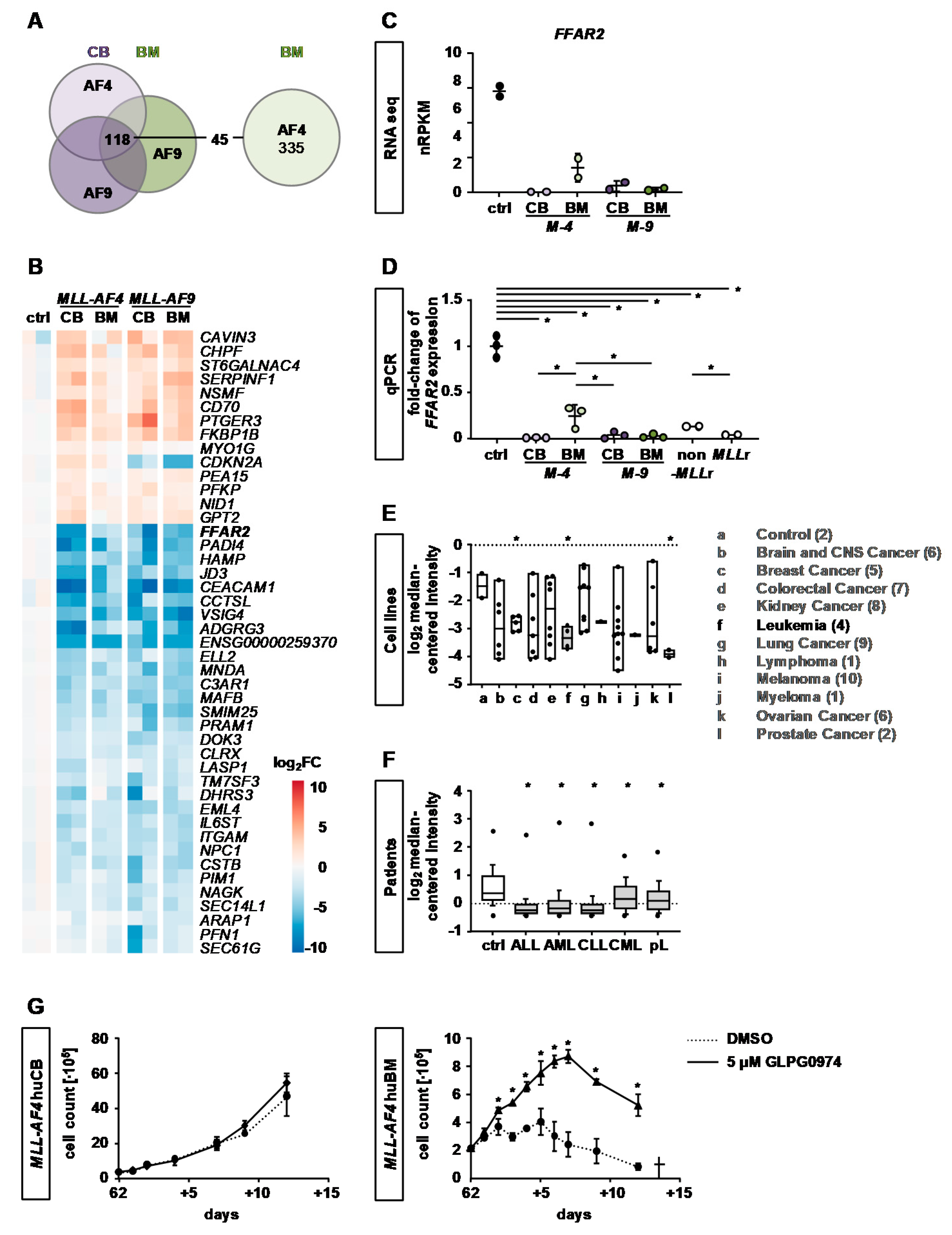
2.4. Identification of Common KMT2Ar Target Genes and Uncovering of FFAR2 as Possible Intrinsic Factor Responsible for Cell Transformation
3. Discussion
4. Materials and Methods
4.1. Human CRISPR/Cas9-KMT2Ar Model and Patient Samples
4.2. Quantitative PCR (qPCR)
4.3. May–Gruenwald–Giemsa Cytospin Staining
4.4. Flow Cytometry
4.5. Cell Proliferation Analysis
4.6. RNA Sequencing and Gene Expression Analyses
4.7. Statistical Analyses
4.8. Data Sharing Statement
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Meyer, C.; Burmeister, T.; Groger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-de-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Gaynon, P.S.; Boyett, J.M.; Chessells, J.M.; Baruchel, A.; Kamps, W.; Silverman, L.B.; Biondi, A.; Harms, D.O.; Vilmer, E.; et al. Outcome of treatment in childhood acute lymphoblastic leukaemia with rearrangements of the 11q23 chromosomal region. Lancet 2002, 359, 1909–1915. [Google Scholar] [CrossRef]
- Eguchi, M.; Eguchi-Ishimae, M.; Greaves, M. The role of the MLL gene in infant leukemia. Int. J. Hematol. 2003, 78, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Frankel, L.S.; Carroll, A.J.; Raimondi, S.C.; Shuster, J.J.; Head, D.R.; Crist, W.M.; Land, V.J.; Pullen, D.J.; Steuber, C.P.; et al. Clinical characteristics and treatment outcome of childhood acute lymphoblastic leukemia with the t(4;11)(q21;q23): A collaborative study of 40 cases. Blood 1991, 77, 440–447. [Google Scholar] [CrossRef]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef] [Green Version]
- Super, H.J.; McCabe, N.R.; Thirman, M.J.; Larson, R.A.; Le Beau, M.M.; Pedersen-Bjergaard, J.; Philip, P.; Diaz, M.O.; Rowley, J.D. Rearrangements of the MLL gene in therapy-related acute myeloid leukemia in patients previously treated with agents targeting DNA-topoisomerase II. Blood 1993, 82, 3705–3711. [Google Scholar] [CrossRef] [Green Version]
- Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Gedman, A.L.; Dang, J.; Nakitandwe, J.; Holmfeldt, L.; Parker, M.; Easton, J.; et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat. Genet. 2015, 47, 330–337. [Google Scholar] [CrossRef]
- Bolouri, H.; Farrar, J.E.; Triche, T., Jr.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Horton, S.J.; Jaques, J.; Woolthuis, C.; van Dijk, J.; Mesuraca, M.; Huls, G.; Morrone, G.; Vellenga, E.; Schuringa, J.J. MLL-AF9-mediated immortalization of human hematopoietic cells along different lineages changes during ontogeny. Leukemia 2013, 27, 1116–1126. [Google Scholar] [CrossRef] [Green Version]
- Okeyo-Owuor, T.; Li, Y.; Patel, R.M.; Yang, W.; Casey, E.B.; Cluster, A.S.; Porter, S.N.; Bryder, D.; Magee, J.A. The efficiency of murine MLL-ENL-driven leukemia initiation changes with age and peaks during neonatal development. Blood Adv. 2019, 3, 2388–2399. [Google Scholar] [CrossRef]
- Barabe, F.; Kennedy, J.A.; Hope, K.J.; Dick, J.E. Modeling the initiation and progression of human acute leukemia in mice. Science 2007, 316, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Buechele, C.; Breese, E.H.; Schneidawind, D.; Lin, C.H.; Jeong, J.; Duque-Afonso, J.; Wong, S.H.; Smith, K.S.; Negrin, R.S.; Porteus, M.; et al. MLL leukemia induction by genome editing of human CD34+ hematopoietic cells. Blood 2015, 126, 1683–1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Wunderlich, M.; Fox, C.; Alvarez, S.; Cigudosa, J.C.; Wilhelm, J.S.; Zheng, Y.; Cancelas, J.A.; Gu, Y.; Jansen, M.; et al. Microenvironment determines lineage fate in a human model of MLL-AF9 leukemia. Cancer Cell 2008, 13, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Secker, K.A.; Keppeler, H.; Duerr-Stoerzer, S.; Schmid, H.; Schneidawind, D.; Hentrich, T.; Schulze-Hentrich, J.M.; Mankel, B.; Fend, F.; Schneidawind, C. Inhibition of DOT1L and PRMT5 promote synergistic anti-tumor activity in a human MLL leukemia model induced by CRISPR/Cas9. Oncogene 2019. [Google Scholar] [CrossRef] [PubMed]
- Schneidawind, C.; Jeong, J.; Schneidawind, D.; Kim, I.S.; Duque-Afonso, J.; Wong, S.H.K.; Iwasaki, M.; Breese, E.H.; Zehnder, J.L.; Porteus, M.; et al. MLL leukemia induction by t(9;11) chromosomal translocation in human hematopoietic stem cells using genome editing. Blood Adv. 2018, 2, 832–845. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Watanabe, T.; Saito, Y.; Kuroki, Y.; Hijikata, A.; Takagi, M.; Tomizawa, D.; Eguchi, M.; Eguchi-Ishimae, M.; Kaneko, A.; et al. Identification of CD34+ and CD34- leukemia-initiating cells in MLL-rearranged human acute lymphoblastic leukemia. Blood 2015, 125, 967–980. [Google Scholar] [CrossRef] [Green Version]
- Baer, M.R.; Stewart, C.C.; Lawrence, D.; Arthur, D.C.; Mrozek, K.; Strout, M.P.; Davey, F.R.; Schiffer, C.A.; Bloomfield, C.D. Acute myeloid leukemia with 11q23 translocations: Myelomonocytic immunophenotype by multiparameter flow cytometry. Leukemia 1998, 12, 317–325. [Google Scholar] [CrossRef] [Green Version]
- Shankavaram, U.T.; Reinhold, W.C.; Nishizuka, S.; Major, S.; Morita, D.; Chary, K.K.; Reimers, M.A.; Scherf, U.; Kahn, A.; Dolginow, D.; et al. Transcript and protein expression profiles of the NCI-60 cancer cell panel: An integromic microarray study. Mol. Cancer Ther. 2007, 6, 820–832. [Google Scholar] [CrossRef] [Green Version]
- Haferlach, T.; Kohlmann, A.; Wieczorek, L.; Basso, G.; Kronnie, G.T.; Bene, M.C.; De Vos, J.; Hernandez, J.M.; Hofmann, W.K.; Mills, K.I.; et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: Report from the International Microarray Innovations in Leukemia Study Group. J. Clin. Oncol. 2010, 28, 2529–2537. [Google Scholar] [CrossRef] [Green Version]
- Hansen, A.H.; Sergeev, E.; Bolognini, D.; Sprenger, R.R.; Ekberg, J.H.; Ejsing, C.S.; McKenzie, C.J.; Rexen Ulven, E.; Milligan, G.; Ulven, T. Discovery of a Potent Thiazolidine Free Fatty Acid Receptor 2 Agonist with Favorable Pharmacokinetic Properties. J. Med. Chem. 2018, 61, 9534–9550. [Google Scholar] [CrossRef]
- Bindels, L.B.; Porporato, P.E.; Ducastel, S.; Sboarina, M.; Neyrinck, A.M.; Dewulf, E.M.; Feron, O.; Lestavel, S.; Cani, P.D.; Staels, B.; et al. Ffar2 expression regulates leukaemic cell growth in vivo. Br. J. Cancer 2017, 117, 1336–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, P.; Oshima, K.; Huang, Y.W.; Agle, K.A.; Drobyski, W.R.; Chen, X.; Zhang, J.; Yearsley, M.M.; Yu, J.; Wang, L.S. Loss of FFAR2 promotes colon cancer by epigenetic dysregulation of inflammation suppressors. Int. J. Cancer 2018, 143, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; Goode, D.L.; Iwasaki, M.; Wei, M.C.; Kuo, H.P.; Zhu, L.; Schneidawind, D.; Duque-Afonso, J.; Weng, Z.; Cleary, M.L. The H3K4-Methyl Epigenome Regulates Leukemia Stem Cell Oncogenic Potential. Cancer Cell 2015, 28, 198–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, C.L.; Kaiser, S.E.; Bolanos, B.; Nowlin, D.; Grantner, R.; Karlicek-Bryant, S.; Feng, J.L.; Jenkinson, S.; Freeman-Cook, K.; Dann, S.G.; et al. Targeting S-adenosylmethionine biosynthesis with a novel allosteric inhibitor of Mat2A. Nat. Chem. Biol. 2017, 13, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Majer, C.R.; Sneeringer, C.J.; Song, J.; Johnston, L.D.; Scott, M.P.; Smith, J.J.; Xiao, Y.; et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 2011, 20, 53–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan-Penebre, E.; Kuplast, K.G.; Majer, C.R.; Boriack-Sjodin, P.A.; Wigle, T.J.; Johnston, L.D.; Rioux, N.; Munchhof, M.J.; Jin, L.; Jacques, S.L.; et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 2015, 11, 432–437. [Google Scholar] [CrossRef]
- Duncan, K.W.; Rioux, N.; Boriack-Sjodin, P.A.; Munchhof, M.J.; Reiter, L.A.; Majer, C.R.; Jin, L.; Johnston, L.D.; Chan-Penebre, E.; Kuplast, K.G.; et al. Structure and Property Guided Design in the Identification of PRMT5 Tool Compound EPZ015666. ACS Med. Chem. Lett. 2016, 7, 162–166. [Google Scholar] [CrossRef] [Green Version]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner. 2014. Available online: https://www.osti.gov/servlets/purl/1241166 (accessed on 24 May 2020).
- Andrew, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 24 May 2020).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Secker, K.-A.; Bruns, L.; Keppeler, H.; Jeong, J.; Hentrich, T.; Schulze-Hentrich, J.M.; Mankel, B.; Fend, F.; Schneidawind, D.; Schneidawind, C. Only Hematopoietic Stem and Progenitor Cells from Cord Blood Are Susceptible to Malignant Transformation by MLL-AF4 Translocations. Cancers 2020, 12, 1487. https://doi.org/10.3390/cancers12061487
Secker K-A, Bruns L, Keppeler H, Jeong J, Hentrich T, Schulze-Hentrich JM, Mankel B, Fend F, Schneidawind D, Schneidawind C. Only Hematopoietic Stem and Progenitor Cells from Cord Blood Are Susceptible to Malignant Transformation by MLL-AF4 Translocations. Cancers. 2020; 12(6):1487. https://doi.org/10.3390/cancers12061487
Chicago/Turabian StyleSecker, Kathy-Ann, Lukas Bruns, Hildegard Keppeler, Johan Jeong, Thomas Hentrich, Julia M. Schulze-Hentrich, Barbara Mankel, Falko Fend, Dominik Schneidawind, and Corina Schneidawind. 2020. "Only Hematopoietic Stem and Progenitor Cells from Cord Blood Are Susceptible to Malignant Transformation by MLL-AF4 Translocations" Cancers 12, no. 6: 1487. https://doi.org/10.3390/cancers12061487
APA StyleSecker, K. -A., Bruns, L., Keppeler, H., Jeong, J., Hentrich, T., Schulze-Hentrich, J. M., Mankel, B., Fend, F., Schneidawind, D., & Schneidawind, C. (2020). Only Hematopoietic Stem and Progenitor Cells from Cord Blood Are Susceptible to Malignant Transformation by MLL-AF4 Translocations. Cancers, 12(6), 1487. https://doi.org/10.3390/cancers12061487