Management of Drug Resistance in Mantle Cell Lymphoma



Abstract
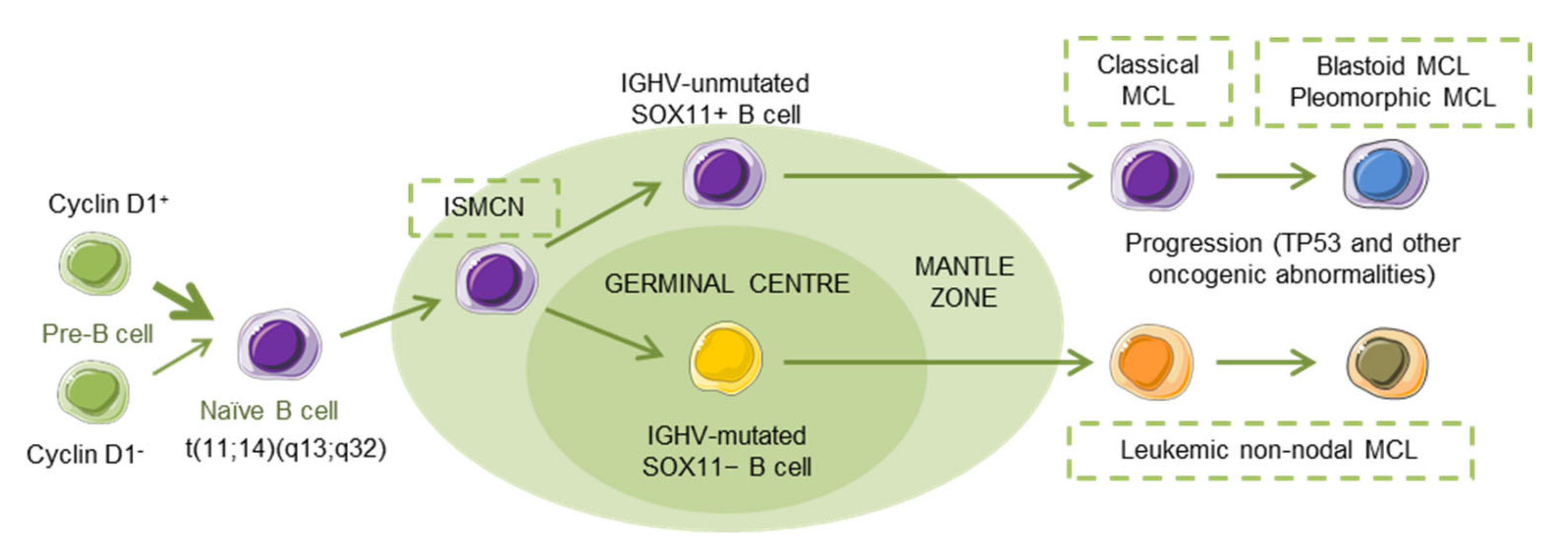
:1. Physiopathology of Mantle Cell Lymphoma
1.1. MCL Subtypes
1.2. MCL Biological Features and Prognostic Factors
1.3. MCL Therapy
2. Molecular Signatures of MCL
2.1. Translocation t(11;14)(q13;q32)
2.2. Recurrent Genomic Mutations
2.3. Deletions of INK4A/ARF (CDKN2A) Locus
2.4. Abnormalities of Signaling Pathways
2.4.1. B-Cell Receptor Signaling
2.4.2. NF-κB Signaling
2.4.3. TLR Signaling
2.4.4. PI3K/AKT/mTOR Signaling Pathway
3. Molecular Mechanisms of Resistance to Standard/Current Therapeutics
3.1. Resistance to BTK and PI3K Inhibitors
3.2. Resistance to Bortezomib and Proteasome Inhibitors
3.3. Resistance to Lenalidomide
3.4. Resistance to Temsirolimus and mTOR Inhibitors
3.5. Resistance to BCL2-Targeting Agents
4. Combination Therapies as Strategies to Overcome Drug Resistance
4.1. Targeting of Environmental Factors
4.1.1. BCR Signaling
4.1.2. Adhesion Molecules
4.1.3. IMiDs
4.2. New Therapeutics Antibodies
4.3. Epigenetic Drugs
4.4. Immune Checkpoint Inhibitors
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Epperla, N.; Hamadani, M.; Fenske, T.S.; Costa, L.J. Incidence and survival trends in mantle cell lymphoma. Br. J. Haematol. 2018, 181, 703–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortelazzo, S.; Ponzoni, M.; Ferreri, A.J.M.; Dreyling, M. Mantle cell lymphoma. Crit. Rev. Oncol. Hematol. 2012, 82, 78–101. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; World Health Organization; International Agency for Research on Cancer. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; WHO: Geneva, Switzerland, 2017; ISBN 9789283244943. [Google Scholar]
- Rule, S. The modern approach to mantle cell lymphoma. Hematol. Oncol. 2019, 37, 66–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jares, P.; Colomer, D.; Campo, E. Molecular pathogenesis of mantle cell lymphoma. J. Clin. Investig. 2012, 122, 3416–3423. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Lee Harris, N.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Jares, P.; Campo, E. Advances in the understanding of mantle cell lymphoma. Br. J. Haematol. 2008, 142, 149–165. [Google Scholar] [CrossRef] [Green Version]
- Mozos, A.; Royo, C.; Hartmann, E.; De Jong, D.; Baró, C.; Valera, A.; Fu, K.; Weisenburger, D.D.; Delabie, J.; Chuang, S.S.; et al. SOX11 expression is highly specific for mantle cell lymphoma and identifies the cyclin D1-negative subtype. Haematologica 2009, 94, 1555–1562. [Google Scholar] [CrossRef] [Green Version]
- Palomero, J.; Vegliante, M.C.; Rodríguez, M.L.; Eguileor, Á.; Castellano, G.; Planas-Rigol, E.; Jares, P.; Ribera-Cortada, I.; Cid, M.C.; Campo, E.; et al. SOX11 promotes tumor angiogenesis through transcriptional regulation of PDGFA in mantle cell lymphoma. Blood 2014, 124, 2235–2247. [Google Scholar] [CrossRef]
- Hoster, E.; Dreyling, M.; Klapper, W.; Gisselbrecht, C.; Van Hoof, A.; Kluin-Nelemans, H.C.; Pfreundschuh, M.; Reiser, M.; Metzner, B.; Einsele, H.; et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008, 111, 558–565. [Google Scholar] [CrossRef]
- Vose, J.M. Mantle cell lymphoma: 2015 update on diagnosis, risk-stratification, and clinical management. Am. J. Hematol. 2015, 90, 739–745. [Google Scholar] [CrossRef]
- Queirós, A.C.; Beekman, R.; Vilarrasa-Blasi, R.; Duran-Ferrer, M.; Clot, G.; Merkel, A.; Raineri, E.; Russiñol, N.; Castellano, G.; Beà, S.; et al. Decoding the DNA methylome of mantle cell lymphoma in the light of the entire B cell cineage. Cancer Cell 2016, 30, 806–821. [Google Scholar] [CrossRef] [Green Version]
- Fernàndez, V.; Salamero, O.; Espinet, B.; Solé, F.; Royo, C.; Navarro, A.; Camacho, F.; Beà, S.; Hartmann, E.; Amador, V.; et al. Genomic and gene expression profiling defines indolent forms of mantle cell lymphoma. Cancer Res. 2010, 70, 1408–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, P.; Chadburn, A.; Christos, P.; Weil, K.; Furman, R.R.; Ruan, J.; Elstrom, R.; Niesvizky, R.; Ely, S.; Diliberto, M.; et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J. Clin. Oncol. 2009, 27, 1209–1213. [Google Scholar] [CrossRef] [PubMed]
- Spurgeon, S.E.; Till, B.G.; Martin, P.; Goy, A.H.; Dreyling, M.P.; Gopal, A.K.; LeBlanc, M.; Leonard, J.P.; Friedberg, J.W.; Baizer, L.; et al. Recommendations for Clinical Trial Development in Mantle Cell Lymphoma. J. Natl. Cancer Inst. 2016, 109, djw263. [Google Scholar] [CrossRef] [Green Version]
- Diamond, B.; Kumar, A. Mantle Cell Lymphoma: Current and Emerging Treatment Strategies and Unanswered Questions. Hematol. Oncol. Clin. N. Am. 2019, 33, 613–626. [Google Scholar] [CrossRef]
- Yan, F.; Gopal, A.K.; Graf, S.A. Targeted drugs as maintenance therapy after autologous stem cell transplantation in patients with mantle cell lymphoma. Pharmaceuticals 2017, 10, 28. [Google Scholar] [CrossRef] [PubMed]
- Kluin-Nelemans, H.C.; Hoster, E.; Hermine, O.; Walewski, J.; Trneny, M.; Geisler, C.H.; Stilgenbauer, S.; Thieblemont, C.; Vehling-Kaiser, U.; Doorduijn, J.K.; et al. Treatment of older patients with mantle-cell lymphoma. N. Engl. J. Med. 2012, 367, 520–531. [Google Scholar] [CrossRef] [Green Version]
- Robak, T.; Jin, J.; Pylypenko, H.; Verhoef, G.; Siritanaratkul, N.; Drach, J.; Raderer, M.; Mayer, J.; Pereira, J.; Tumyan, G.; et al. Frontline bortezomib, rituximab, cyclophosphamide, doxorubicin, and prednisone (VR-CAP) versus rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) in transplantation-ineligible patients with newly diagnosed mantle cell lymphoma: Final overall survival results of a randomised, open-label, phase 3 study. Lancet Oncol. 2018, 19, 1449–1458. [Google Scholar]
- Atilla, E.; Atilla, P.A.; Demirer, T. Current treatment strategies in relapsed/refractory mantle cell lymphoma: Where are we now? Int. J. Hematol. 2017, 105, 257–264. [Google Scholar] [CrossRef]
- Vaughn, J.E.; Sorror, M.L.; Storer, B.E.; Chauncey, T.R.; Pulsipher, M.A.; Maziarz, R.T.; Maris, M.B.; Hari, P.; Laport, G.G.; Franke, G.N.; et al. Long-term sustained disease control in patients with mantle cell lymphoma with or without active disease after treatment with allogeneic hematopoietic cell transplantation after nonmyeloablative conditioning. Cancer 2015, 121, 3709–3716. [Google Scholar] [CrossRef]
- Rule, S.; Cook, G.; Russell, N.H.; Hunter, A.; Robinson, S.; Morley, N.; Sureda, A.; Patrick, P.; Clifton-Hadley, L.; Adedayo, T.; et al. Allogeneic stem cell transplantation as part of front line therapy for Mantle cell lymphoma. Br. J. Haematol. 2019, 184, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.I.; Bernstein, S.H.; Kahl, B.S.; Djulbegovic, B.; Robertson, M.J.; De Vos, S.; Epner, E.; Krishnan, A.; Leonard, J.P.; Lonial, S.; et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J. Clin. Oncol. 2006, 24, 4867–4874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, G.; Herbrecht, R.; Romaguera, J.; Verhoef, G.; Crump, M.; Gisselbrecht, C.; Laurell, A.; Offner, F.; Strahs, A.; Berkenblit, A.; et al. Phase III study to evaluate temsirolimus compared with investigator’s choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J. Clin. Oncol. 2009, 27, 3822–3829. [Google Scholar] [CrossRef] [PubMed]
- Goy, A.; Sinha, R.; Williams, M.E.; Besisik, S.K.; Drach, J.; Ramchandren, R.; Zhang, L.; Cicero, S.; Fu, T.; Witzig, T.E. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: Phase II MCL-001 (EMERGE) study. J. Clin. Oncol. 2013, 31, 3688–3695. [Google Scholar] [CrossRef]
- Wang, M.L.; Rule, S.; Martin, P.; Goy, A.; Auer, R.; Jurczak, B.S.K.W.; Advani, R.H.; Romaguera, J.E.; Williams, M.E.; Barrientos, J.C.; et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 2013, 369, 507–516. [Google Scholar] [CrossRef] [Green Version]
- Kane, R.C.; Dagher, R.; Farrell, A.; Ko, C.W.; Sridhara, R.; Justice, R.; Pazdur, R. Bortezomib for the treatment of mantle cell lymphoma. Clin. Cancer Res. 2007, 13, 5291–5294. [Google Scholar] [CrossRef] [Green Version]
- Bouabdallah, K.; Ribrag, V.; Terriou, L.; Soria, J.C.; Delarue, R. Temsirolimus in the treatment of mantle cell lymphoma: Frequency and management of adverse effects. Curr. Opin. Oncol. 2013, 25, S1–S12. [Google Scholar] [CrossRef]
- Desai, M.; Newberry, K.; Zhishuo, Z.; Wang, M.; Zhang, L. Lenalidomide in relapsed or refractory mantle cell lymphoma: Overview and perspective. Ther. Adv. Hematol. 2014, 5, 91–101. [Google Scholar] [CrossRef]
- Wang, M.L.; Blum, K.A.; Martin, P.; Goy, A.; Auer, R.; Kahl, B.S.; Jurczak, W.; Advani, R.H.; Romaguera, J.E.; Williams, M.E.; et al. Long-term follow-up of MCL patients treated with single-agent ibrutinib: Updated safety and efficacy results. Blood 2015, 126, 739–745. [Google Scholar] [CrossRef] [Green Version]
- Kochenderfer, J.N.; Feldman, S.A.; Zhao, Y.; Xu, H.; Black, M.A.; Morgan, R.A.; Wilson, W.H.; Rosenberg, S.A. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J. Immunother. 2009, 32, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell therapy in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Leonard, J.P.; LaCasce, A.S.; Smith, M.R.; Noy, A.; Chirieac, L.R.; Rodig, S.J.; Yu, J.Q.; Vallabhajosula, S.; Schoder, H.; English, P.; et al. Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood 2012, 119, 4597–4607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furtado, M.; Dyer, M.J.S.; Johnson, R.; Berrow, M.; Rule, S. Ofatumumab monotherapy in relapsed/refractory mantle cell lymphoma—A phase II trial. Br. J. Haematol. 2014, 165, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Morschhauser, F.A.; Cartron, G.; Thieblemont, C.; Solal-Céligny, P.; Haioun, C.; Bouabdallah, R.; Feugier, P.; Bouabdallah, K.; Asikanius, E.; Lei, G.; et al. Obinutuzumab (GA101) monotherapy in relapsed/refractory diffuse large B-cell lymphoma or mantle-cell lymphoma: Results from the phase II GAUGUIN study. J. Clin. Oncol. 2013, 31, 2912–2919. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.S.; Roberts, A.W.; Seymour, J.F.; Pagel, J.M.; Kahl, B.S.; Wierda, W.G.; Puvvada, S.; Kipps, T.J.; Anderson, M.A.; Salem, A.H.; et al. Phase I first-in-human study of venetoclax in patients with relapsed or refractory non-hodgkin lymphoma. J. Clin. Oncol. 2017, 35, 826–833. [Google Scholar] [CrossRef] [Green Version]
- Kahl, B.S.; Spurgeon, S.E.; Furman, R.R.; Flinn, I.W.; Coutre, S.E.; Brown, J.R.; Benson, D.M.; Byrd, J.C.; Peterman, S.; Cho, Y.; et al. A phase 1 study of the PI3Kδ inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood 2014, 123, 3398–3405. [Google Scholar] [CrossRef] [Green Version]
- Heider, U.; Kaiser, M.; Sterz, J.; Zavrski, I.; Jakob, C.; Fleissner, C.; Eucker, J.; Possinger, K.; Sezer, O. Histone deacetylase inhibitors reduce VEGF production and induce growth suppression and apoptosis in human mantle cell lymphoma. Eur. J. Haematol. 2006, 76, 42–50. [Google Scholar] [CrossRef]
- Evens, A.M.; Balasubramanian, S.; Vose, J.M.; Harb, W.; Gordon, L.I.; Langdon, R.; Sprague, J.; Sirisawad, M.; Mani, C.; Yue, J.; et al. A phase I/II multicenter, open-label study of the oral histone deacetylase inhibitor abexinostat in relapsed/refractory lymphoma. Clin. Cancer Res. 2016, 22, 1059–1066. [Google Scholar] [CrossRef] [Green Version]
- Renner, C.; Zinzani, P.L.; Gressin, R.; Klingbiel, D.; Dietrich, P.Y.; Hitz, F.; Bargetzi, M.; Mingrone, W.; Martinelli, G.; Trojan, A.; et al. A multicenter phase II trial (SAKK 36/06) of single-agent everolimus (RAD001) in patients with relapsed or refractory mantle cell lymphoma. Haematologica 2012, 97, 1085–1091. [Google Scholar] [CrossRef] [Green Version]
- Cheah, C.Y.; Seymour, J.F.; Wang, M.L. Mantle cell lymphoma. J. Clin. Oncol. 2016, 34, 1256–1269. [Google Scholar] [CrossRef]
- Zhang, J.; Jima, D.; Moffitt, A.B.; Liu, Q.; Czader, M.; Hsi, E.D.; Fedoriw, Y.; Dunphy, C.H.; Richards, K.L.; Gill, J.I.; et al. The genomic landscape of mantle cell lymphoma is related to the epigenetically determined chromatin state of normal B cells. Blood 2014, 123, 2988–2996. [Google Scholar] [CrossRef] [PubMed]
- Bea, S.; Valdes-Mas, R.; Navarro, A.; Salaverria, I.; Martin-Garcia, D.; Jares, P.; Gine, E.; Pinyol, M.; Royo, C.; Nadeu, F.; et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 18250–18255. [Google Scholar] [CrossRef] [Green Version]
- Sherr, C.J. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004, 18, 2699–2711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenwald, A. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell 2003, 3, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, A.; Sandoval, N.; Das, M.; Pillai, R.; Chen, L.; Chen, R.W.; Amin, H.M.; Wang, M.; Marcucci, G.; Weisenburger, D.D.; et al. CCND1 mutations increase protein stability and promote ibrutinib resistance in mantle cell lymphoma. Oncotarget 2016, 7, 73558–73572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiemann, K.; Alluin, J.V.; Honegger, A.; Chomchan, P.; Gaur, S.; Yun, Y.; Forman, S.J.; Rossi, J.J.; Chen, R.W. siRNAs targeting cyclin D1 and cyclin D2 enhance the cytotoxicity of chemotherapeutic agents in mantle cell lymphoma cell lines. Leuk. Lymphoma 2013, 52, 2148–2154. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Zhang, W.; Wang, J.; Liu, Y.; An, R.; Jing, H. Genomic landscape and prognostic analysis of mantle cell lymphoma. Cancer Gene Ther. 2018, 25, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Greiner, T.C.; Dasgupta, C.; Ho, V.V.; Weisenburger, D.D.; Smith, L.M.; Lynch, J.C.; Vose, J.; Fu, K.; Armitage, J.O.; Braziel, R.M.; et al. Mutation and genomic deletion status of ataxia telangiectasia mutated (ATM) and p53 confer specific gene expression profiles in mantle cell lymphoma. Proc. Natl. Acad. Sci. USA 2006, 103, 2352–2357. [Google Scholar] [CrossRef] [Green Version]
- Eskelund, C.W.; Dahl, C.; Hansen, J.W.; Westman, M.; Kolstad, A.; Pedersen, L.B.; Montano-Almendras, C.P.; Husby, S.; Freiburghaus, C.; Ek, S.; et al. TP53 mutations identify younger mantle cell lymphoma patients who do not benefit from intensive chemoimmunotherapy. Blood 2017, 130, 1903–1910. [Google Scholar] [CrossRef] [Green Version]
- Aukema, S.M.; Hoster, E.; Rosenwald, A.; Canoni, D.; Delfau-Larue, M.-H.; Rymkiewicz, G.; Thorns, C.; Hartmann, S.; Kluin-Nelemans, H.C.; Hermine, O.; et al. Expression of TP53 is associated with the outcome of MCL independent of MIPI and Ki-67 in trials of the European MCL Network. Blood 2018, 131, 417–420. [Google Scholar] [CrossRef]
- Kridel, R.; Meissner, B.; Rogic, S.; Boyle, M.; Telenius, A.; Woolcock, B.; Gunarwardana, J.; Jenkins, C.; Cochrane, C.; Ben-Neriah, S.; et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood 2012, 119, 1963–1971. [Google Scholar] [CrossRef]
- Meissner, B.; Kridel, R.; Lim, R.S.; Rogic, S.; Tse, K.; Scott, D.W.; Moore, R.; Mungall, A.J.; Marra, M.A.; Connors, J.M.; et al. The E3 ubiquitin ligase UBR5 is recurrently mutated in mantle cell lymphoma. Blood 2013, 121, 3161–3164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahal, R.; Frick, M.; Romero, R.; Korn, J.M.; Kridel, R.; Chan, F.C.; Meissner, B.; Bhang, H.; Ruddy, D.; Kauffmann, A.; et al. Pharmacological and genomic profiling identifies NF-kappaB-targeted treatment strategies for mantle cell lymphoma. Nat. Med. 2013, 20, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; de Miranda, N.F.C.C.; Chen, L.; Wasik, A.M.; Mansouri, L.; Jurczak, W.; Galazka, K.; Dlugosz-Danecka, M.; Machaczka, M.; Zhang, H.; et al. Genetic heterogeneity in primary and relapsed mantle cell lymphomas: Impact of recurrent CARD11 mutations. Oncotarget 2016, 7, 38180–38190. [Google Scholar] [CrossRef]
- Cheng, S.; Guo, A.; Lu, P.; Ma, J.; Coleman, M.; Wang, Y.L. Functional characterization of BTKC481S mutation that confers ibrutinib resistance: Exploration of alternative kinase inhibitors. Leukemia 2015, 29, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Chiron, D.; Liberto, M.D.; Martin, P.; Huang, X.; Sharman, J.; Blecua, P.; Mathew, S.; Vijay, P.; Eng, K.; Ali, S.; et al. Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov. 2014, 4, 1022–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.; Li, L.; Pinnix, C.; Dabaja, B.; Nomie, K.; Lam, L.; Wang, M. ATM mutation and radiosensitivity: An opportunity in the therapy of mantle cell lymphoma. Crit. Rev. Oncol. Hematol. 2016, 107, 14–19. [Google Scholar] [CrossRef]
- Beà, S.; Salaverria, I.; Armengol, L.; Pinyol, M.; Fernandez, V.; Hartmann, E.M.; Jares, P.; Amador, V.; Hernandez, L.; Navarro, A.; et al. Uniparental disomies, homozygous deletions, amplifications, and target genes in mantle cell lymphoma revealed by integrative high-resolution whole-genome profiling. Blood 2009, 113, 3059–3069. [Google Scholar] [CrossRef]
- Hutter, G.; Scheubner, M.; Zimmermann, Y.; Kalla, J.; Katzenberger, T.; Hübler, K.; Roth, S.; Hiddemann, W.; Ott, G.; Dreyling, M. Differential effect of epigenetic alterations and genomic deletions of CDK inhibitors [p16(INK4a),p15(INK4b),p14(ARF)] in mantle cell lymphoma. Genes. Chromosomes Cancer 2005, 45, 203–210. [Google Scholar] [CrossRef]
- Kawamata, N.; Ogawa, S.; Gueller, S.; Ross, S.H.; Huynh, T.; Chen, J.; Chang, A.; Nabavi-Nouis, S.; Megrabian, N.; Siebert, R. Identified hidden genomic changes in mantle cell lymphoma using high-resolution single nucleotide polymorphism genomic array. Exp. Hematol. 2009, 37, 937–946. [Google Scholar] [CrossRef] [Green Version]
- Beà, S.; Tort, F.; Pinyol, M.; Puig, X.; Hernandez, L.; Hernandez, S.; Fernandez, P.L.; van Lohuizen, M.; Colomer, D.; Campo, E. BMI-1 gene amplification and overexpression in hematological malignancies occur mainly in mantle cell lymphomas. Cancer Res. 2001, 61, 2409–2412. [Google Scholar] [PubMed]
- Jardin, F.; Picquenot, J.M.; Parmentier, F.; Ruminy, P.; Cornic, M.; Penther, D.; Bertrand, P.; Lanic, H.; Cassuto, O.; Humbrecht, C.; et al. Detection of gene copy number aberrations in mantle cell lymphoma by a single quantitative multiplex PCR assay: Clinicopathological relevance and prognosis value. Br. J. Haematol. 2009, 146, 607–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, J.A.; Wiestner, A. Targeting B cell receptor signalling in cancer: Preclinical and clinical advances. Nat. Rev. Cancer 2018, 18, 148–167. [Google Scholar] [CrossRef] [PubMed]
- Herrera, A.F.; Jacobsen, E.D. Ibrutinib for the treatment of mantle cell lymphoma. Clin. Cancer Res. 2014, 20, 5365–5371. [Google Scholar] [CrossRef] [Green Version]
- Hadzidimitriou, A.; Agathangelidis, A.; Darzentas, N.; Murray, F.; Delfau-Larue, M.H.; Pedersen, L.B.; Lopez, A.N.; Dagklis, A.; Rombout, P.; Beldjord, K.; et al. Is there a role for antigen selection in mantle cell lymphoma? Immunogenetic support from a series of 807 cases. Blood 2011, 118, 3088–3095. [Google Scholar] [CrossRef] [Green Version]
- Chang, B.Y.; Francesco, M.; De Rooij, M.F.M.; Magadala, P.; Steggerda, S.M.; Huang, M.M.; Kuil, A.; Herman, S.E.M.; Chang, S.; Pals, S.T.; et al. Egress of CD19+CD5+ cells into peripheral blood following treatment with the Bruton tyrosine kinase inhibitor ibrutinib in mantle cell lymphoma patients. Blood 2013, 122, 2412–2424. [Google Scholar] [CrossRef]
- Pighi, C.; Gu, T.L.; Dalai, I.; Barbi, S.; Parolini, C.; Bertolaso, A.; Pedron, S.; Parisi, A.; Ren, J.; Cecconi, D.; et al. Phospho-proteomic analysis of mantle cell lymphoma cells suggests a pro-survival role of B-cell receptor signaling. Cell. Oncol. 2011, 34, 141–153. [Google Scholar] [CrossRef] [Green Version]
- Boyd, R.S.; Jukes-Jones, R.; Walewska, R.; Brown, D.; Dyer, M.J.S.; Cain, K. Protein profiling of plasma membranes defines aberrant signaling pathways in mantle cell lymphoma. Mol. Cell. Proteom. 2009, 8, 1501–1515. [Google Scholar] [CrossRef] [Green Version]
- Myklebust, J.H.; Brody, J.; Kohrt, H.E.; Kolstad, A.; Czerwinski, D.K.; Wälchli, S.; Green, M.R.; Trøen, G.; Liestø, K.; Beiske, K.; et al. Distinct patterns of B-cell receptor signaling in non-Hodgkin lymphomas identified by single-cell profiling. Blood 2017, 129, 759–770. [Google Scholar] [CrossRef]
- Advani, R.H.; Buggy, J.J.; Sharman, J.P.; Smith, S.M.; Boyd, T.E.; Grant, B.; Kolibaba, K.S.; Furman, R.R.; Rodriguez, S.; Chang, B.Y.; et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J. Clin. Oncol. 2013, 31, 88–94. [Google Scholar] [CrossRef]
- Saba, N.S.; Liu, D.; Herman, S.E.M.; Underbayev, C.; Tian, X.; Behrend, D.; Weniger, M.A.; Skarzynski, M.; Gyamfi, J.; Fontan, L.; et al. Pathogenic role of B-cell receptor signaling and canonical NF-kB activation in mantle cell lymphoma. Blood 2016, 128, 82–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaji, S.; Ahmed, M.; Lorence, E.; Yan, F.; Nomie, K.; Wang, M. NF-κB signaling and its relevance to the treatment of mantle cell lymphoma. J. Hematol. Oncol. 2018, 11, 83. [Google Scholar] [CrossRef] [PubMed]
- Mastorci, K.; Muraro, E.; Pasini, E. Toll-Like receptor 1/2 and 5 ligands enhance the expression of cyclin D1 and D3 and induce proliferation in mantle cell lymphoma. PLoS ONE 2016, 11, e0153823. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhao, Y.; Qian, J.; Sun, L.; Lu, Y.; Li, H.; Li, Y.; Yang, J.; Cai, Z.; Yi, Q. Toll-like receptor-4 signaling in mantle cell lymphoma: Effects on tumor growth and immune evasion. Cancer 2013, 119, 782–791. [Google Scholar] [CrossRef] [Green Version]
- Akhter, A.; Street, L.; Ghosh, S.; Burns, B.F.; Elyamany, G.; Shabani-Rad, M.-T.; Stewart, D.A.; Mansoor, A. Concomitant high expression of Toll-like receptor (TLR) and B-cell receptor (BCR) signalling molecules has clinical implications in mantle cell lymphoma. Hematol. Oncol. 2015, 35, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Rudelius, M.; Pittaluga, S.; Nishizuka, S.; Pham, T.H.T.; Fend, F.; Jaffe, E.S.; Quintanilla-Martinez, L.; Raffeld, M. Constitutive activation of Akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood 2006, 108, 1668–1676. [Google Scholar] [CrossRef] [Green Version]
- Tabe, Y.; Jin, L.; Konopleva, M.; Shikami, M.; Kimura, S.; Andreeff, M.; Raffeld, M.; Miida, T. Class IA phosphatidylinositol 3-kinase inhibition inhibits cell growth and proliferation in mantle cell lymphoma. Acta Haematol. 2013, 131, 59–69. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, H.H.; Liu, X.; Nunez-Cruz, S.; Jillab, M.; Melnikov, O.; Nath, K.; Glickson, J. ROR1/CD19 receptor complex promotes growth of mantle cell lymphoma cells independently of the B cell receptor-BTK signaling pathway. J. Immunol. 2019, 203, 2043–2048. [Google Scholar] [CrossRef]
- Dreyling, M.; Ferrero, S.; on behalf of European Mantle Cell Lymphoma Network. The role of targeted treatment in mantle cell lymphoma: Is transplant dead or alive? Haematologica 2016, 101, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.P.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17, 1–23. [Google Scholar]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Lu, P.; Guo, A.; Cheng, S.; Zong, H.; Martin, P.; Coleman, M.; Wang, Y.L. Characterization of ibrutinib-sensitive and -resistant mantle lymphoma cells. Br. J. Haematol. 2014, 166, 849–861. [Google Scholar] [CrossRef]
- Martin, P.; Maddocks, K.; Leonard, J.P.; Ruan, J.; Goy, A.; Wagner-Johnston, N.; Rule, S.; Advani, R.; Iberri, D.; Phillips, T.; et al. Postibrutinib outcomes in patients with mantle cell lymphoma. Blood 2016, 127, 1559–1563. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Rule, S.; Zinzani, P.L.; Goy, A.; Casasnovas, O.; Smith, S.D.; Damaj, G.; Dooduijn, J.K.; Lamy, T.; Morschhauser, F.; et al. Durable response with single-agent acalabrutinib in patients with relapsed or refractory mantle cell lymphoma. Leukemia 2019, 373, 2762–2766. [Google Scholar] [CrossRef] [Green Version]
- Witzig, T.E.; Inwards, D. Acalabrutinib for mantle cell lymphoma. Blood 2019, 133, 2564–2570. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.J.; Goodell, L.; Glod, J.; Gelinas, C.; Rabson, A.B.; Strair, R.K. Mesenchymal stromal cells protect mantle cell lymphoma cells from spontaneous and drug-induced apoptosis through secretion of B-cell activating factor and activation of the canonical and non-canonical nuclear factor ~B pathways. Haematologica 2012, 97, 1255–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Lwin, T.; Silva, A.; Shah, B.; Tao, J.; Fang, B.; Zhang, L.; Fu, K.; Bi, C.; Li, J.; et al. Unification of de novo and acquired ibrutinib resistance in mantle cell lymphoma. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Chiron, D.; Martin, P.; Liberto, M.D.; Huang, X.; Ely, S.; Lannutti, B.J.; Leonard, J.P.; Mason, C.E.; Chen-Kiang, S. Induction of prolonged early G 1arrest by CDK4/CDK6 inhibition reprograms lymphoma cells for durable PI3Kδ inhibition through PIK3IP1. Cell Cycle 2013, 12, 1892–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woyach, J.A.; Furman, R.R.; Liu, T.-M.; Ozer, H.G.; Zapatka, M.; Ruppert, A.S.; Xue, L.; Li, D.H.-H.; Steggerda, S.M.; Versele, M.; et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N. Engl. J. Med. 2014, 370, 2286–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, P.; Kanagal-Shamanna, R.; Zhang, S.; Ahmed, M.; Ghorab, A.; Zhang, L.; Ok, C.Y.; Li, S.; Hagemeister, F.; Zeng, D.; et al. Long-term outcomes and mutation profiling of patients with mantle cell lymphoma (MCL) who discontinued ibrutinib. Br. J. Haematol. 2018, 183, 578–587. [Google Scholar] [CrossRef] [Green Version]
- Col, J.D.; Zancai, P.; Terrin, L.; Guidoboni, M.; Ponzoni, M.; Pavan, A.; Spina, M.; Bergamin, S.; Rizzo, S.; Tirelli, U.; et al. Distinct functional significance of Akt and mTOR constitutive activation in mantle cell lymphoma. Blood 2008, 111, 5142–5151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Psyrri, A.; Papageorgiou, S.; Liakata, E.; Scorilas, A.; Rontogianni, D.; Kontos, C.K.; Argyriou, P.; Pectasides, D.; Harhalakis, N.; Pappa, V.; et al. Phosphatidylinositol 3′-kinase catalytic subunit α gene amplification contributes to the pathogenesis of mantle cell lymphoma. Clin. Cancer Res. 2009, 15, 5724–5732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyengar, S.; Clear, A.; Bödör, C.; Maharaj, L.; Lee, A.; Calaminici, M.; Matthews, J.; Iqbal, S.; Auer, R.; Gribben, J.; et al. P110α-mediated constitutive PI3K signaling limits the efficacy of p110δ-selective inhibition in mantle cell lymphoma, particularly with multiple relapse. Blood 2013, 121, 2274–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compagno, M.; Wang, Q.; Pighi, C.; Cheong, T.-C.; Meng, F.-L.; Poggio, T.; Yeap, L.-S.; Karaca, E.; Blasco, R.B.; Langellotto, F.; et al. Phosphatidylinositol 3-kinase $δ$ blockade increases genomic instability in B cells. Nature 2017, 542, 489–493. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Yao, Y.; Zhang, S.; Liu, Y.; Guo, H.; Ahmed, M.; Bell, T.; Zhang, H.; Han, G.; Lorence, E.; et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci. Transl. Med. 2019, 11, eaau1167. [Google Scholar] [CrossRef]
- Guan, J.; Huang, D.; Yakimchuk, K.; Okret, S. p110a Inhibition Overcomes Stromal Cell{\textendash} Mediated Ibrutinib Resistance in Mantle Cell Lymphoma. Mol. Cancer Ther. 2018, 17, 1090–1100. [Google Scholar] [CrossRef] [Green Version]
- Spriano, F.; Tarantelli, C.; Gaudio, E.; Gerlach, M.M.; Priebe, V.; Cascione, L.; Bernasconi, E.; Targa, A.; Mascia, M.; Dirnhofer, S.; et al. Single and combined BTK and PI3K$δ$ inhibition with acalabrutinib and ACP-319 in pre-clinical models of aggressive lymphomas. Br. J. Haematol. 2019, 187, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Perez-Galan, P.; Mora-Jensen, H.; Weniger, M.A.; Shaffer, A.L., III; Rizzatti, E.G.; Chapman, C.M.; Mo, C.C.; Stennett, L.S.; Rader, C.; Liu, P.; et al. Bortezomib resistance in mantle cell lymphoma is associated with plasmacytic differentiation. Blood 2011, 117, 542–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Santamarta, M.; Quinet, G.; Reyes-Garau, D.; Sola, B.; Roué, G.; Rodriguez, M.S. Resistance to the proteasome inhibitors: Lessons from multiple myeloma and mantle cell lymphoma. Adv. Exp. Med. Biol. 2020, 1233, 153–174. [Google Scholar] [PubMed]
- Pérez-Galán, P.; Roué, G.; Villamor, N.; Campo, E.; Colomer, D. The BH3-mimetic GX15-070 synergizes with bortezomib in mantle cell lymphoma by enhancing Noxa-mediated activation of Bak. Blood 2007, 109, 4441–4449. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.T.; Young, K.H.; Kahl, B.S.; Markovina, S.; Miyamoto, S. Prevalence of bortezomib-resistant constitutive NF-kappaB activity in mantle cell lymphoma. Mol. Cancer 2008, 7, 14–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manni, S.; Brancalion, A.; Mandato, E.; Tubi, L.Q.; Colpo, A.; Pizzi, M.; Cappellesso, R.; Zaffino, F.; Maggio, S.A.D.; Cabrelle, A.; et al. Protein kinase CK2 inhibition down modulates the NF-κB and STAT3 survival pathways, enhances the cellular proteotoxic stress and synergistically boosts the cytotoxic effect of bortezomib on multiple myeloma and mantle cell lymphoma cells. PLoS ONE 2013, 8, e75280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Galan, P.; Roué, G.; Villamor, N.; Montserrat, E.; Campo, E.; Colomer, D. The proteasome inhibitor bortezomib induces apoptosis in mantle-cell lymphoma through generation of ROS and Noxa activation independent of p53 status. Blood 2006, 107, 257–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weniger, M.A.; Rizzatti, E.G.; Pérez-galán, P. Treatment-induced oxidative stress and cellular antioxidant capacity determine response to bortezomib in mantle cell lymphoma. Clin. Cancer Res. 2011, 17, 5101–5112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelebart, P.; Anand, M.; Armanious, H.; Peters, A.C.; Dien Bard, J.; Amin, H.M.; Lai, R. Constitutive activation of the Wnt canonical pathway in mantle cell lymphoma. Blood 2008, 112, 5171–5179. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Tilló, E.; Fanlo, L.; Siles, L.; Montes-Moreno, S.; Moros, A.; Chiva-Blanch, G.; Estruch, R.; Martinez, A.; Colomer, D.; Orffy, B.G.; et al. The EMT activator ZEB1 promotes tumor growth and determines differential response to chemotherapy in mantle cell lymphoma. Cell Death Differ. 2014, 21, 247–257. [Google Scholar] [CrossRef]
- Wang, Q.; Mora-Jensen, H.; Weniger, M.A.; Perez-Galan, P.; Wolford, C.; Hai, T.; Ron, D.; Chen, W.; Trenkle, W.; Wiestner, A. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc. Natl. Acad. Sci. USA 2009, 16, 2200–2205. [Google Scholar] [CrossRef] [Green Version]
- Roué, G.; Pérez-Galán, P.; Mozos, A.; López-Guerra, M.; Xargay-Torrent, S.; Rosich, L.; Saborit-Villarroya, I.; Normant, E.; Campo, E.; Colomer, D. The Hsp90 inhibitor IPI-504 overcomes bortezomib resistance in mantle cell lymphoma in vitro and in vivo by down-regulation of the prosurvival ER chaperone BiP/Grp78. Blood 2011, 117, 1270–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heine, S.; Kleih, M.; Giménez, N.; Böpple, K.; Ott, G.; Colomer, D.; Aulitzky, W.E.; van der Kuip, H.; Silkenstedt, E. Cyclin D1-CDK4 activity drives sensitivity to bortezomib in mantle cell lymphoma by blocking autophagy-mediated proteolysis of NOXA. J. Hematol. Oncol. 2018, 11, 112. [Google Scholar] [CrossRef]
- Moros, A.; Rodríguez, V.; Saborit-Villarroya, I.; Montraveta, A.; Balsas, P.; Sandy, P.; Martínez, A.; Wiestner, A.; Normant, E.; Campo, E.; et al. Synergistic antitumor activity of lenalidomide with the BET bromodomain inhibitor CPI203 in bortezomib-resistant mantle cell lymphoma. Leukemia 2014, 28, 2049–2059. [Google Scholar] [CrossRef] [PubMed]
- Vegliante, M.C.; Palomero, J.; Pérez-Galán, P.; Roué, G.; Castellano, G.; Navarro, A.; Clot, G.; Moros, A.; Suárez-Cisneros, H.; Beà, S.; et al. SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive mantle cell lymphoma. Blood 2013, 121, 2175–2185. [Google Scholar] [CrossRef]
- Desai, S.; Maurin, M.; Smith, M.A.; Bolick, S.C.E.; Dessureault, S.; Tao, J.; Sotomayor, E.; Wright, K.L. PRDM1 is required for mantle cell lymphoma response to bortezomib. Mol. Cancer Res. 2010, 8, 907–918. [Google Scholar] [CrossRef] [Green Version]
- Vose, J.M.; Habermann, T.M.; Czuczman, M.S.; Zinzani, P.L.; Reeder, C.B.; Tuscano, J.M.; Lossos, I.S.; Li, J.; Pietronigro, D.; Witzig, T.E. Single-agent lenalidomide is active in patients with relapsed or refractory aggressive non-Hodgkin lymphoma who received prior stem cell transplantation. Br. J. Haematol. 2013, 162, 639–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinzani, P.L.; Vose, J.M.; Czuczman, M.S.; Reeder, C.B.; Haioun, C.; Polikoff, J.; Tilly, H.; Zhang, L.; Prandi, K.; Li, J.; et al. Long-term follow-up of lenalidomide in relapsed/refractory mantle cell lymphoma: Subset analysis of the NHL-003 study. Ann. Oncol. 2013, 24, 2892–2897. [Google Scholar] [CrossRef] [PubMed]
- Gribben, J.G.; Fowler, N.; Morschhauser, F. Mechanisms of action of lenalidomide in B-cell non-hodgkin lymphoma. J. Clin. Oncol. 2015, 33, 2803–2811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagner, P.R.; Chiu, H.; Ortiz, M.; Apollonio, B.; Wang, M.; Couto, S.; Waldman, M.F.; Flynt, E.; Ramsay, A.G.; Trotter, M.; et al. Activity of lenalidomide in mantle cell lymphoma can be explained by NK cell-mediated cytotoxicity. Br. J. Haematol. 2017, 179, 399–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Girona, A.; Mendy, D.; Ito, T.; Miller, K.; Gandhi, A.K.; Kang, J.; Karasawa, S.; Carmel, G.; Jackson, P.; Abbasian, M.; et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 2019, 26, 2326–2335. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wu, K.; Bai, W.; Cui, X.; Chen, Y.; Xie, Y.; Xie, Y. Synergistic cytotoxicity of lenalidomide and dexamethasone in mantle cell lymphoma via cereblon-dependent targeting of the IL-6/STAT3/PI3K axis. EBioMedicine 2017, 20, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Moros, A.; Bustany, S.; Cahu, J.; Saborit-Villarroya, I.; Martínez, A.; Colomer, D.; Sola, B.; Roué, G. Antitumoral activity of lenalidomide in in vitro and in vivo models of mantle cell lymphomainvolves the destabilization of cyclin D1/p27KIP1 Complexes. Clin. Cancer Res. 2014, 20, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Sordo-Bahamonde, C.; Vitale, M.; Lorenzo-Herrero, S.; López-Soto, A.; Gonzalez, S. Mechanisms of resistance to NK cell immunotherapy. Cancers 2020, 12, 893. [Google Scholar] [CrossRef] [Green Version]
- Rosich, L.; Xargay-Torrent, S.; López-Guerra, M.; Campo, E.; Colomer, D.; Roué, G. Counteracting autophagy overcomes resistance to everolimus in mantle cell lymphoma. Clin. Cancer Res. 2012, 18, 5278–5289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosich, L.; Colomer, D.; Roué, G. Autophagy controls everolimus (RAD001) activity in mantle cell lymphoma. Autophagy 2013, 9, 115–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mestre-Escorihuela, C.; Rubio-Moscardo, F.; Richter, J.A.; Siebert, R.; Climent, J.; Fresquet, V.; Beltran, E.; Agirre, X.; Marugan, I.; Marín, M.; et al. Homozygous deletions localize novel tumor suppressor genes in B-cell lymphomas. Blood 2007, 109, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Tagawa, H.; Karnan, S.; Suzuki, R.; Matsuo, K.; Zhang, X.; Ota, A.; Morishima, Y.; Nakamura, S.; Seto, M. Genome-wide array-based CGH for mantle cell lymphoma: Identification of homozygous deletions of the proapoptotic gene BIM. Oncogene 2005, 24, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Prukova, D.; Andera, L.; Nahacka, Z.; Karolova, J.; Svaton, M.; Klanova, M.; Havranek, O.; Soukup, J.; Svobodova, K.; Zemanova, Z.; et al. Cotargeting of BCL2 with venetoclax and MCL1 with S63845 is synthetically lethal in vivo in relapsed mantle cell lymphoma. Clin. Cancer Res. 2019, 25, 4455–4465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahir, S.K.; Smith, M.L.; Hessler, P.; Rapp, L.R.; Idler, K.B.; Park, C.H.; Leverson, J.D.; Lam, L.T. Potential mechanisms of resistance to venetoclax and strategies to circumvent it. BMC Cancer 2017, 17, 399. [Google Scholar] [CrossRef]
- Li, Y.; Bouchlaka, M.N.; Wolff, J.; Grindle, K.M.; Lu, L.; Qian, S.; Zhong, X.; Pflum, N.; Jobin, P.; Kahl, B.S.; et al. FBXO10 deficiency and BTK activation upregulate BCL2 expression in mantle cell lymphoma. Oncogene 2016, 35, 6223–6234. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Ren, Y.; Lawlor, M.; Shah, B.D.; Park, P.M.C.; Lwin, T.; Wang, X.; Liu, K.; Wang, M.; Gao, J.; et al. BCL2 amplicon loss and transcriptional remodeling drives ABT-199 resistance in B cell lymphoma models. Cancer Cell 2019, 35, 752–766. [Google Scholar] [CrossRef]
- Agarwal, R.; Chan, Y.-C.; Tam, C.S.; Hunter, T.; Vassiliadis, D.; Teh, C.E.; Thijssen, R.; Yeh, P.; Wong, S.Q.; Ftouni, S.; et al. Dynamic molecular monitoring reveals that SWI-SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat. Med. 2018, 25, 119–129. [Google Scholar] [CrossRef]
- Tucker, D.; Rule, S. Novel agents in mantle cell lymphoma. Expert Rev. Anticancer. Ther. 2017, 17, 491–506. [Google Scholar] [CrossRef]
- Ruan, J.; Martin, P.; Shah, B.; Schuster, S.J.; Smith, S.M.; Furman, R.R.; Christos, P.; Rodriguez, A.; Svoboda, J.; Lewis, J.; et al. Lenalidomide plus rituximab as initial treatment for mantle-cell Lymphoma. N. Engl. J. Med. 2015, 373, 1835–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.E.; Li, H.; Smith, M.R.; Gascoyne, R.D.; Paietta, E.M.; Yang, D.T.; Advani, R.H.; Horning, S.J.; Kahl, B.S. Phase 2 study of VcR-CVAD with maintenance rituximab for untreated mantle cell lymphoma: An Eastern Cooperative Oncology Group study (E1405). Blood 2014, 123, 1665–1673. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Thieblemont, C.; Morschhauser, F.; Flinn, I.; Friedberg, J.W.; Amorim, S.; Hivert, B.; Westin, J.; Vermeulen, J.; Bandyopadhyay, N.; et al. Combination of ibrutinib with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) for treatment-naive patients with CD20-positive B-cell non-Hodgkin lymphoma: A non-randomised, phase 1b study. Lancet Oncol. 2014, 15, 1019–1026. [Google Scholar] [CrossRef]
- Maddocks, K.; Christian, B.; Jaglowski, S.; Flynn, J.; Jones, J.A.; Porcu, P.; Wei, L.; Jenkins, C.; Lozanski, G.; Byrd, J.C.; et al. A phase 1/1b study of rituximab, bendamustine, and ibrutinib in patients with untreated and relapsed/refractory non-Hodgkin lymphoma. Blood 2015, 125, 242–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.L.; Lee, H.; Chuang, H.; Wagner-Bartak, N.; Hagemeister, F.; Westin, J.; Fayad, L.; Samaniego, F.; Turturro, F.; Oki, Y.; et al. Ibrutinib in combination with rituximab in relapsed or refractory mantle cell lymphoma: A single-centre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 48–56. [Google Scholar] [CrossRef]
- Tam, C.S.; Anderson, M.A.; Pott, C.; Agarwal, R.; Handunnetti, S.; Hicks, R.J.; Burbury, K.; Turner, G.; Di Iulio, J.; Bressel, M.; et al. Ibrutinib plus venetoclax for the treatment of mantle-cell lymphoma. N. Engl. J. Med. 2018, 378, 1211–1223. [Google Scholar] [CrossRef]
- Allen, C.D.C.; Okada, T.; Cyster, J.G. Germinal-Center Organization and Cellular Dynamics. Immunity 2007, 27, 190–202. [Google Scholar] [CrossRef] [Green Version]
- Kurtova, A.V.; Tamayo, A.T.; Ford, R.J.; Burger, J.A. Mantle cell lymphoma cells express high levels of CXCR4, CXCR5, and VLA-4 (CD49d): Importance for interactions with the stromal microenvironment and specific targeting. Blood 2009, 113, 4604–4613. [Google Scholar] [CrossRef]
- Pérez-Galán, P.; Roué, G. The challenge of the microenvironment in mantle cell lymphoma. In Hodgkin and Non-Hodgkin Lymphomas Seen Through Their Microenvironment: Impact on Diagnosis, Prognosis and Innovative Therapy (Volume 2); Future Medicine Ltd.: London, UK, 2015; pp. 84–97. [Google Scholar]
- Richardson, S.J.; Eve, H.E.; Copplestone, J.A.; Dyer, M.J.; Rule, S.A.J. Activity of thalidomide and lenalidomide in mantle cell lymphoma. Acta Haematol. 2009, 123, 21–29. [Google Scholar] [CrossRef]
- Damaj, G.; Lefrère, F.; Delarue, R.; Varet, B.; Furman, R.; Hermine, O. Thalidomide therapy induces response in relapsed mantle cell lymphoma. Leukemia 2003, 17, 1914–1915. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Fayad, L.; Wagner-Bartak, N.; Zhang, L.; Hagemeister, F.; Neelapu, S.S.; Samaniego, F.; McLaughlin, P.; Fanale, M.; Younes, A.; et al. Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: A phase 1/2 clinical trial. Lancet Oncol. 2012, 13, 716–723. [Google Scholar] [CrossRef]
- Ruan, J.; Martin, P.; Christos, P.; Cerchietti, L.; Tam, W.; Shah, B.; Schuster, S.J.; Rodriguez, A.; Hyman, D.; Calvo-Vidal, M.N.; et al. Five-year follow-up of lenalidomide plus rituximab as initial treatment of mantle cell lymphoma. Blood 2018, 132, 2016–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, V.A.; Jung, S.H.; Johnson, J.; Lacasce, A.; Blum, K.A.; Bartlett, N.L.; Pitcher, B.N.; Cheson, B.D. Therapy with bortezomib plus lenalidomide for relapsed/refractory mantle cell lymphoma: Final results of a phase II trial (CALGB 50501). Leuk. Lymphoma 2015, 56, 958–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albertsson-Lindblad, A.; Kolstad, A.; Laurell, A.; Räty, R.; Grønbæk, K.; Sundberg, J.; Pedersen, L.B.; Ralfkiær, E.; Karjalainen-Lindsberg, M.L.; Sundström, C.; et al. Lenalidomide-bendamustine-rituximab in patients older than 65 years with untreated mantle cell lymphoma. Blood 2016, 128, 1814–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barth, M.J.; Mavis, C.; Czuczman, M.S.; Hernandez-Ilizaliturri, F.J. Ofatumumab Exhibits Enhanced in Vitro and in Vivo Activity Compared to Rituximab in Preclinical Models of Mantle Cell Lymphoma. Clin. Cancer Res. 2015, 21, 4391–4397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niederfellner, G.; Lammens, A.; Mundigl, O.; Georges, G.J.; Schaefer, W.; Schwaiger, M.; Franke, A.; Wiechmann, K.; Jenewein, S.; Slootstra, J.W.; et al. Epitope characterization and crystal structure of GA101 provide insights into the molecular basis for type I/II distinction of CD20 antibodies. Blood 2011, 118, 358–367. [Google Scholar] [CrossRef] [Green Version]
- Chiron, D.; Bellanger, C.; Papin, A.; Tessoulin, B.; Dousset, C.; Maiga, S.; Moreau, A.; Esbelin, J.; Trichet, V.; Chen-Kiang, S.; et al. Rational targeted therapies to overcome microenvironment-dependent expansion of mantle cell lymphoma. Blood 2016, 128, 2808–2818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legouill, S.; Beldi-Ferchiou, A.; Cacheux, V.; Alcantara, M.; Salles, G.; Canioni, D.; Bodet-Milin, C.; Delwail, V.; Gastinne, T.; Thieblemont, C.; et al. Obitunuzumab plus DHAP followed by autologous stem cell transplantation plus obitunuzumab maintenance provides a high MRD response rate in untreated MCL patients. Results of LYMA-101 trial—A LYSA study. HemaSphere 2019, 3, 3. [Google Scholar] [CrossRef]
- Le Gouill, S.; Morschhauser, F.; Bouabdallah, K.; Cartron, G.; Casasnovas, R.-O.; Milpied, N.-J.; Gastinne, T.; Davies, A.J.; Herbaux, C.; Chiron, D.; et al. Ibrutinib plus obinutuzumab and venetoclax in relapsed/refractory mantle cells lymphoma patients. Results of the OASIS phase I clinical trial. Blood 2018, 132, 4158. [Google Scholar] [CrossRef]
- Zhou, X.; Steinhardt, M.J.; Düll, J.; Krummenast, F.; Danhof, S.; Meckel, K.; Nickel, K.; Grathwohl, D.; Leicht, H.B.; Rosenwald, A.; et al. Obinutuzumab and venetoclax induced complete remission in a patient with ibrutinib-resistant non-nodal leukemic mantle cell lymphoma. Eur. J. Haematol. 2020, 104, 352–355. [Google Scholar] [CrossRef]
- Kolibaba, K.; Burke, J.M.; Brooks, H.D.; Mahadevan, D.; Melear, J.; Farber, C.M.; Fanning, S.R.; Schreeder, M.T.; Boccia, R.V.; Sportelli, P.; et al. Ublituximab (TG-1101), a novel glycoengineered anti-CD20 monoclonal antibody, in combination with ibrutinib is highly active in patients with relapsed and/or refractory mantle cell lymphoma; Results of a phase II trial. Blood 2015, 126, 3980. [Google Scholar] [CrossRef]
- Lunning, M.; Vose, J.; Nastoupil, L.; Fowler, N.; Burger, J.A.; Wierda, W.G.; Schreeder, M.T.; Siddiqi, T.; Flowers, C.R.; Cohen, J.B.; et al. Ublituximab and umbralisib in relapsed/refractory B-cell non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood 2019, 134, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Stein, R.; Mattes, M.J.; Cardillo, T.M.; Hansen, H.J.; Chang, C.H.; Burton, J.; Govindan, S.; Goldenberg, D.M. CD74: A new candidate target for the immunotherapy of B-cell neoplasms. Clin. Cancer Res. 2007, 13, 5556s–5563s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alinari, L.; Yu, B.; Christian, B.A.; Yan, F.; Shin, J.; Lapalombella, R.; Hertlein, E.; Lustberg, M.E.; Quinion, C.; Zhang, X.; et al. Combination anti-CD74 (milatuzumab) and anti-CD20 (rituximab) monoclonal antibody therapy has in vitro and in vivo activity in mantle cell lymphoma. Blood 2011, 117, 4530–4541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goebeler, M.-E.; Knop, S.; Viardot, A.; Kufer, P.; Topp, M.S.; Einsele, H.; Noppeney, R.; Hess, G.; Kallert, S.; Mackensen, A.; et al. Bispecific T-cell engager (BiTE) antibody construct Blinatumomab for the treatment of patients with relapsed/refractory non-Hodgkin lymphoma: Final results from a phase I study. J. Clin. Oncol. 2016, 34, 1104–1111. [Google Scholar] [CrossRef] [PubMed]
- Poh, C.; Frankel, P.; Ruel, C.; Abedi, M.; Schwab, E.; Costello, C.L.; Zain, J.; Budde, L.E.; William, B.M.; Foss, F.M.; et al. Blinatumomab/lenalidomide in relapsed/refractory non-Hodgkin’s lymphoma: A phase I California Cancer Consortium study of safety, efficacy and immune correlative analysis. Blood 2019, 134, 760. [Google Scholar] [CrossRef]
- Ribeiro, M.L.; Reyes-Garau, D.; Armengol, M.; Fernández-Serrano, M.; Roué, G. Recent advances in the targeting of epigenetic regulators in b-cell non-hodgkin lymphoma. Front. Genet. 2019, 10, 986. [Google Scholar] [CrossRef]
- Chaturvedi, N.K.; Hatch, N.D.; Sutton, G.L.; Kling, M.; Vose, J.M.; Joshi, S.S. A novel approach to eliminate therapy-resistant mantle cell lymphoma: Synergistic effects of Vorinostat with Palbociclib. Leuk. Lymphoma 2019, 60, 1214–1223. [Google Scholar] [CrossRef]
- Yazbeck, V.; Shafer, D.; Perkins, E.B.; Coppola, D.; Sokol, L.; Richards, K.L.; Shea, T.; Ruan, J.; Parekh, S.; Strair, R.; et al. A Phase II Trial of bortezomib and vorinostat in mantle Cell lymphoma and diffuse large B-cell lymphoma. Clin. Lymphoma Myeloma Leuk. 2018, 18, 569–575.e1. [Google Scholar] [CrossRef]
- Sun, B.; Shah, B.; Fiskus, W.; Qi, J.; Rajapakshe, K.; Coarfa, C.; Li, L.; Devaraj, S.G.T.; Sharma, S.; Zhang, L.; et al. Synergistic activity of BET protein antagonist-based combinations in mantle cell lymphoma cells sensitive or resistant to ibrutinib. Blood 2015, 126, 1565–1574. [Google Scholar] [CrossRef]
- Spurgeon, S.E.; Sharma, K.; Claxton, D.F.; Ehmann, C.; Pu, J.; Shimko, S.; Stewart, A.; Subbiah, N.; Palmbach, G.; LeBlanc, F.; et al. Phase 1–2 study of vorinostat (SAHA), cladribine and rituximab (SCR) in relapsed B-cell non-Hodgkin lymphoma and previously untreated mantle cell lymphoma. Br. J. Haematol. 2019, 186, 845–854. [Google Scholar] [CrossRef]
- Puvvada, S.D.; Guillen-Rodriguez, J.; Kumar, A.; Inclán, L.; Heard, K.; Rivera, X.I.; Anwer, F.; Schatz, J.H.; Mahadevan, D.; Persky, D.O. Phase 2 open-label study of bortezomib, cladribine, and rituximab in advanced, newly diagnosed, and relapsed/refractory mantle-cell and indolent lymphomas. Clin. Lymphoma Myeloma Leuk. 2018, 18, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Harrington, B.K.; Wheeler, E.; Hornbuckle, K.; Shana’ah, A.Y.; Youssef, Y.; Smith, L.; Hassan, Q.; Klamer, B.; Zhang, X.; Long, M.; et al. Modulation of immune checkpoint molecule expression in mantle cell lymphoma. Leuk. Lymphoma 2019, 60, 2498–2507. [Google Scholar] [CrossRef] [PubMed]
- Pritchett, J.C.; Ansell, S.M. Immunologic treatment strategies in mantle cell lymphoma: Checkpoint inhibitors, chimeric antigen receptor (CAR) T-cells, and bispecific T-cell engager (BiTE) molecules. Ann. Lymphoma 2019, 3, 1–7. [Google Scholar] [CrossRef]
- Witkowska, M.; Smolewski, P. Immune checkpoint inhibitors to treat malignant lymphomas. J. Immunol. Res. 2018, 2018, 1982423. [Google Scholar] [CrossRef] [Green Version]
- Kauder, S.E.; Kuo, T.C.; Chen, A.; Harrabi, O.; Rocha, S.S.; Doyle, L.; Bollini, S.; Han, B.; Sangalang, E.R.B.; Sim, J.; et al. ALX148 is a high affinity Sirpα fusion protein that blocks CD47, enhances the activity of anti-cancer antibodies and checkpoint inhibitors, and has a favorable safety profile in preclinical models. Blood 2017, 130, 112. [Google Scholar]
- Kim, T.M.; Lakhani, N.; Gainor, J.; Kamdar, M.; Fanning, P.; Squifflet, P.; Jin, F.; Wan, H.; Pons, J.; Randolph, S.S.; et al. A Phase 1 sstudy of ALX148, a CD47 blocker, in combination with rituximab in patients with non-Hodgkin lymphoma. Blood 2019, 134, 1953. [Google Scholar] [CrossRef]
- Wasiuk, A.; Testa, J.; Weidlick, J.; Sisson, C.; Vitale, L.; Widger, J.; Crocker, A.; Thomas, L.J.; Goldstein, J.; Marsh, H.C.; et al. CD27-mediated regulatory T cell depletion and effector T cell costimulation both contribute to antitumor efficacy. J. Immunol. 2017, 199, 4110–4123. [Google Scholar] [CrossRef] [Green Version]
- Villasboas, J.C.; Reeder, C.B.; Tun, H.W.; Bartlett, N.L.; Sharon, E.; Laplant, B.; Adjei, A.A.; Kline, J.; Hernandez-Ilizaliturri, F.J.; Awan, F.T.; et al. The DIAL study (Dual Immunomodulation in Aggressive Lymphoma): A randomized phase 2 study of CDX-1127 (varlilumab) in combination with nivolumab in patients with relapsed or refractory aggressive B-cell lymphomas (NCI 10089/NCT03038672). Blood 2019, 134, 1591. [Google Scholar] [CrossRef]
- Gopal, A.K.; Levy, R.; Houot, R.; Patel, S.P.; Popplewell, L.; Jacobson, C.; Mu, X.J.; Deng, S.; Ching, K.A.; Chen, Y.; et al. First-in-human study of utomilumab, a 4-1BB/CD137 agonist, in combination with rituximab in patients with follicular and other CD20+ non-Hodgkin ymphomas. Clin. Cancer Res. 2020, in press. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Gene | Frequency (Range) * | Protein Function | References |
|---|---|---|---|
| ATM | 38–50% | DNA repair DNA damage response | [42,43,48,49,53,54] |
| CCND1 | 16–35% | Cell cycle regulation | [42,43,48,52,53,54] |
| TP53 | 14–31% | DNA damage response Cell cycle regulation | [42,43,48,49,53,54] |
| MLL2 | 14–20% | Epigenetic regulator (HMT) | [42,43] |
| MLL3 | 16% | Epigenetic regulator (HMT) | [42,54] |
| WHSC1 | 7–31% | Epigenetic regulator (HMT) | [42,43,54] |
| BIRC3 | 6–9% | Apoptosis regulator through TRAF2 | [42,43,54] |
| NOTCH1 | 5–16% | NOTCH survival pathway | [42,43,52,53,54] |
| NOTCH2 | 5–6% | NOTCH survival pathway | [42,48] |
| TRAF2 | 7% | NF-κB pathway | [54] |
| UBR5 | 7–18% | Proteasome degradation(E3 ligase) | [42,53,54] |
| RB1 | nd | Cell cycle regulation | [42] |
| SMARCA4 | nd | Chromatin modifier | [42] |
| CARD11 | 5% | NF-κB pathway | [55] |
| Drug Combination | Targets | Study Number | Efficiency |
|---|---|---|---|
| Obinutuzumab + Venetoclax + Ibrutinib | CD20, BCL2, BTK | NCT02558816 | No results available |
| Ibrutinib + Lenalidomide + Rituximab | BTK, CRBN/CD20 | NCT02446236 | No results available |
| Alisertib + Bortezomib + Rituximab | Aurora A kinase, 20S proteasome, CD20 | NCT01695941 | No results available |
| Ibrutinib + Bortezomib | BTK, 20S proteasome | NCT02356458 | No results available |
| Rituximab + Bendamustine + Ibrutinib | CD20, alkylating agent, BTK | NCT01479842 | No results available |
| Lenalidomide + Ibrutinib | CRBN, BTK | NCT01955499 | No results available |
| BKM120 + Rituximab | PI3K, CD20 | NCT02049541 | No results available |
| Entospletinib + Obinutuzumab | SYK, CD20 | NCT03010358 | No results available |
| Everolimus + Lenalidomide | mTOR, CRBN | NCT01075321 | 9.8% CR, 19.5% PR, 39% SD, 29.3% progression |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roué, G.; Sola, B. Management of Drug Resistance in Mantle Cell Lymphoma. Cancers 2020, 12, 1565. https://doi.org/10.3390/cancers12061565
Roué G, Sola B. Management of Drug Resistance in Mantle Cell Lymphoma. Cancers. 2020; 12(6):1565. https://doi.org/10.3390/cancers12061565
Chicago/Turabian StyleRoué, Gaël, and Brigitte Sola. 2020. "Management of Drug Resistance in Mantle Cell Lymphoma" Cancers 12, no. 6: 1565. https://doi.org/10.3390/cancers12061565
APA StyleRoué, G., & Sola, B. (2020). Management of Drug Resistance in Mantle Cell Lymphoma. Cancers, 12(6), 1565. https://doi.org/10.3390/cancers12061565