Cisplatin Resistance in Testicular Germ Cell Tumors: Current Challenges from Various Perspectives




Abstract
:1. Introduction: A Brief Overview over Testicular Germ Cell Tumors
2. The Clinical Impact of a Cisplatin-Resistant Disease
3. Models for Studying Cisplatin Resistance Biology
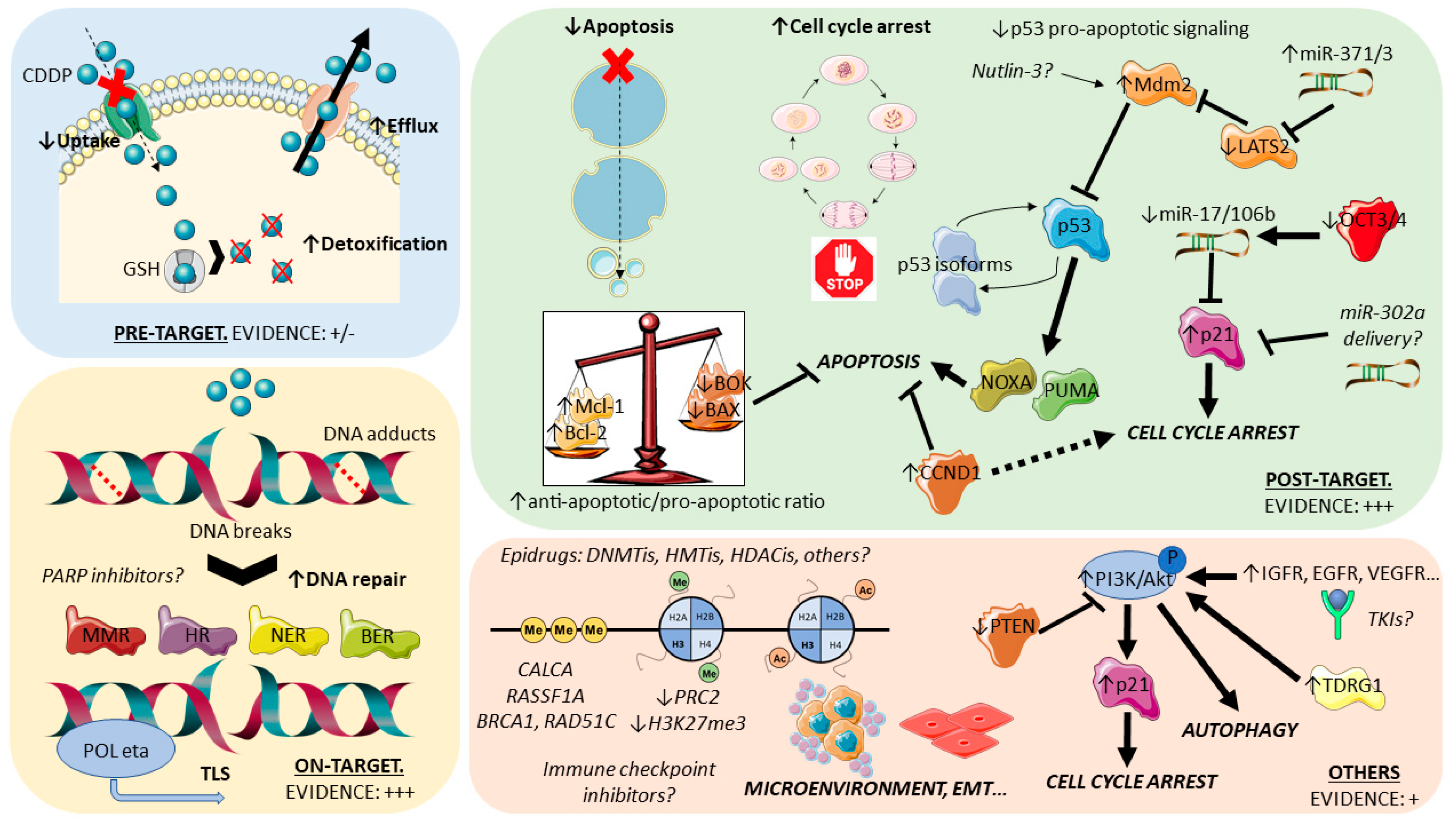
4. Dissecting Cisplatin Resistance Mechanisms
4.1. General Overview and Insights from Various Tumor Models
4.2. TGCTs, Cisplatin Resistance, and DNA Damage Repair Systems
4.3. TGCTs, Cisplatin Resistance, the p53/Mdm2 Axis, and Apoptosis Initiation
4.4. TGCTs, Cisplatin Resistance, and Epigenetics
4.5. Seeking for Relevant Mutations/Copy Number Alterations Related to Cisplatin Resistance
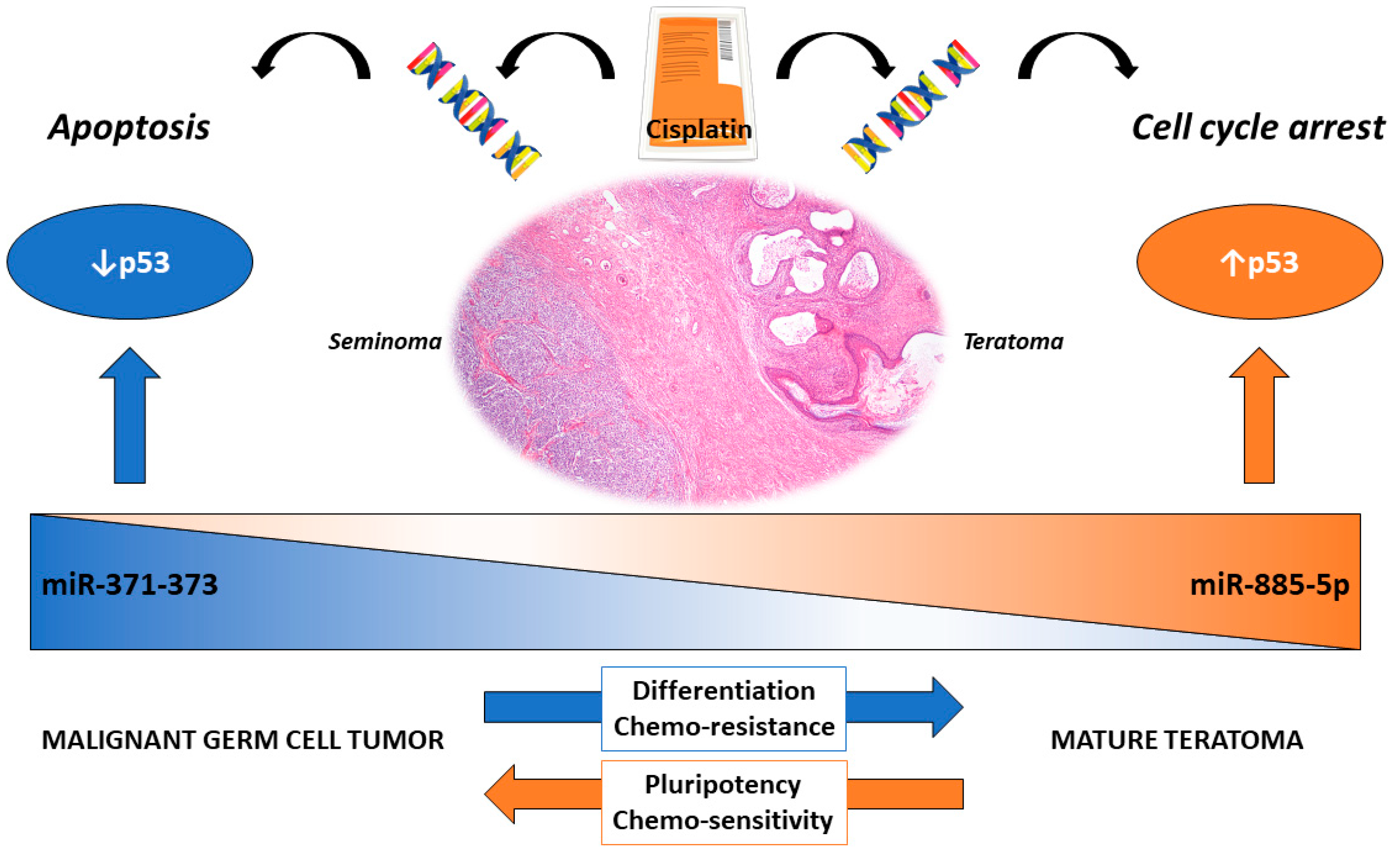
5. Differentiation-Dependent Cisplatin Resistance: Specificities of Teratoma
6. Seeking for Novel Treatments Options for Cisplatin-Resistant Diseases
6.1. Immunotherapies
6.2. Epidrugs
6.3. Others
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Znaor, A.; Skakkebaek, N.E.; Rajpert-De Meyts, E.; Laversanne, M.; Kulis, T.; Gurney, J.; Sarfati, D.; McGlynn, K.A.; Bray, F. Testicular cancer incidence predictions in Europe 2010–2035: A rising burden despite population ageing. Int. J. Cancer 2019, 147, 820–828. [Google Scholar] [CrossRef]
- Fedorova, V.A.; Kadyrova, R.A.; Slita, A.V.; Muryleva, A.A.; Petrova, P.R.; Kovalskaya, A.V.; Lobov, A.N.; Zileeva, Z.R.; Tsypyshev, D.O.; Borisevich, S.S.; et al. Antiviral activity of amides and carboxamides of quinolizidine alkaloid (-)-cytisine against human influenza virus A (H1N1) and parainfluenza virus type 3. Nat. Prod. Res. 2019, 1–9. [Google Scholar] [CrossRef]
- Oosterhuis, J.W.; Looijenga, L.H.J. Human germ cell tumours from a developmental perspective. Nat. Rev. Cancer 2019, 19, 522–537. [Google Scholar] [CrossRef]
- Berney, D.M.; Looijenga, L.H.; Idrees, M.; Oosterhuis, J.W.; Rajpert-De Meyts, E.; Ulbright, T.M.; Skakkebaek, N.E. Germ cell neoplasia in situ (GCNIS): Evolution of the current nomenclature for testicular pre-invasive germ cell malignancy. Histopathology 2016, 69, 7–10. [Google Scholar] [CrossRef]
- Lobo, J.; Costa, A.L.; Vilela-Salgueiro, B.; Rodrigues, A.; Guimaraes, R.; Cantante, M.; Lopes, P.; Antunes, L.; Jeronimo, C.; Henrique, R. Testicular germ cell tumors: Revisiting a series in light of the new WHO classification and AJCC staging systems, focusing on challenges for pathologists. Hum. Pathol. 2018, 82, 113–124. [Google Scholar] [CrossRef]
- Giuliano, C.J.; Freemantle, S.J.; Spinella, M.J. Testicular Germ Cell Tumors: A Paradigm for the Successful Treatment of Solid Tumor Stem Cells. Curr. Cancer Ther. Rev. 2006, 2, 255–270. [Google Scholar] [CrossRef]
- Moul, J.W.; Dodge, R.K.; Robertson, J.E.; Paulson, D.F.; Walther, P.J. The impact of the “cisplatin era” of treatment on survival in testicular cancer. World J. Urol. 1991, 9, 45–50. [Google Scholar] [CrossRef]
- Allen, J.C.; Kirschner, A.; Scarpato, K.R.; Morgans, A.K. Current Management of Refractory Germ Cell Tumors and Future Directions. Curr. Oncol. Rep. 2017, 19, 8. [Google Scholar] [CrossRef]
- Dieckmann, K.P.; Richter-Simonsen, H.; Kulejewski, M.; Ikogho, R.; Zecha, H.; Anheuser, P.; Pichlmeier, U.; Isbarn, H. Testicular Germ-Cell Tumours: A Descriptive Analysis of Clinical Characteristics at First Presentation. Urol. Int. 2018, 100, 409–419. [Google Scholar] [CrossRef]
- Lobo, J.; Stoop, H.; Gillis, A.J.M.; Looijenga, L.H.J.; Oosterhuis, W. Interobserver Agreement in Vascular Invasion Scoring and the Added Value of Immunohistochemistry for Vascular Markers to Predict Disease Relapse in Stage I Testicular Nonseminomas. Am. J. Surg. Pathol. 2019, 43, 1711–1719. [Google Scholar] [CrossRef]
- El Charif, O.; Mapes, B.; Trendowski, M.R.; Wheeler, H.E.; Wing, C.; Dinh, P.C., Jr.; Frisina, R.D.; Feldman, D.R.; Hamilton, R.J.; Vaughn, D.J.; et al. Clinical and Genome-wide Analysis of Cisplatin-induced Tinnitus Implicates Novel Ototoxic Mechanisms. Clin. Cancer Res. 2019, 25, 4104–4116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, Y.; Nakamura, T.; Nakanishi, H.; Oishi, M.; Hongo, F.; Okihara, K.; Mizutani, S.; Kuroda, J.; Ukimura, O. Therapy-related acute myeloid leukemia and myelodysplastic syndrome among refractory germ cell tumor patients. Int. J. Urol. 2018, 25, 678–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvammen, O.; Myklebust, T.A.; Solberg, A.; Moller, B.; Klepp, O.H.; Fossa, S.D.; Tandstad, T. Long-term Relative Survival after Diagnosis of Testicular Germ Cell Tumor. Cancer Epidemiol. Prev. Biomark. 2016, 25, 773–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokemeyer, C.; Kollmannsberger, C.; Harstrick, A.; Beyer, J.; Gerl, A.; Casper, J.; Metzner, B.; Hartmann, J.T.; Schmoll, H.J.; Kanz, L. Treatment of patients with cisplatin-refractory testicular germ-cell cancer. German Testicular Cancer Study Group (GTCSG). Int. J. Cancer 1999, 83, 848–851. [Google Scholar] [CrossRef]
- Bakardjieva-Mihaylova, V.; Skvarova Kramarzova, K.; Slamova, M.; Svaton, M.; Rejlova, K.; Zaliova, M.; Dobiasova, A.; Fiser, K.; Stuchly, J.; Grega, M.; et al. Molecular Basis of Cisplatin Resistance in Testicular Germ Cell Tumors. Cancers (Basel) 2019, 11, 1316. [Google Scholar] [CrossRef] [Green Version]
- Oing, C.; Skowron, M.A.; Bokemeyer, C.; Nettersheim, D. Epigenetic treatment combinations to effectively target cisplatin-resistant germ cell tumors: Past, present, and future considerations. Andrology 2019, 7, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Fazal, Z.; Freemantle, S.J.; Spinella, M.J. Mechanisms of cisplatin sensitivity and resistance in testicular germ cell tumors. Cancer Drug Resist. 2019, 2, 580–594. [Google Scholar] [CrossRef] [Green Version]
- Oing, C.; Seidel, C.; Bokemeyer, C. Therapeutic approaches for refractory germ cell cancer. Expert Rev. Anticancer Ther. 2018, 18, 389–397. [Google Scholar] [CrossRef]
- Kalavska, K.; Conteduca, V.; De Giorgi, U.; Mego, M. Molecular Mechanisms of Resistance in Testicular Germ Cell Tumors-clinical Implications. Curr. Cancer Drug Targets 2018, 18, 967–978. [Google Scholar] [CrossRef]
- Schmidtova, S.; Kalavska, K.; Kucerova, L. Molecular Mechanisms of Cisplatin Chemoresistance and Its Circumventing in Testicular Germ Cell Tumors. Curr. Oncol. Rep. 2018, 20, 88. [Google Scholar] [CrossRef]
- Oing, C.; Alsdorf, W.H.; von Amsberg, G.; Oechsle, K.; Bokemeyer, C. Platinum-refractory germ cell tumors: An update on current treatment options and developments. World J. Urol. 2017, 35, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, C.; Honecker, F. Cisplatin resistance in germ cell tumours: Models and mechanisms. Andrology 2015, 3, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Koster, R.; van Vugt, M.A.; Timmer-Bosscha, H.; Gietema, J.A.; de Jong, S. Unravelling mechanisms of cisplatin sensitivity and resistance in testicular cancer. Expert Rev. Mol. Med. 2013, 15, e12. [Google Scholar] [CrossRef] [PubMed]
- Lorch, A.; Beyer, J.; Bascoul-Mollevi, C.; Kramar, A.; Einhorn, L.H.; Necchi, A.; Massard, C.; De Giorgi, U.; Flechon, A.; International Prognostic Factors Study Group. Prognostic factors in patients with metastatic germ cell tumors who experienced treatment failure with cisplatin-based first-line chemotherapy. J. Clin. Oncol. 2010, 28, 4906–4911. [Google Scholar] [CrossRef]
- Verrill, C.; Yilmaz, A.; Srigley, J.R.; Amin, M.B.; Comperat, E.; Egevad, L.; Ulbright, T.M.; Tickoo, S.K.; Berney, D.M.; Epstein, J.I.; et al. Reporting and Staging of Testicular Germ Cell Tumors: The International Society of Urological Pathology (ISUP) Testicular Cancer Consultation Conference Recommendations. Am. J. Surg. Pathol. 2017, 41, e22–e32. [Google Scholar] [CrossRef]
- Honecker, F.; Aparicio, J.; Berney, D.; Beyer, J.; Bokemeyer, C.; Cathomas, R.; Clarke, N.; Cohn-Cedermark, G.; Daugaard, G.; Dieckmann, K.P.; et al. ESMO Consensus Conference on testicular germ cell cancer: Diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, 1658–1686. [Google Scholar] [CrossRef]
- Koychev, D.; Oechsle, K.; Bokemeyer, C.; Honecker, F. Treatment of patients with relapsed and/or cisplatin-refractory metastatic germ cell tumours: An update. Int. J. Androl. 2011, 34, e266–e273. [Google Scholar] [CrossRef]
- Pico, J.L.; Rosti, G.; Kramar, A.; Wandt, H.; Koza, V.; Salvioni, R.; Theodore, C.; Lelli, G.; Siegert, W.; Horwich, A.; et al. A randomised trial of high-dose chemotherapy in the salvage treatment of patients failing first-line platinum chemotherapy for advanced germ cell tumours. Ann. Oncol. 2005, 16, 1152–1159. [Google Scholar] [CrossRef]
- Lorch, A.; Bascoul-Mollevi, C.; Kramar, A.; Einhorn, L.; Necchi, A.; Massard, C.; De Giorgi, U.; Flechon, A.; Margolin, K.; Lotz, J.P.; et al. Conventional-dose versus high-dose chemotherapy as first salvage treatment in male patients with metastatic germ cell tumors: Evidence from a large international database. J. Clin. Oncol. 2011, 29, 2178–2184. [Google Scholar] [CrossRef]
- Albers, P.; Ganz, A.; Hannig, E.; Miersch, W.D.; Muller, S.C. Salvage surgery of chemorefractory germ cell tumors with elevated tumor markers. J. Urol. 2000, 164, 381–384. [Google Scholar] [CrossRef]
- Mayer, F.; Wermann, H.; Albers, P.; Stoop, H.; Gillis, A.J.; Hartmann, J.T.; Bokemeyer, C.C.; Oosterhuis, J.W.; Looijenga, L.H.; Honecker, F. Histopathological and molecular features of late relapses in non-seminomas. BJU Int. 2011, 107, 936–943. [Google Scholar] [CrossRef] [PubMed]
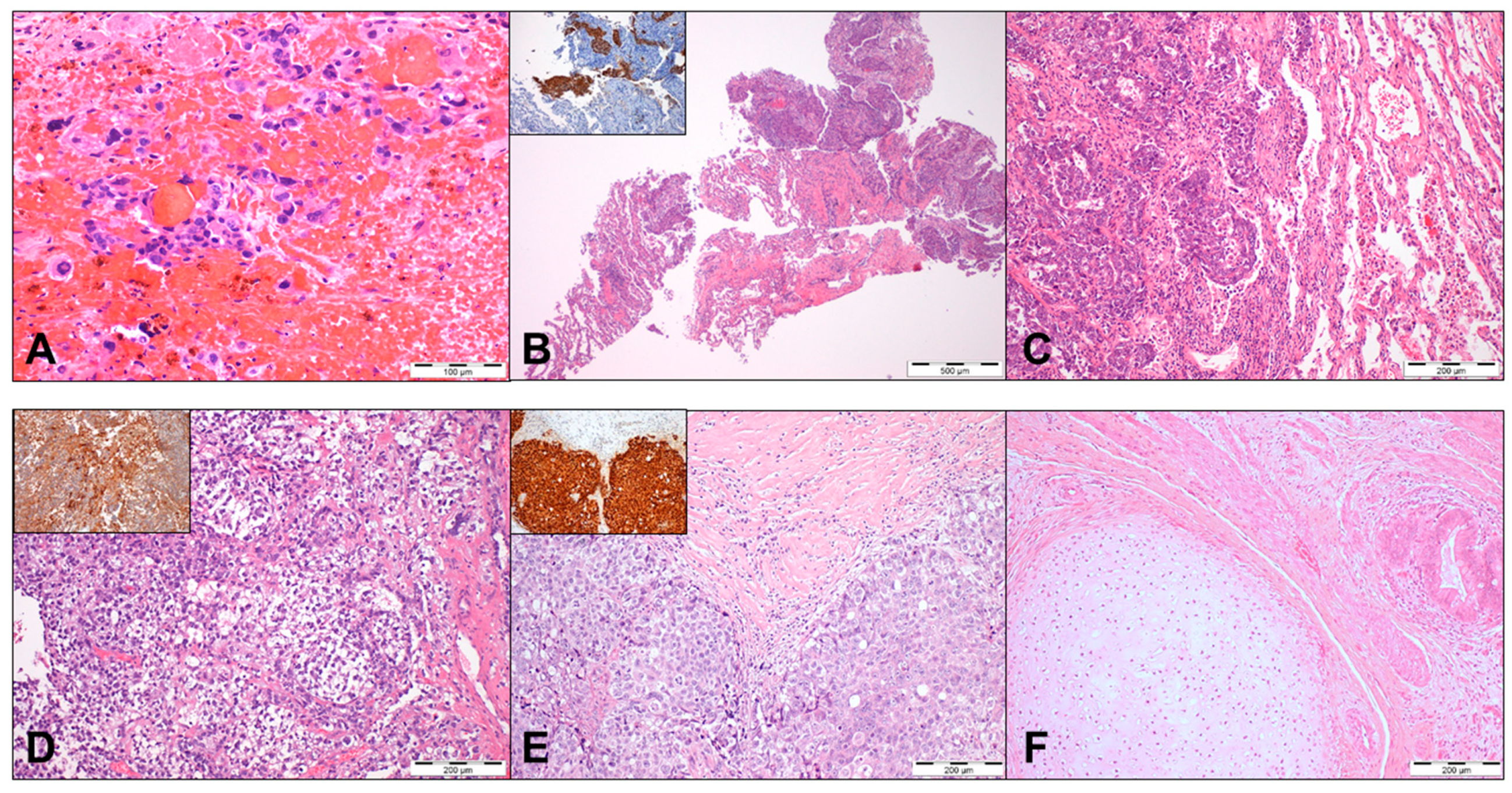
- Ronchi, A.; Pagliuca, F.; Franco, R. Testicular germ cell tumors: The changing role of the pathologist. Ann. Transl. Med. 2019, 7, S204. [Google Scholar] [CrossRef] [PubMed]
- Verrill, C.; Perry-Keene, J.; Srigley, J.R.; Zhou, M.; Humphrey, P.A.; Lopez-Beltran, A.; Egevad, L.; Ulbright, T.M.; Tickoo, S.K.; Epstein, J.I.; et al. Intraoperative Consultation and Macroscopic Handling: The International Society of Urological Pathology (ISUP) Testicular Cancer Consultation Conference Recommendations. Am. J. Surg. Pathol. 2018, 42, e33–e43. [Google Scholar] [CrossRef] [PubMed]
- Honecker, F.; Wermann, H.; Mayer, F.; Gillis, A.J.; Stoop, H.; van Gurp, R.J.; Oechsle, K.; Steyerberg, E.; Hartmann, J.T.; Dinjens, W.N.; et al. Microsatellite instability, mismatch repair deficiency, and BRAF mutation in treatment-resistant germ cell tumors. J. Clin. Oncol. 2009, 27, 2129–2136. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Rodrigues, A.; Guimaraes, R.; Cantante, M.; Lopes, P.; Mauricio, J.; Oliveira, J.; Jeronimo, C.; Henrique, R. Detailed Characterization of Immune Cell Infiltrate and Expression of Immune Checkpoint Molecules PD-L1/CTLA-4 and MMR Proteins in Testicular Germ Cell Tumors Disclose Novel Disease Biomarkers. Cancers (Basel) 2019, 11, 1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, F.; Gillis, A.J.; Dinjens, W.; Oosterhuis, J.W.; Bokemeyer, C.; Looijenga, L.H. Microsatellite instability of germ cell tumors is associated with resistance to systemic treatment. Cancer Res. 2002, 62, 2758–2760. [Google Scholar]
- Loveday, C.; Litchfield, K.; Proszek, P.Z.; Cornish, A.J.; Santo, F.; Levy, M.; Macintyre, G.; Holryod, A.; Broderick, P.; Dudakia, D.; et al. Genomic landscape of platinum resistant and sensitive testicular cancers. Nat. Commun. 2020, 11, 2189. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Shih, J.; Hollern, D.P.; Wang, L.; Bowlby, R.; Tickoo, S.K.; Thorsson, V.; Mungall, A.J.; Newton, Y.; Hegde, A.M.; et al. Integrated Molecular Characterization of Testicular Germ Cell Tumors. Cell Rep. 2018, 23, 3392–3406. [Google Scholar] [CrossRef] [PubMed]
- Kita, H.; Okamoto, K.; Kushima, R.; Kawauchi, A.; Chano, T. Dimethyl sulfoxide induces chemotherapeutic resistance in the treatment of testicular embryonal carcinomas. Oncol. Lett. 2015, 10, 661–666. [Google Scholar] [CrossRef]
- Lobo, J.; Gillis, A.J.M.; van den Berg, A.; Dorssers, L.C.J.; Belge, G.; Dieckmann, K.P.; Roest, H.P.; van der Laan, L.J.W.; Gietema, J.; Hamilton, R.J.; et al. Identification and Validation Model for Informative Liquid Biopsy-Based microRNA Biomarkers: Insights from Germ Cell Tumor In Vitro, In Vivo and Patient-Derived Data. Cells 2019, 8, 1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piulats, J.M.; Vidal, A.; Garcia-Rodriguez, F.J.; Munoz, C.; Nadal, M.; Moutinho, C.; Martinez-Iniesta, M.; Mora, J.; Figueras, A.; Guino, E.; et al. Orthoxenografts of Testicular Germ Cell Tumors Demonstrate Genomic Changes Associated with Cisplatin Resistance and Identify PDMP as a Resensitizing Agent. Clin. Cancer Res. 2018, 24, 3755–3766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidtova, S.; Kalavska, K.; Gercakova, K.; Cierna, Z.; Miklikova, S.; Smolkova, B.; Buocikova, V.; Miskovska, V.; Durinikova, E.; Burikova, M.; et al. Disulfiram Overcomes Cisplatin Resistance in Human Embryonal Carcinoma Cells. Cancers (Basel) 2019, 11, 1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, H.; Nooter, K.; Boersma, A.W.; van Wingerden, K.E.; Looijenga, L.H.; Jochemsen, A.G.; Stoter, G. Distinct p53-independent apoptotic cell death signalling pathways in testicular germ cell tumour cell lines. Int. J. Cancer 1999, 81, 620–628. [Google Scholar] [CrossRef]
- Port, M.; Glaesener, S.; Ruf, C.; Riecke, A.; Bokemeyer, C.; Meineke, V.; Honecker, F.; Abend, M. Micro-RNA expression in cisplatin resistant germ cell tumor cell lines. Mol. Cancer 2011, 10, 52. [Google Scholar] [CrossRef] [Green Version]
- Nitzsche, B.; Gloesenkamp, C.; Schrader, M.; Hoffmann, B.; Zengerling, F.; Balabanov, S.; Honecker, F.; Hopfner, M. Anti-tumour activity of two novel compounds in cisplatin-resistant testicular germ cell cancer. Br. J. Cancer 2012, 107, 1853–1863. [Google Scholar] [CrossRef] [Green Version]
- De Jong, J.; Stoop, H.; Gillis, A.J.; Hersmus, R.; van Gurp, R.J.; van de Geijn, G.J.; van Drunen, E.; Beverloo, H.B.; Schneider, D.T.; Sherlock, J.K.; et al. Further characterization of the first seminoma cell line TCam-2. Genes Chromosomes Cancer 2008, 47, 185–196. [Google Scholar] [CrossRef]
- Mizuno, Y.; Gotoh, A.; Kamidono, S.; Kitazawa, S. Establishment and characterization of a new human testicular germ cell tumor cell line (TCam-2). Nihon Hinyokika Gakkai Zasshi 1993, 84, 1211–1218. [Google Scholar] [CrossRef] [Green Version]
- Timmerman, D.M.; Lobo, J.; Gillis, A.; Remmers, T.; Schmidtova, S.; Kalavska, K.; Hulleman, E.; Oing, C.; Dorssers, L.; Ulbright, T.; et al. Sequential 12q- and 3p-amplification is associated with cisplatin resistance in male germ cell cancers. 2020; in preparation. [Google Scholar]
- Schaffrath, J.; Schmoll, H.J.; Voigt, W.; Muller, L.P.; Muller-Tidow, C.; Mueller, T. Efficacy of targeted drugs in germ cell cancer cell lines with differential cisplatin sensitivity. PLoS ONE 2017, 12, e0178930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmer-Bosscha, H.; Timmer, A.; Meijer, C.; de Vries, E.G.; de Jong, B.; Oosterhuis, J.W.; Mulder, N.H. cis-Diamminedichloroplatinum(ii) resistance in vitro and in vivo in human embryonal carcinoma cells. Cancer Res. 1993, 53, 5707–5713. [Google Scholar] [PubMed]
- Mueller, T.; Mueller, L.P.; Luetzkendorf, J.; Voigt, W.; Simon, H.; Schmoll, H.J. Loss of Oct-3/4 expression in embryonal carcinoma cells is associated with induction of cisplatin resistance. Tumor Biol. 2006, 27, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.; Powles, T.; Shamash, J.; Veerupillai, A.; McGrowder, E.; Noel, E.; Lu, Y.J.; Oliver, T.; Joel, S. The relative activity of cisplatin, oxaliplatin and satraplatin in testicular germ cell tumour sensitive and resistant cell lines. Cancer Chemother. Pharmacol. 2009, 64, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.; Mueller, L.P.; Holzhausen, H.J.; Witthuhn, R.; Albers, P.; Schmoll, H.J. Histological evidence for the existence of germ cell tumor cells showing embryonal carcinoma morphology but lacking OCT4 expression and cisplatin sensitivity. Histochem. Cell Biol. 2010, 134, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Umezu, T.; Sakurai, M.; Kajiyama, H.; Yamamoto, E.; Ino, K.; Nawa, A.; Kikkawa, F. Establishment of cisplatin-resistant ovarian yolk sac tumor cells and investigation of the mechanism of cisplatin resistance using this cell line. Gynecol. Obstet. Invest. 2011, 71, 104–111. [Google Scholar] [CrossRef]
- Minucci, S.; Horn, V.; Bhattacharyya, N.; Russanova, V.; Ogryzko, V.V.; Gabriele, L.; Howard, B.H.; Ozato, K. A histone deacetylase inhibitor potentiates retinoid receptor action in embryonal carcinoma cells. Proc. Natl. Acad. Sci. USA 1997, 94, 11295–11300. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.; Kumar, S.; Tchounwou, P.B. Cisplatin-Based Chemotherapy of Human Cancers. J. Cancer Sci. Ther. 2019, 11, 4. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef] [Green Version]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.H.; Chang, J.Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int. J. Mol. Sci. 2019, 20, 4136. [Google Scholar] [CrossRef] [Green Version]
- Mayer, F.; Honecker, F.; Looijenga, L.H.; Bokemeyer, C. Towards an understanding of the biological basis of response to cisplatin-based chemotherapy in germ-cell tumors. Ann. Oncol. 2003, 14, 825–832. [Google Scholar] [CrossRef]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, F.; Stoop, H.; Scheffer, G.L.; Scheper, R.; Oosterhuis, J.W.; Looijenga, L.H.; Bokemeyer, C. Molecular determinants of treatment response in human germ cell tumors. Clin. Cancer Res. 2003, 9, 767–773. [Google Scholar] [PubMed]
- Chen, H.H.; Kuo, M.T. Role of glutathione in the regulation of Cisplatin resistance in cancer chemotherapy. Met. Drugs 2010, 2010, 430939. [Google Scholar] [CrossRef] [PubMed]
- Masters, J.R.; Thomas, R.; Hall, A.G.; Hogarth, L.; Matheson, E.C.; Cattan, A.R.; Lohrer, H. Sensitivity of testis tumour cells to chemotherapeutic drugs: Role of detoxifying pathways. Eur. J. Cancer 1996, 32A, 1248–1253. [Google Scholar] [CrossRef]
- Koropatnick, J.; Kloth, D.M.; Kadhim, S.; Chin, J.L.; Cherian, M.G. Metallothionein expression and resistance to cisplatin in a human germ cell tumor cell line. J. Pharmacol. Exp. Ther. 1995, 275, 1681–1687. [Google Scholar]
- Meijer, C.; Timmer, A.; De Vries, E.G.; Groten, J.P.; Knol, A.; Zwart, N.; Dam, W.A.; Sleijfer, D.T.; Mulder, N.H. Role of metallothionein in cisplatin sensitivity of germ-cell tumours. Int. J. Cancer 2000, 85, 777–781. [Google Scholar] [CrossRef]
- Bloom, J.C.; Loehr, A.R.; Schimenti, J.C.; Weiss, R.S. Germline genome protection: Implications for gamete quality and germ cell tumorigenesis. Andrology 2019, 7, 516–526. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, A.K.; Han, C.; Zhao, R.; Cui, T.; Dai, Y.; Mao, C.; Zhao, W.; Zhang, X.; Yu, J.; Wang, Q.E. Enhanced expression of DNA polymerase eta contributes to cisplatin resistance of ovarian cancer stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, 4411–4416. [Google Scholar] [CrossRef] [Green Version]
- Kersemaekers, A.M.; Mayer, F.; Molier, M.; van Weeren, P.C.; Oosterhuis, J.W.; Bokemeyer, C.; Looijenga, L.H. Role of P53 and MDM2 in treatment response of human germ cell tumors. J. Clin. Oncol. 2002, 20, 1551–1561. [Google Scholar] [CrossRef]
- Michaud, W.A.; Nichols, A.C.; Mroz, E.A.; Faquin, W.C.; Clark, J.R.; Begum, S.; Westra, W.H.; Wada, H.; Busse, P.M.; Ellisen, L.W.; et al. Bcl-2 blocks cisplatin-induced apoptosis and predicts poor outcome following chemoradiation treatment in advanced oropharyngeal squamous cell carcinoma. Clin. Cancer Res. 2009, 15, 1645–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.H.; He, W.S.; Nong, L.; Zhu, Q.Y.; Hu, K.; Zhang, R.G.; Huang, L.L.; Zhu, F.; Wu, G. Acquired cisplatin resistance in human lung adenocarcinoma cells is associated with enhanced autophagy. Cancer Biother. Radiopharm. 2010, 25, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shen, X. Heat shock protein 27 protects L929 cells from cisplatin-induced apoptosis by enhancing Akt activation and abating suppression of thioredoxin reductase activity. Clin. Cancer Res. 2007, 13, 2855–2864. [Google Scholar] [CrossRef] [Green Version]
- Peng, D.; Wei, J.; Gan, Y.; Yang, J.; Jiang, X.; Kitazawa, R.; Xiang, Y.; Dai, Y.; Tang, Y. Testis developmental related gene 1 regulates the chemosensitivity of seminoma TCam-2 cells to cisplatin via autophagy. J. Cell Mol. Med. 2019, 23, 7773–7784. [Google Scholar] [CrossRef] [PubMed]
- Juliachs, M.; Munoz, C.; Moutinho, C.A.; Vidal, A.; Condom, E.; Esteller, M.; Graupera, M.; Casanovas, O.; Germa, J.R.; Villanueva, A.; et al. The PDGFRbeta-AKT pathway contributes to CDDP-acquired resistance in testicular germ cell tumors. Clin. Cancer Res. 2014, 20, 658–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaisman, A.; Varchenko, M.; Umar, A.; Kunkel, T.A.; Risinger, J.I.; Barrett, J.C.; Hamilton, T.C.; Chaney, S.G. The role of hMLH1, hMSH3, and hMSH6 defects in cisplatin and oxaliplatin resistance: Correlation with replicative bypass of platinum-DNA adducts. Cancer Res. 1998, 58, 3579–3585. [Google Scholar]
- Olasz, J.; Mandoky, L.; Geczi, L.; Bodrogi, I.; Csuka, O.; Bak, M. Influence of hMLH1 methylation, mismatch repair deficiency and microsatellite instability on chemoresistance of testicular germ-cell tumors. Anticancer Res. 2005, 25, 4319–4324. [Google Scholar]
- Lobo, J.; Pinto, C.; Pinheiro, M.; Lobo, F.; Sousa, N.; Lopes, P.; Looijenga, L.H.; Jeronimo, C.; Teixeira, M.R.; Henrique, R. Widening the spectrum of Lynch syndrome: First report of testicular seminoma attributable to MSH2 loss. Histopathology 2020, 76, 486–489. [Google Scholar] [CrossRef]
- Rudolph, C.; Melau, C.; Nielsen, J.E.; Vile Jensen, K.; Liu, D.; Pena-Diaz, J.; Rajpert-De Meyts, E.; Rasmussen, L.J.; Jorgensen, A. Involvement of the DNA mismatch repair system in cisplatin sensitivity of testicular germ cell tumours. Cell Oncol. 2017, 40, 341–355. [Google Scholar] [CrossRef]
- Velasco, A.; Riquelme, E.; Schultz, M.; Wistuba, I.I.; Villarroel, L.; Pizarro, J.; Berlin, A.; Ittmann, M.; Koh, M.S.; Leach, F.S. Mismatch repair gene expression and genetic instability in testicular germ cell tumor. Cancer Biol. Ther. 2004, 3, 977–982. [Google Scholar] [CrossRef] [Green Version]
- Carcano, F.M.; Lengert, A.H.; Vidal, D.O.; Scapulatempo Neto, C.; Queiroz, L.; Marques, H.; Baltazar, F.; Berardinelli, G.N.; Martinelli, C.M.; da Silva, E.C.; et al. Absence of microsatellite instability and BRAF (V600E) mutation in testicular germ cell tumors. Andrology 2016, 4, 866–872. [Google Scholar] [CrossRef] [Green Version]
- Bassett, E.; Vaisman, A.; Tropea, K.A.; McCall, C.M.; Masutani, C.; Hanaoka, F.; Chaney, S.G. Frameshifts and deletions during in vitro translesion synthesis past Pt-DNA adducts by DNA polymerases beta and eta. DNA Repair 2002, 1, 1003–1016. [Google Scholar] [CrossRef]
- Welsh, C.; Day, R.; McGurk, C.; Masters, J.R.; Wood, R.D.; Koberle, B. Reduced levels of XPA, ERCC1 and XPF DNA repair proteins in testis tumor cell lines. Int. J. Cancer 2004, 110, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Koberle, B.; Masters, J.R.; Hartley, J.A.; Wood, R.D. Defective repair of cisplatin-induced DNA damage caused by reduced XPA protein in testicular germ cell tumours. Curr. Biol. 1999, 9, 273–276. [Google Scholar] [CrossRef] [Green Version]
- Cierna, Z.; Miskovska, V.; Roska, J.; Jurkovicova, D.; Pulzova, L.B.; Sestakova, Z.; Hurbanova, L.; Machalekova, K.; Chovanec, M.; Rejlekova, K.; et al. Increased levels of XPA might be the basis of cisplatin resistance in germ cell tumours. BMC Cancer 2020, 20, 17. [Google Scholar] [CrossRef]
- Mendoza, J.; Martinez, J.; Hernandez, C.; Perez-Montiel, D.; Castro, C.; Fabian-Morales, E.; Santibanez, M.; Gonzalez-Barrios, R.; Diaz-Chavez, J.; Andonegui, M.A.; et al. Association between ERCC1 and XPA expression and polymorphisms and the response to cisplatin in testicular germ cell tumours. Br. J. Cancer 2013, 109, 68–75. [Google Scholar] [CrossRef]
- Awuah, S.G.; Riddell, I.A.; Lippard, S.J. Repair shielding of platinum-DNA lesions in testicular germ cell tumors by high-mobility group box protein 4 imparts cisplatin hypersensitivity. Proc. Natl. Acad. Sci. USA 2017, 114, 950–955. [Google Scholar] [CrossRef] [Green Version]
- Koberle, B.; Roginskaya, V.; Zima, K.S.; Masters, J.R.; Wood, R.D. Elevation of XPA protein level in testis tumor cells without increasing resistance to cisplatin or UV radiation. Mol. Carcinog. 2008, 47, 580–586. [Google Scholar] [CrossRef]
- Honecker, F.; Mayer, F.; Stoop, H.; Oosterhuis, J.W.; Koch, S.; Bokemeyer, C.; Looijenga, L.H. Xeroderma pigmentosum group a protein and chemotherapy resistance in human germ cell tumors. Lab. Investig. 2003, 83, 1489–1495. [Google Scholar] [CrossRef] [Green Version]
- Koberle, B.; Payne, J.; Grimaldi, K.A.; Hartley, J.A.; Masters, J.R. DNA repair in cisplatin-sensitive and resistant human cell lines measured in specific genes by quantitative polymerase chain reaction. Biochem. Pharmacol. 1996, 52, 1729–1734. [Google Scholar] [CrossRef]
- Robertson, K.A.; Bullock, H.A.; Xu, Y.; Tritt, R.; Zimmerman, E.; Ulbright, T.M.; Foster, R.S.; Einhorn, L.H.; Kelley, M.R. Altered expression of Ape1/ref-1 in germ cell tumors and overexpression in NT2 cells confers resistance to bleomycin and radiation. Cancer Res. 2001, 61, 2220–2225. [Google Scholar]
- Cavallo, F.; Graziani, G.; Antinozzi, C.; Feldman, D.R.; Houldsworth, J.; Bosl, G.J.; Chaganti, R.S.; Moynahan, M.E.; Jasin, M.; Barchi, M. Reduced proficiency in homologous recombination underlies the high sensitivity of embryonal carcinoma testicular germ cell tumors to Cisplatin and poly (adp-ribose) polymerase inhibition. PLoS ONE 2012, 7, e51563. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Mego, M.; Cierna, Z.; Svetlovska, D.; Macak, D.; Machalekova, K.; Miskovska, V.; Chovanec, M.; Usakova, V.; Obertova, J.; Babal, P.; et al. PARP expression in germ cell tumours. J. Clin. Pathol. 2013, 66, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Cavallo, F.; Feldman, D.R.; Barchi, M. Revisiting DNA damage repair, p53-mediated apoptosis and cisplatin sensitivity in germ cell tumors. Int. J. Dev. Biol. 2013, 57, 273–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riou, G.; Barrois, M.; Prost, S.; Terrier, M.J.; Theodore, C.; Levine, A.J. The p53 and mdm-2 genes in human testicular germ-cell tumors. Mol. Carcinog. 1995, 12, 124–131. [Google Scholar] [CrossRef]
- Bartkova, J.; Rajpert-De Meyts, E.; Skakkebaek, N.E.; Lukas, J.; Bartek, J. DNA damage response in human testes and testicular germ cell tumours: Biology and implications for therapy. Int. J. Androl. 2007, 30, 282–291; discussion 291. [Google Scholar] [CrossRef]
- Guillou, L.; Estreicher, A.; Chaubert, P.; Hurlimann, J.; Kurt, A.M.; Metthez, G.; Iggo, R.; Gray, A.C.; Jichlinski, P.; Leisinger, H.J.; et al. Germ cell tumors of the testis overexpress wild-type p53. Am. J. Pathol. 1996, 149, 1221–1228. [Google Scholar]
- Gutekunst, M.; Oren, M.; Weilbacher, A.; Dengler, M.A.; Markwardt, C.; Thomale, J.; Aulitzky, W.E.; van der Kuip, H. p53 hypersensitivity is the predominant mechanism of the unique responsiveness of testicular germ cell tumor (TGCT) cells to cisplatin. PLoS ONE 2011, 6, e19198. [Google Scholar] [CrossRef]
- Gutekunst, M.; Mueller, T.; Weilbacher, A.; Dengler, M.A.; Bedke, J.; Kruck, S.; Oren, M.; Aulitzky, W.E.; van der Kuip, H. Cisplatin hypersensitivity of testicular germ cell tumors is determined by high constitutive Noxa levels mediated by Oct-4. Cancer Res. 2013, 73, 1460–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houldsworth, J.; Xiao, H.; Murty, V.V.; Chen, W.; Ray, B.; Reuter, V.E.; Bosl, G.J.; Chaganti, R.S. Human male germ cell tumor resistance to cisplatin is linked to TP53 gene mutation. Oncogene 1998, 16, 2345–2349. [Google Scholar] [CrossRef] [Green Version]
- Bagrodia, A.; Lee, B.H.; Lee, W.; Cha, E.K.; Sfakianos, J.P.; Iyer, G.; Pietzak, E.J.; Gao, S.P.; Zabor, E.C.; Ostrovnaya, I.; et al. Genetic Determinants of Cisplatin Resistance in Patients With Advanced Germ Cell Tumors. J. Clin. Oncol. 2016, 34, 4000–4007. [Google Scholar] [CrossRef]
- Di Pietro, A.; Koster, R.; Boersma-van Eck, W.; Dam, W.A.; Mulder, N.H.; Gietema, J.A.; de Vries, E.G.; de Jong, S. Pro- and anti-apoptotic effects of p53 in cisplatin-treated human testicular cancer are cell context-dependent. Cell Cycle 2012, 11, 4552–4562. [Google Scholar] [CrossRef] [Green Version]
- Grande, L.; Bretones, G.; Rosa-Garrido, M.; Garrido-Martin, E.M.; Hernandez, T.; Fraile, S.; Botella, L.; de Alava, E.; Vidal, A.; Garcia del Muro, X.; et al. Transcription factors Sp1 and p73 control the expression of the proapoptotic protein NOXA in the response of testicular embryonal carcinoma cells to cisplatin. J. Biol. Chem. 2012, 287, 26495–26505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, U.; Moll-Rocek, J.; Moll, U.M.; Dobbelstein, M. Endogenous retrovirus drives hitherto unknown proapoptotic p63 isoforms in the male germ line of humans and great apes. Proc. Natl. Acad. Sci. USA 2011, 108, 3624–3629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voorhoeve, P.M.; le Sage, C.; Schrier, M.; Gillis, A.J.; Stoop, H.; Nagel, R.; Liu, Y.P.; van Duijse, J.; Drost, J.; Griekspoor, A.; et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell 2006, 124, 1169–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, S.; Muhlenberg, T.; Leahy, M.; Hoiczyk, M.; Gauler, T.; Schuler, M.; Looijenga, L. Therapeutic potential of Mdm2 inhibition in malignant germ cell tumours. Eur. Urol. 2010, 57, 679–687. [Google Scholar] [CrossRef]
- Koster, R.; Timmer-Bosscha, H.; Bischoff, R.; Gietema, J.A.; de Jong, S. Disruption of the MDM2-p53 interaction strongly potentiates p53-dependent apoptosis in cisplatin-resistant human testicular carcinoma cells via the Fas/FasL pathway. Cell Death Dis. 2011, 2, e148. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Cheng, Q.; Li, Z.; Chen, J. p53 inactivation by MDM2 and MDMX negative feedback loops in testicular germ cell tumors. Cell Cycle 2010, 9, 1411–1420. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Velasco, A.; Duran, I.; Garcia, E.; Taron, M.; Ballestin, C.; Castellanos, D.; Cortes-Funes, H.; Paz-Ares, L. Biological markers of cisplatin resistance in advanced testicular germ cell tumours. Clin. Transl. Oncol. 2012, 14, 452–457. [Google Scholar] [CrossRef]
- Mandoky, L.; Szende, B.; Geczi, L.; Bodrogi, I.; Kasler, M.; Bak, M. Apoptosis regulation and spontaneous apoptosis index of testicular germ cell tumors are associated with differentiation and resistance to systemic treatment. Anticancer Res. 2008, 28, 1641–1649. [Google Scholar] [PubMed]
- Lobo, J.; Alzamora, M.A.; Guimaraes, R.; Cantante, M.; Lopes, P.; Braga, I.; Mauricio, J.; Jeronimo, C.; Henrique, R. p53 and MDM2 expression in primary and metastatic testicular germ cell tumors: Association with clinical outcome. Andrology 2020. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Mejia-Hernandez, J.O.; Vijayakumaran, R.; Keam, S.P.; Haupt, Y. The long and the short of it: The MDM4 tail so far. J. Mol. Cell Biol. 2019, 11, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Nappi, L.; Annala, M.; Vandekerkhove, G.; Fazli, L.; Gleave, M.; Chi, K.N.; Kollmannsberger, C.K.; Wyatt, A.W. Molecular dissection of primary mediastinal germ cell tumors. J. Clin. Oncol. 2017, 35, 417. [Google Scholar] [CrossRef]
- Ronchi, A.; Cozzolino, I.; Montella, M.; Panarese, I.; Zito Marino, F.; Rossetti, S.; Chieffi, P.; Accardo, M.; Facchini, G.; Franco, R. Extragonadal germ cell tumors: Not just a matter of location. A review about clinical, molecular and pathological features. Cancer Med. 2019, 8, 6832–6840. [Google Scholar] [CrossRef]
- Lu, C.; Riedell, P.; Miller, C.A.; Hagemann, I.S.; Westervelt, P.; Ozenberger, B.A.; O’Laughlin, M.; Magrini, V.; Demeter, R.T.; Duncavage, E.J.; et al. A common founding clone with TP53 and PTEN mutations gives rise to a concurrent germ cell tumor and acute megakaryoblastic leukemia. Mol. Case Stud. 2016, 2, a000687. [Google Scholar] [CrossRef] [Green Version]
- Chu, J.; Shi, Z.; Jiao, Y.; Han, Z.; Dou, Q.; Ye, J.; Cui, X. B-cell lymphoma 2 ovarian killer suppresses testicular cancer cell malignant behavior, but plays a role in platinum resistance. Anticancer Drugs 2018, 29, 839–846. [Google Scholar] [CrossRef]
- Sano, M.; Nakanishi, Y.; Yagasaki, H.; Honma, T.; Oinuma, T.; Obana, Y.; Suzuki, A.; Nemoto, N. Overexpression of anti-apoptotic Mcl-1 in testicular germ cell tumours. Histopathology 2005, 46, 532–539. [Google Scholar] [CrossRef]
- Chresta, C.M.; Masters, J.R.; Hickman, J.A. Hypersensitivity of human testicular tumors to etoposide-induced apoptosis is associated with functional p53 and a high Bax:Bcl-2 ratio. Cancer Res. 1996, 56, 1834–1841. [Google Scholar]
- Burger, H.; Nooter, K.; Boersma, A.W.; Kortland, C.J.; Stoter, G. Expression of p53, Bcl-2 and Bax in cisplatin-induced apoptosis in testicular germ cell tumour cell lines. Br. J. Cancer 1998, 77, 1562–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltaci, S.; Orhan, D.; Turkolmez, K.; Yesilli, C.; Beduk, Y.; Tulunay, O. P53, bcl-2 and bax immunoreactivity as predictors of response and outcome after chemotherapy for metastatic germ cell testicular tumours. BJU Int. 2001, 87, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Noel, E.E.; Yeste-Velasco, M.; Mao, X.; Perry, J.; Kudahetti, S.C.; Li, N.F.; Sharp, S.; Chaplin, T.; Xue, L.; McIntyre, A.; et al. The association of CCND1 overexpression and cisplatin resistance in testicular germ cell tumors and other cancers. Am. J. Pathol. 2010, 176, 2607–2615. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.L.; Lobo, J.; Jeronimo, C.; Henrique, R. The epigenetics of testicular germ cell tumors: Looking for novel disease biomarkers. Epigenomics 2017, 9, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Koul, S.; McKiernan, J.M.; Narayan, G.; Houldsworth, J.; Bacik, J.; Dobrzynski, D.L.; Assaad, A.M.; Mansukhani, M.; Reuter, V.E.; Bosl, G.J.; et al. Role of promoter hypermethylation in Cisplatin treatment response of male germ cell tumors. Mol. Cancer 2004, 3, 16. [Google Scholar] [CrossRef] [Green Version]
- Markulin, D.; Vojta, A.; Samarzija, I.; Gamulin, M.; Beceheli, I.; Jukic, I.; Maglov, C.; Zoldos, V.; Fucic, A. Association between RASSF1A Promoter Methylation and Testicular Germ Cell Tumor: A Meta-analysis and a Cohort Study. Cancer Genom. Proteom. 2017, 14, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Ellinger, J.; Albers, P.; Perabo, F.G.; Muller, S.C.; von Ruecker, A.; Bastian, P.J. CpG island hypermethylation of cell-free circulating serum DNA in patients with testicular cancer. J. Urol. 2009, 182, 324–329. [Google Scholar] [CrossRef]
- Martinelli, C.; Lengert, A.V.H.; Carcano, F.M.; Silva, E.C.A.; Brait, M.; Lopes, L.F.; Vidal, D.O. MGMT and CALCA promoter methylation are associated with poor prognosis in testicular germ cell tumor patients. Oncotarget 2017, 8, 50608–50617. [Google Scholar] [CrossRef] [Green Version]
- Beyrouthy, M.J.; Garner, K.M.; Hever, M.P.; Freemantle, S.J.; Eastman, A.; Dmitrovsky, E.; Spinella, M.J. High DNA methyltransferase 3B expression mediates 5-aza-deoxycytidine hypersensitivity in testicular germ cell tumors. Cancer Res. 2009, 69, 9360–9366. [Google Scholar] [CrossRef] [Green Version]
- Wongtrakoongate, P.; Li, J.; Andrews, P.W. Aza-deoxycytidine induces apoptosis or differentiation via DNMT3B and targets embryonal carcinoma cells but not their differentiated derivatives. Br. J. Cancer 2014, 110, 2131–2138. [Google Scholar] [CrossRef] [Green Version]
- Biswal, B.K.; Beyrouthy, M.J.; Hever-Jardine, M.P.; Armstrong, D.; Tomlinson, C.R.; Christensen, B.C.; Marsit, C.J.; Spinella, M.J. Acute hypersensitivity of pluripotent testicular cancer-derived embryonal carcinoma to low-dose 5-aza deoxycytidine is associated with global DNA Damage-associated p53 activation, anti-pluripotency and DNA demethylation. PLoS ONE 2012, 7, e53003. [Google Scholar] [CrossRef]
- Wermann, H.; Stoop, H.; Gillis, A.J.; Honecker, F.; van Gurp, R.J.; Ammerpohl, O.; Richter, J.; Oosterhuis, J.W.; Bokemeyer, C.; Looijenga, L.H. Global DNA methylation in fetal human germ cells and germ cell tumours: Association with differentiation and cisplatin resistance. J. Pathol. 2010, 221, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Fazal, Z.; Corbet, A.K.; Bikorimana, E.; Rodriguez, J.C.; Khan, E.M.; Shahid, K.; Freemantle, S.J.; Spinella, M.J. Epigenetic Remodeling through Downregulation of Polycomb Repressive Complex 2 Mediates Chemotherapy Resistance in Testicular Germ Cell Tumors. Cancers (Basel) 2019, 11, 796. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, T.H.; Liu, Y.R.; Chang, T.Y.; Liang, M.L.; Chen, H.H.; Wang, H.W.; Yen, Y.; Wong, T.T. Global DNA methylation analysis reveals miR-214-3p contributes to cisplatin resistance in pediatric intracranial nongerminomatous malignant germ cell tumors. Neuro-Oncology 2018, 20, 519–530. [Google Scholar] [CrossRef]
- Liu, L.; Lian, J.; Zhang, H.; Tian, H.; Liang, M.; Yin, M.; Sun, F. MicroRNA-302a sensitizes testicular embryonal carcinoma cells to cisplatin-induced cell death. J. Cell Physiol. 2013, 228, 2294–2304. [Google Scholar] [CrossRef]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 9, 3970. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J.; Berger, S.L. The interplay between epigenetic changes and the p53 protein in stem cells. Genes Dev. 2017, 31, 1195–1201. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Perez-Burgos, L.; Placek, B.J.; Sengupta, R.; Richter, M.; Dorsey, J.A.; Kubicek, S.; Opravil, S.; Jenuwein, T.; Berger, S.L. Repression of p53 activity by Smyd2-mediated methylation. Nature 2006, 444, 629–632. [Google Scholar] [CrossRef]
- Shi, X.; Kachirskaia, I.; Yamaguchi, H.; West, L.E.; Wen, H.; Wang, E.W.; Dutta, S.; Appella, E.; Gozani, O. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol. Cell 2007, 27, 636–646. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Dou, Z.; Sammons, M.A.; Levine, A.J.; Berger, S.L. Lysine methylation represses p53 activity in teratocarcinoma cancer cells. Proc. Natl. Acad. Sci. USA 2016, 113, 9822–9827. [Google Scholar] [CrossRef] [Green Version]
- Dorssers, L.C.J.; Gillis, A.J.M.; Stoop, H.; van Marion, R.; Nieboer, M.M.; van Riet, J.; van de Werken, H.J.G.; Oosterhuis, J.W.; de Ridder, J.; Looijenga, L.H.J. Molecular heterogeneity and early metastatic clone selection in testicular germ cell cancer development. Br. J. Cancer 2019, 120, 444–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, M.T.; Lenkiewicz, E.; Malasi, S.; Stanton, M.; Slack, J.; Andrews, P.; Pagliaro, L.; Bryce, A.H. Clonal analyses of refractory testicular germ cell tumors. PLoS ONE 2019, 14, e0213815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Necchi, A.; Bratslavsky, G.; Corona, R.J.; Chung, J.H.; Millis, S.Z.; Elvin, J.A.; Vergilio, J.A.; Suh, J.; Ramkissoon, S.; Severson, E.; et al. Genomic Characterization of Testicular Germ Cell Tumors Relapsing After Chemotherapy. Eur. Urol. Focus 2020, 6, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.M.; Buckley, C.; Holley, S.; Menias, C.O. Teratomas: A multimodality review. Curr. Probl. Diagn. Radiol. 2012, 41, 210–219. [Google Scholar] [CrossRef]
- David, S.; Andras, F.; Endre, K.; Balint, K.; Arpad, K.; Csaba, P.; Karoly, S.; Tamas, T. More Cases of Benign Testicular Teratomas are Detected in Adults than in Children. A Clinicopathological Study of 543 Testicular Germ Cell Tumor Cases. Pathol. Oncol. Res. 2017, 23, 513–517. [Google Scholar] [CrossRef]
- Anheuser, P.; Kranz, J.; Stolle, E.; Hoflmayer, D.; Buscheck, F.; Muhlstadt, S.; Lock, G.; Dieckmann, K.P. Testicular epidermoid cysts: A reevaluation. BMC Urol. 2019, 19, 52. [Google Scholar] [CrossRef]
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2016, 70, 93–105. [Google Scholar] [CrossRef]
- Korkola, J.E.; Houldsworth, J.; Bosl, G.J.; Chaganti, R.S. Molecular events in germ cell tumours: Linking chromosome-12 gain, acquisition of pluripotency and response to cisplatin. BJU Int. 2009, 104, 1334–1338. [Google Scholar] [CrossRef]
- Van Echten, J.; Sleijfer, D.T.; Wiersema, J.; Schraffordt Koops, H.; Oosterhuis, J.W.; de Jong, B. Cytogenetics of primary testicular nonseminoma, residual mature teratoma, and growing teratoma lesion in individual patients. Cancer Genet. Cytogenet. 1997, 96, 1–6. [Google Scholar] [CrossRef]
- Einhorn, L.H.; Foster, R.S. What are the indications for postchemotherapy retroperitoneal lymph node dissection? Ann. Oncol. 2014, 25, 301–303. [Google Scholar] [CrossRef]
- Donohue, J.P.; Fox, E.P.; Williams, S.D.; Loehrer, P.J.; Ulbright, T.M.; Einhorn, L.H.; Weathers, T.D. Persistent cancer in postchemotherapy retroperitoneal lymph-node dissection: Outcome analysis. World J. Urol. 1994, 12, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Almstrup, K.; Lobo, J.; Morup, N.; Belge, G.; Rajpert-De Meyts, E.; Looijenga, L.H.J.; Dieckmann, K.P. Application of miRNAs in the diagnosis and monitoring of testicular germ cell tumours. Nat. Rev. Urol. 2020, 17, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Nettersheim, D.; Arndt, I.; Sharma, R.; Riesenberg, S.; Jostes, S.; Schneider, S.; Holzel, M.; Kristiansen, G.; Schorle, H. The cancer/testis-antigen PRAME supports the pluripotency network and represses somatic and germ cell differentiation programs in seminomas. Br. J. Cancer 2016, 115, 454–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nettersheim, D.; Gillis, A.; Biermann, K.; Looijenga, L.H.; Schorle, H. The seminoma cell line TCam-2 is sensitive to HDAC inhibitor depsipeptide but tolerates various other chemotherapeutic drugs and loss of NANOG expression. Genes Chromosomes Cancer 2011, 50, 1033–1042. [Google Scholar] [CrossRef]
- Skotheim, R.I.; Lind, G.E.; Monni, O.; Nesland, J.M.; Abeler, V.M.; Fossa, S.D.; Duale, N.; Brunborg, G.; Kallioniemi, O.; Andrews, P.W.; et al. Differentiation of human embryonal carcinomas in vitro and in vivo reveals expression profiles relevant to normal development. Cancer Res. 2005, 65, 5588–5598. [Google Scholar] [CrossRef] [Green Version]
- Honecker, F.; Rohlfing, T.; Harder, S.; Braig, M.; Gillis, A.J.; Glaesener, S.; Barett, C.; Bokemeyer, C.; Buck, F.; Brummendorf, T.H.; et al. Proteome analysis of the effects of all-trans retinoic acid on human germ cell tumor cell lines. J. Proteom. 2014, 96, 300–313. [Google Scholar] [CrossRef]
- Taylor-Weiner, A.; Zack, T.; O’Donnell, E.; Guerriero, J.L.; Bernard, B.; Reddy, A.; Han, G.C.; AlDubayan, S.; Amin-Mansour, A.; Schumacher, S.E.; et al. Genomic evolution and chemoresistance in germ-cell tumours. Nature 2016, 540, 114–118. [Google Scholar] [CrossRef]
- Koster, R.; di Pietro, A.; Timmer-Bosscha, H.; Gibcus, J.H.; van den Berg, A.; Suurmeijer, A.J.; Bischoff, R.; Gietema, J.A.; de Jong, S. Cytoplasmic p21 expression levels determine cisplatin resistance in human testicular cancer. J. Clin. Investig. 2010, 120, 3594–3605. [Google Scholar] [CrossRef] [Green Version]
- Looijenga, L.H.; Stoop, H.; de Leeuw, H.P.; de Gouveia Brazao, C.A.; Gillis, A.J.; van Roozendaal, K.E.; van Zoelen, E.J.; Weber, R.F.; Wolffenbuttel, K.P.; van Dekken, H.; et al. POU5F1 (OCT3/4) identifies cells with pluripotent potential in human germ cell tumors. Cancer Res. 2003, 63, 2244–2250. [Google Scholar]
- Moasser, M.M.; Motzer, R.J.; Khoo, K.S.; Lyn, P.; Murphy, B.A.; Bosl, G.J.; Dmitrovsky, E. All-trans retinoic acid for treating germ cell tumors. In vitro activity and results of a phase II trial. Cancer 1995, 76, 680–686. [Google Scholar] [CrossRef]
- Pierpont, T.M.; Lyndaker, A.M.; Anderson, C.M.; Jin, Q.; Moore, E.S.; Roden, J.L.; Braxton, A.; Bagepalli, L.; Kataria, N.; Hu, H.Z.; et al. Chemotherapy-Induced Depletion of OCT4-Positive Cancer Stem Cells in a Mouse Model of Malignant Testicular Cancer. Cell Rep. 2017, 21, 1896–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paffenholz, P.; Pfister, D.; Matveev, V.; Heidenreich, A. Diagnosis and management of the growing teratoma syndrome: A single-center experience and review of the literature. Urol. Oncol. 2018, 36, 529.e23–529.e30. [Google Scholar] [CrossRef] [PubMed]
- Logothetis, C.J.; Samuels, M.L.; Trindade, A.; Johnson, D.E. The growing teratoma syndrome. Cancer 1982, 50, 1629–1635. [Google Scholar] [CrossRef]
- Hiester, A.; Nettersheim, D.; Nini, A.; Lusch, A.; Albers, P. Management, Treatment, and Molecular Background of the Growing Teratoma Syndrome. Urol. Clin. North Am. 2019, 46, 419–427. [Google Scholar] [CrossRef]
- Michalski, W.; Jonska-Gmyrek, J.; Poniatowska, G.; Kucharz, J.; Stelmasiak, P.; Nietupski, K.; Sadowska, M.; Demkow, T.; Wiechno, P. Testicular teratomas: A growing problem? Med. Oncol. 2018, 35, 153. [Google Scholar] [CrossRef]
- Scheckel, C.J.; Kosiorek, H.E.; Butterfield, R.; Ho, T.H.; Hilal, T. Germ Cell Tumors with Malignant Somatic Transformation: A Mayo Clinic Experience. Oncol. Res. Treat. 2019, 42, 95–100. [Google Scholar] [CrossRef]
- Necchi, A.; Colecchia, M.; Nicolai, N.; Piva, L.; Catanzaro, M.; Biasoni, D.; Torelli, T.; Stagni, S.; Paolini, B.; Milani, A.; et al. Towards the definition of the best management and prognostic factors of teratoma with malignant transformation: A single-institution case series and new proposal. BJU Int. 2011, 107, 1088–1094. [Google Scholar] [CrossRef]
- Giannatempo, P.; Pond, G.R.; Sonpavde, G.; Albany, C.; Loriot, Y.; Sweeney, C.J.; Salvioni, R.; Colecchia, M.; Nicolai, N.; Raggi, D.; et al. Treatment and Clinical Outcomes of Patients with Teratoma with Somatic-Type Malignant Transformation: An International Collaboration. J. Urol. 2016, 196, 95–100. [Google Scholar] [CrossRef]
- Colecchia, M.; Necchi, A.; Paolini, B.; Nicolai, N.; Salvioni, R. Teratoma with somatic-type malignant components in germ cell tumors of the testis: A clinicopathologic analysis of 40 cases with outcome correlation. Int. J. Surg. Pathol. 2011, 19, 321–327. [Google Scholar] [CrossRef]
- Dashti, N.K.; Jimenez, R.E. Somatic-type malignancy in germ cell tumors. In Pathology and Biology of Human Germ Cell Tumors; Springer: Berlin/Heidelberg, Germany, 2017; pp. 419–439. [Google Scholar] [CrossRef]
- Zong, X.; Yang, J.X.; Zhang, Y.; Cao, D.Y.; Shen, K.; You, Y.; Guo, L.N. Postchemotherapy sarcoma as a somatic-type malignancy derived from the gonadal yolk sac tumor in a patient with 46, XY pure gonadal dysgenesis. Onco Targets Ther. 2019, 12, 2365–2372. [Google Scholar] [CrossRef] [Green Version]
- Singla, N.; Lafin, J.T.; Ghandour, R.A.; Kaffenberger, S.; Amatruda, J.F.; Bagrodia, A. Genetics of testicular germ cell tumors. Curr. Opin. Urol. 2019, 29, 344–349. [Google Scholar] [CrossRef]
- Chovanec, M.; De Giorgi, U.; Mego, M. Immune-Related Concepts in Biology and Treatment of Germ-Cell Tumors. Adv. Urol. 2018, 2018, 3718165. [Google Scholar] [CrossRef] [Green Version]
- Chovanec, M.; Mardiak, J.; Mego, M. Immune mechanisms and possible immune therapy in testicular germ cell tumours. Andrology 2019, 7, 479–486. [Google Scholar] [CrossRef] [Green Version]
- Fankhauser, C.D.; Curioni-Fontecedro, A.; Allmann, V.; Beyer, J.; Tischler, V.; Sulser, T.; Moch, H.; Bode, P.K. Frequent PD-L1 expression in testicular germ cell tumors. Br. J. Cancer 2015, 113, 411–413. [Google Scholar] [CrossRef] [Green Version]
- Cierna, Z.; Mego, M.; Miskovska, V.; Machalekova, K.; Chovanec, M.; Svetlovska, D.; Hainova, K.; Rejlekova, K.; Macak, D.; Spanik, S.; et al. Prognostic value of programmed-death-1 receptor (PD-1) and its ligand 1 (PD-L1) in testicular germ cell tumors. Ann. Oncol. 2016, 27, 300–305. [Google Scholar] [CrossRef]
- Chovanec, M.; Cierna, Z.; Miskovska, V.; Machalekova, K.; Svetlovska, D.; Kalavska, K.; Rejlekova, K.; Spanik, S.; Kajo, K.; Babal, P.; et al. Prognostic role of programmed-death ligand 1 (PD-L1) expressing tumor infiltrating lymphocytes in testicular germ cell tumors. Oncotarget 2017, 8, 21794–21805. [Google Scholar] [CrossRef] [Green Version]
- Adra, N.; Einhorn, L.H.; Althouse, S.K.; Ammakkanavar, N.R.; Musapatika, D.; Albany, C.; Vaughn, D.; Hanna, N.H. Phase II trial of pembrolizumab in patients with platinum refractory germ-cell tumors: A Hoosier Cancer Research Network Study GU14-206. Ann. Oncol. 2018, 29, 209–214. [Google Scholar] [CrossRef]
- Necchi, A.; Giannatempo, P.; Raggi, D.; Mariani, L.; Colecchia, M.; Fare, E.; Monopoli, F.; Calareso, G.; Ali, S.M.; Ross, J.S.; et al. An Open-label Randomized Phase 2 study of Durvalumab Alone or in Combination with Tremelimumab in Patients with Advanced Germ Cell Tumors (APACHE): Results from the First Planned Interim Analysis. Eur. Urol. 2019, 75, 201–203. [Google Scholar] [CrossRef]
- Mego, M.; Svetlovska, D.; Chovanec, M.; Reckova, M.; Rejlekova, K.; Obertova, J.; Palacka, P.; Sycova-Mila, Z.; De Giorgi, U.; Mardiak, J. Phase II study of avelumab in multiple relapsed/refractory germ cell cancer. Investig. New Drugs 2019, 37, 748–754. [Google Scholar] [CrossRef]
- Oing, C.; Bokemeyer, C. Biological basis and early clinical results of immunotherapy for cisplatin-resistant germ cell cancer. Curr. Opin. Urol. 2018, 28, 479–484. [Google Scholar] [CrossRef]
- Zschabitz, S.; Lasitschka, F.; Jager, D.; Grullich, C. Activity of immune checkpoint inhibition in platinum refractory germ-cell tumors. Ann. Oncol. 2016, 27, 1356–1360. [Google Scholar] [CrossRef] [PubMed]
- Semaan, A.; Haddad, F.G.; Eid, R.; Kourie, H.R.; Nemr, E. Immunotherapy: Last bullet in platinum refractory germ cell testicular cancer. Future Oncol. 2019, 15, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Oing, C.; Verem, I.; Mansour, W.Y.; Bokemeyer, C.; Dyshlovoy, S.; Honecker, F. 5-Azacitidine Exerts Prolonged Pro-Apoptotic Effects and Overcomes Cisplatin-Resistance in Non-Seminomatous Germ Cell Tumor Cells. Int. J. Mol. Sci. 2018, 20, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albany, C.; Hever-Jardine, M.P.; von Herrmann, K.M.; Yim, C.Y.; Tam, J.; Warzecha, J.M.; Shin, L.; Bock, S.E.; Curran, B.S.; Chaudhry, A.S.; et al. Refractory testicular germ cell tumors are highly sensitive to the second generation DNA methylation inhibitor guadecitabine. Oncotarget 2017, 8, 2949–2959. [Google Scholar] [CrossRef] [Green Version]
- Steele, N.; Finn, P.; Brown, R.; Plumb, J.A. Combined inhibition of DNA methylation and histone acetylation enhances gene re-expression and drug sensitivity in vivo. Br. J. Cancer 2009, 100, 758–763. [Google Scholar] [CrossRef] [Green Version]
- Roth, B.J.; Elson, P.; Sledge, G.W.; Einhorn, L.H.; Trump, D.L. 5-Azacytidine (NSC 102816) in refractory germ cell tumors. Investig. New Drugs 1993, 11, 201–202. [Google Scholar] [CrossRef]
- Matei, D.; Ghamande, S.; Roman, L.; Alvarez Secord, A.; Nemunaitis, J.; Markham, M.J.; Nephew, K.P.; Jueliger, S.; Oganesian, A.; Naim, S.; et al. A Phase I Clinical Trial of Guadecitabine and Carboplatin in Platinum-Resistant, Recurrent Ovarian Cancer: Clinical, Pharmacokinetic, and Pharmacodynamic Analyses. Clin. Cancer Res. 2018, 24, 2285–2293. [Google Scholar] [CrossRef] [Green Version]
- Beyer, U.; Kronung, S.K.; Leha, A.; Walter, L.; Dobbelstein, M. Comprehensive identification of genes driven by ERV9-LTRs reveals TNFRSF10B as a re-activatable mediator of testicular cancer cell death. Cell Death Differ. 2016, 23, 64–75. [Google Scholar] [CrossRef] [Green Version]
- Nettersheim, D.; Jostes, S.; Fabry, M.; Honecker, F.; Schumacher, V.; Kirfel, J.; Kristiansen, G.; Schorle, H. A signaling cascade including ARID1A, GADD45B and DUSP1 induces apoptosis and affects the cell cycle of germ cell cancers after romidepsin treatment. Oncotarget 2016, 7, 74931–74946. [Google Scholar] [CrossRef]
- Jostes, S.; Nettersheim, D.; Fellermeyer, M.; Schneider, S.; Hafezi, F.; Honecker, F.; Schumacher, V.; Geyer, M.; Kristiansen, G.; Schorle, H. The bromodomain inhibitor JQ1 triggers growth arrest and apoptosis in testicular germ cell tumours in vitro and in vivo. J. Cell Mol. Med. 2017, 21, 1300–1314. [Google Scholar] [CrossRef]
- Kurz, L.; Miklyaeva, A.; Skowron, M.A.; Overbeck, N.; Poschmann, G.; Becker, T.; Eul, K.; Kurz, T.; Schonberger, S.; Calaminus, G.; et al. ARID1A Regulates Transcription and the Epigenetic Landscape via POLE and DMAP1 while ARID1A Deficiency or Pharmacological Inhibition Sensitizes Germ Cell Tumor Cells to ATR Inhibition. Cancers (Basel) 2020, 12, 905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobo, J.; Jeronimo, C.; Henrique, R. Targeting the Immune system and Epigenetic Landscape of Urological Tumors. Int. J. Mol. Sci. 2020, 21, 829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenner, M.; Oing, C.; Dieing, A.; Gauler, T.; Oechsle, K.; Lorch, A.; Hentrich, M.; Kopp, H.G.; Bokemeyer, C.; Honecker, F. Everolimus in patients with multiply relapsed or cisplatin refractory germ cell tumors: Results of a phase II, single-arm, open-label multicenter trial (RADIT) of the German Testicular Cancer Study Group. J. Cancer Res. Clin. Oncol. 2019, 145, 717–723. [Google Scholar] [CrossRef]
- Oing, C.; Hentrich, M.; Lorch, A.; Glaser, D.; Rumpold, H.; Ochsenreither, S.; Richter, S.; Dieing, A.; Zschabitz, S.; Pereira, R.R.; et al. Treatment of refractory germ-cell tumours with single-agent cabazitaxel: A German Testicular Cancer Study Group case series. J. Cancer Res. Clin. Oncol. 2020, 146, 449–455. [Google Scholar] [CrossRef]
- Burger, A.M.; Double, J.A.; Newell, D.R. Inhibition of telomerase activity by cisplatin in human testicular cancer cells. Eur. J. Cancer 1997, 33, 638–644. [Google Scholar] [CrossRef]
- Cressey, T.R.; Tilby, M.J.; Newell, D.R. Decreased telomerase activity is not a reliable indicator of chemosensitivity in testicular cancer cell lines. Eur. J. Cancer 2002, 38, 586–593. [Google Scholar] [CrossRef]
- Skowron, M.A.; Vermeulen, M.; Winkelhausen, A.; Becker, T.K.; Bremmer, F.; Petzsch, P.; Schonberger, S.; Calaminus, G.; Kohrer, K.; Albers, P.; et al. CDK4/6 inhibition presents as a therapeutic option for paediatric and adult germ cell tumours and induces cell cycle arrest and apoptosis via canonical and non-canonical mechanisms. Br. J. Cancer 2020. [Google Scholar] [CrossRef]
- Castillo-Avila, W.; Piulats, J.M.; Garcia Del Muro, X.; Vidal, A.; Condom, E.; Casanovas, O.; Mora, J.; Germa, J.R.; Capella, G.; Villanueva, A.; et al. Sunitinib inhibits tumor growth and synergizes with cisplatin in orthotopic models of cisplatin-sensitive and cisplatin-resistant human testicular germ cell tumors. Clin. Cancer Res. 2009, 15, 3384–3395. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| In Vitro Models | ||
|---|---|---|
| Cell Line | Features | Resistant Clone and References |
| NCCIT | Primary mediastinal. Adult male. Mixed nonseminoma (with embryonal carcinoma). p53-mutated [43]. | NCCIT-R [42,44] |
| 2102Ep | Primary testicular. Adult male. Mixed nonseminoma (with embryonal carcinoma). Wild-type p53 [43]. | 2102Ep-R [44,45] |
| NTera-2 | Primary testicular. Adult male. Embryonal carcinoma. Wild-type p53 [43]. | NT2-R [42,44] |
| TCam-2 | Primary testicular. Adult male. Seminoma. BRAF V600E mutation [46,47]. | TCam-2-R [48] |
| Others less commonly used | NOY-1-R (yolk sac tumor), H12.1RA, H12.1D, 1411HP, 1777NRpmet, GCT44 (mixed tumor), Tera-CP, 833K-R (embryonal carcinoma), GCT27-R, SuSa-R (teratoma), and P19 (mouse-derived and embryonal carcinoma). | [48,49,50,51,52,53,54,55] |
| In vivo models | ||
| Injection of resistant cell lines. | [42,53] | |
| Implantation of human tissue samples. | [41] | |
| Drug | Main Mechanism/Drug Class | Main Findings | Reference |
|---|---|---|---|
| Cabazitaxel | Chemotherapy; Taxane (microtubule inhibitor) | Clinical study. Limited activity in heavily treated refractory GCTs. | [195] |
| Dissulfiram | ALDH inhibitor (cancer stem cell marker) | Pre-clinical study. Synergistic antitumor effect (+ cisplatin) in an in vivo model. Restores sensitivity to cisplatin. | [42] |
| Everolimus | mTOR inhibitor | Clinical study. Limited activity in heavily treated refractory GCTs. | [194] |
| Sunitinib | RTK inhibitor (including VEGFR) | Pre-clinical study. Antitumor effect (including a reduced vasculature) in an in vivo model, including of a cisplatin resistant disease. | [199] |
| Avelumab | Immunotherapy; PD-L1 inhibitor | Clinical study. Well-tolerated but limited activity in heavily treated refractory GCTs. | [180] |
| Pembrolizumab | Immunotherapy; PD-1 inhibitor | Clinical study. Well-tolerated but limited activity in heavily treated refractory GCTs. | [178] |
| Durvalumab and Tremelimumab | Immunotherapy; anti-PD-L1 and anti-CTLA4 | Clinical study. Partial response and stable disease in 2 patients; remaining with disease progression. | [179] |
| 5-azacytidine | Epigenetics; Demethylating agent (nucleoside analog) | Pre-clinical study. Proapoptotic effect at low nM concentrations and overcomes cisplatin resistance. | [184] |
| Decitabine | Epigenetics; Demethylating agent (nucleoside analog) | Pre-clinical study. Causes DNA damage, promotes p53 and p21 activation, downregulates pluripotency factors, and activates ATM. Restores sensitivity to cisplatin. | [129,131] |
| Guadecitabine | Epigenetics; Demethylating agent (nucleoside analog) | Pre-clinical study. Restores sensitivity to cisplatin in in vivo models. | [185] |
| JQ1 | Epigenetics; BET inhibitor | Pre-clinical study. Induces apoptosis, more prominent in resistant clones compared to parental. | [191] |
| C63 and BRD-K98645985 | Epigenetics; ARID1A (chromatin remodeler) inhibitor | Pre-clinical study. ARID1A inhibitors sensitizes cisplatin-resistant cells to ATR inhibition. | [192] |
| Romidepsin | Epigenetics; HDAC inhibitor | Pre-clinical study. Antitumor effect, including in cisplatin-resistant cells. | [190] |
| Olaparib | DNA repair; PARP inhibitor | Pre-clinical study. Antitumor effect as monotherapy and sensitization to cisplatin. | [92] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lobo, J.; Jerónimo, C.; Henrique, R. Cisplatin Resistance in Testicular Germ Cell Tumors: Current Challenges from Various Perspectives. Cancers 2020, 12, 1601. https://doi.org/10.3390/cancers12061601
Lobo J, Jerónimo C, Henrique R. Cisplatin Resistance in Testicular Germ Cell Tumors: Current Challenges from Various Perspectives. Cancers. 2020; 12(6):1601. https://doi.org/10.3390/cancers12061601
Chicago/Turabian StyleLobo, João, Carmen Jerónimo, and Rui Henrique. 2020. "Cisplatin Resistance in Testicular Germ Cell Tumors: Current Challenges from Various Perspectives" Cancers 12, no. 6: 1601. https://doi.org/10.3390/cancers12061601
APA StyleLobo, J., Jerónimo, C., & Henrique, R. (2020). Cisplatin Resistance in Testicular Germ Cell Tumors: Current Challenges from Various Perspectives. Cancers, 12(6), 1601. https://doi.org/10.3390/cancers12061601