Overcoming Platinum and PARP-Inhibitor Resistance in Ovarian Cancer


Abstract
:1. Introduction
2. Alterations in DNA Damage Repair Can Drive Treatment Resistance
2.1. HRD Conveys Sensitivity to Platinum and PARPi
2.2. Reactivation of HR Is a Mechanism of Acquired Resistance
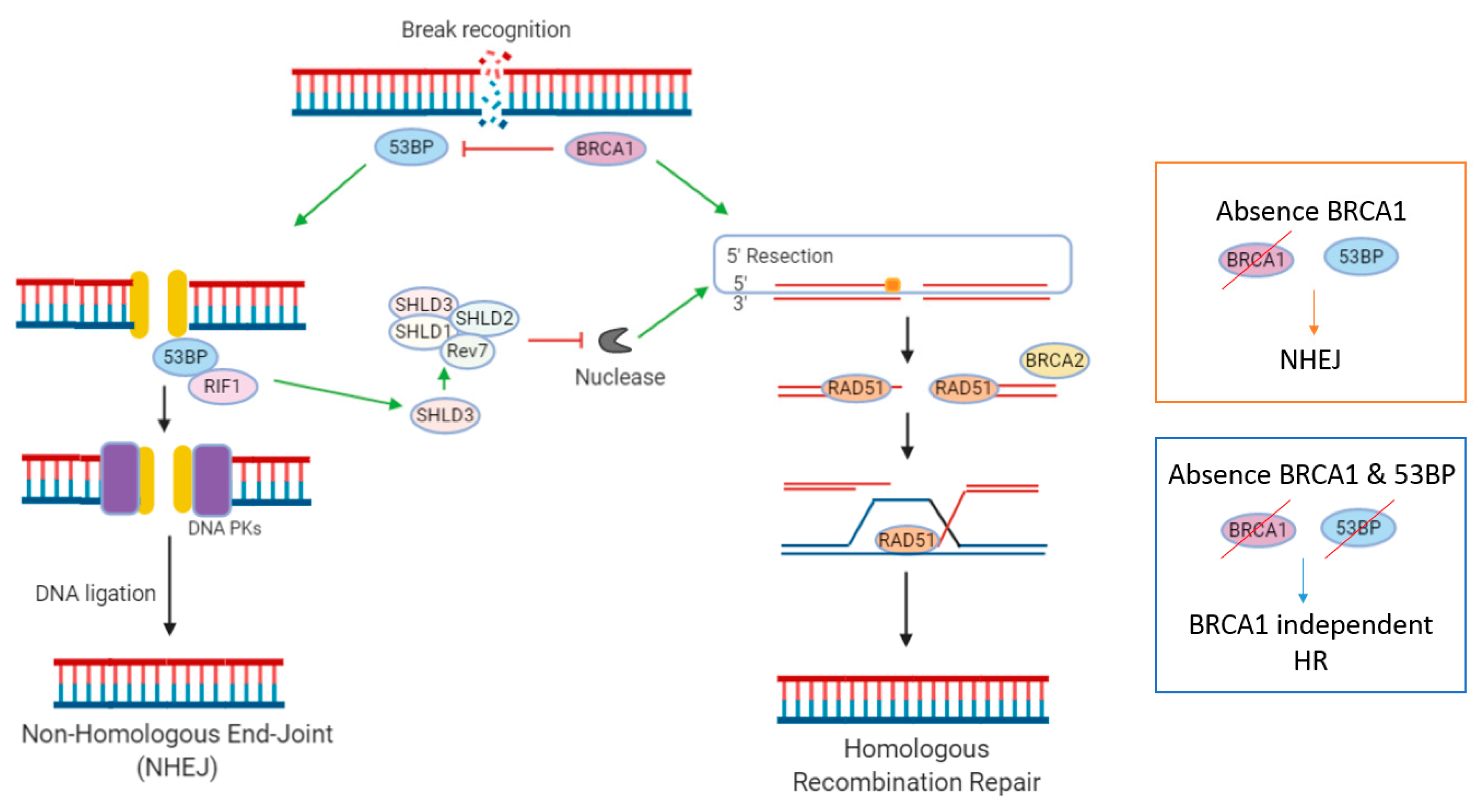
2.3. Non-Homologous End Joining (NHEJ)
2.4. Nucleotide Excision Repair
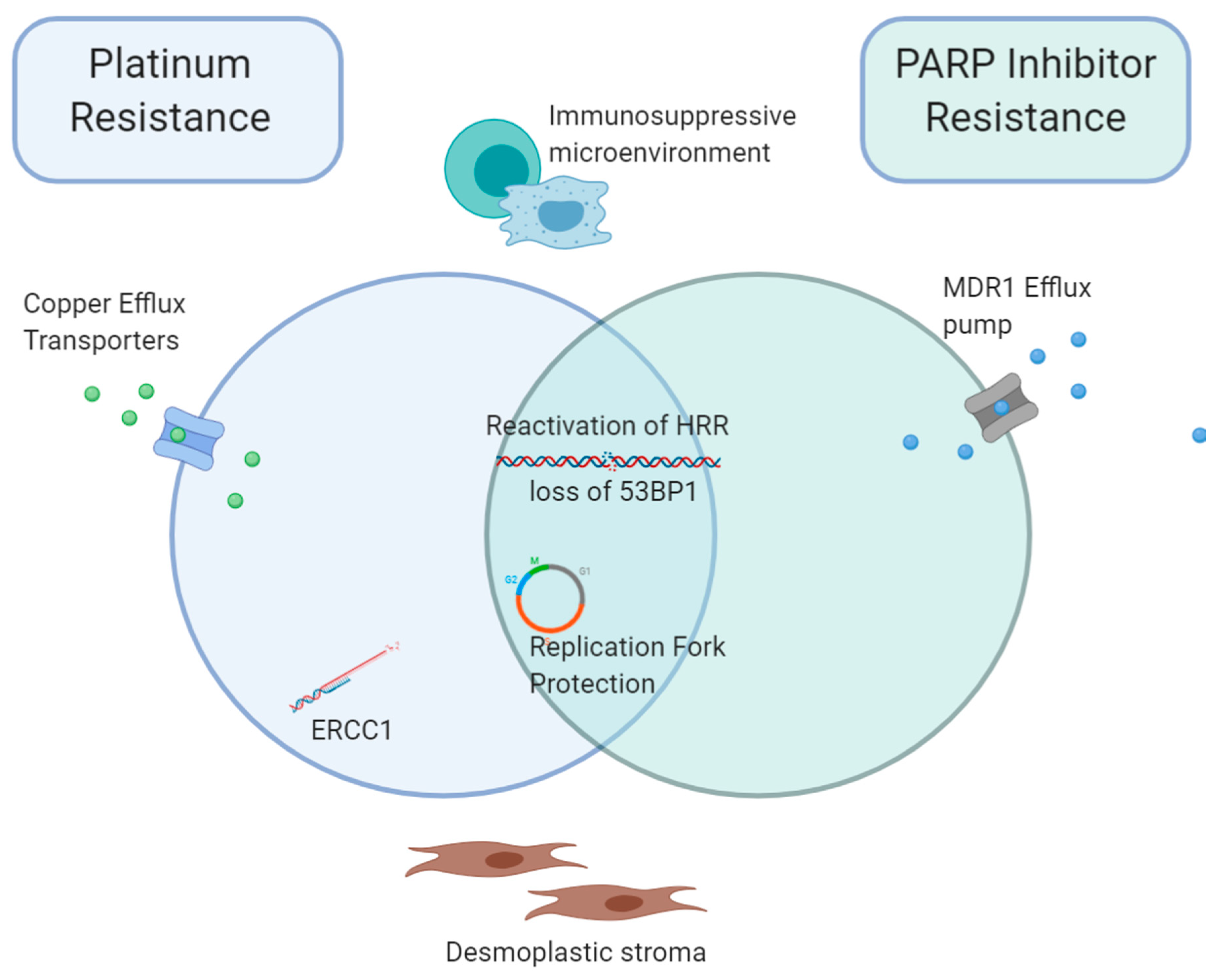
2.5. Replication Fork Protection
2.6. Reduced Cellular Availability of Drugs
3. Immunosuppressive Tumour Microenvironment
4. Targeting Molecular Vulnerabilities to Overcome Treatment Resistance
4.1. Targeting ATR
4.2. Epigenetic Resensitisation
4.3. Cell Cycle Checkpoint Inhibitors
4.4. BET Inhibitors
4.5. Anti-Angiogenic Therapies
4.6. G-Quadruplex Stabilisation
5. Overlap between Acquired Platinum and PARPi Resistance Mechanisms
6. Biomarkers of Resistance to PARPi and Platinum
7. Conclusions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackledge, G.; Lawton, F.; Redman, C.; Kelly, K. Response of patients in phase II studies of chemotherapy in ovarian cancer: Implications for patient treatment and the design of phase II trials. Br. J. Cancer 1989, 59, 650–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gore, M.E.; Fryatt, I.; Wiltshaw, E.; Dawson, T. Treatment of relapsed carcinoma of the ovary with cisplatin or carboplatin following initial treatment with these compounds. Gynecol. Oncol. 1990, 36, 207–211. [Google Scholar] [CrossRef]
- Markman, M.; Rothman, R.; Hakes, T.; Reichman, B.; Hoskins, W.; Rubin, S.; Jones, W.; Almadrones, L.; Lewis, J.L., Jr. Second-line platinum therapy in patients with ovarian cancer previously treated with cisplatin. J. Clin. Oncol. 1991, 9, 389–393. [Google Scholar] [CrossRef]
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial ovarian cancer. Lancet 2019, 393, 1240–1253. [Google Scholar] [CrossRef] [Green Version]
- Madariaga, A.; Rustin, G.J.S.; Buckanovich, R.J.; Trent, J.C.; Oza, A.M. Wanna Get Away? Maintenance Treatments and Chemotherapy Holidays in Gynecologic Cancers. Am. Soc. Clin. Oncol. Educ. Book Am. Soc. Clin. Oncol. Annu. Meet. 2019, 39, e152–e166. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Gonzalez-Martin, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [Green Version]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Perol, D.; Gonzalez-Martin, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Maenpaa, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Eng. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [Green Version]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- Tomao, F.; Bardhi, E.; Di Pinto, A.; Sassu, C.M.; Biagioli, E.; Petrella, M.C.; Palaia, I.; Muzii, L.; Colombo, N.; Panici, P.B. Parp inhibitors as maintenance treatment in platinum sensitive recurrent ovarian cancer: An updated meta-analysis of randomized clinical trials according to BRCA mutational status. Cancer Treat. Rev. 2019, 80, 101909. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Li, X.; Li, W.; Bai, H.; Zhang, Z. PARP inhibitors in ovarian cancer: Sensitivity prediction and resistance mechanisms. J. Cell. Mol. Med. 2019, 23, 2303–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, T.A.; Plummer, R.; Azad, N.S.; Helleday, T. The DNA Damaging Revolution: PARP Inhibitors and Beyond. Am. Soc. Clin. Oncol. Educ. Book Am. Soc. Clin. Oncol. Annu. Meet. 2019, 39, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.-A.; Mooij, T.M.; Roos-Blom, M.-J.; Jervis, S.; Van Leeuwen, F.E.; Milne, R.L.; Andrieu, N. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [Green Version]
- McCuaig, J.M.; Stockley, T.L.; Shaw, P.; Fung-Kee-Fung, M.; Altman, A.D.; Bentley, J.; Bernardini, M.Q.; Cormier, B.; Hirte, H.; Kieser, K.; et al. Evolution of genetic assessment for BRCA-associated gynaecologic malignancies: A Canadian multisociety roadmap. J. Med. Genet. 2018, 55, 571–577. [Google Scholar] [CrossRef]
- Gee, M.E.; Faraahi, Z.; McCormick, A.; Edmondson, R.J. DNA damage repair in ovarian cancer: Unlocking the heterogeneity. J. Ovarian Res. 2018, 11, 50. [Google Scholar] [CrossRef] [Green Version]
- Kubelac, P.; Genestie, C.; Auguste, A.; Mesnage, S.; Le Formal, A.; Pautier, P.; Gouy, S.; Morice, P.; Bentivegna, E.; Maulard, A.; et al. Changes in DNA Damage Response Markers with Treatment in Advanced Ovarian Cancer. Cancers 2020, 12, 707. [Google Scholar] [CrossRef] [Green Version]
- Damia, G.; Broggini, M. Platinum Resistance in Ovarian Cancer: Role of DNA Repair. Cancers 2019, 11, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murai, J.; Shar-yin, N.H.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, T.; Zhang, Z.; Payne, S.H.; Zhang, B.; McDermott, J.E.; Zhou, J.Y.; Petyuk, V.A.; Chen, L.; Ray, D.; et al. Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 2016, 166, 755–765. [Google Scholar] [CrossRef] [Green Version]
- Trenner, A.; Sartori, A.A. Harnessing DNA Double-Strand Break Repair for Cancer Treatment. Front. Oncol. 2019, 9, 1388. [Google Scholar] [CrossRef]
- Madariaga, A.; Lheureux, S.; Oza, A.M. Tailoring Ovarian Cancer Treatment: Implications of BRCA1/2 Mutations. Cancers 2019, 11, 416. [Google Scholar] [CrossRef] [Green Version]
- Hoppe, M.M.; Sundar, R.; Tan, D.S.P.; Jeyasekharan, A.D. Biomarkers for Homologous Recombination Deficiency in Cancer. J. Natl. Cancer Inst. 2018, 110, 704–713. [Google Scholar] [CrossRef] [Green Version]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.; Zander, S.A.; Derksen, P.W.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef] [Green Version]
- Hay, T.; Matthews, J.R.; Pietzka, L.; Lau, A.; Cranston, A.; Nygren, A.O.; Douglas-Jones, A.; Smith, G.C.; Martin, N.M.; O’Connor, M.; et al. Poly(ADP-ribose) polymerase-1 inhibitor treatment regresses autochthonous Brca2/p53-mutant mammary tumors in vivo and delays tumor relapse in combination with carboplatin. Cancer Res. 2009, 69, 3850–3855. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.K.; Harrell, M.I.; Oza, A.M.; Oaknin, A.; Ray-Coquard, I.; Tinker, A.V.; Helman, E.; Radke, M.R.; Say, C.; Vo, L.T.; et al. BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2019, 9, 210–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayor, P.; Gay, L.M.; Lele, S.; Elvin, J.A. BRCA1 reversion mutation acquired after treatment identified by liquid biopsy. Gynecol. Oncol. Rep. 2017, 21, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Comino-Mendez, I.; de Bruijn, I.; Tian, L.; Meisel, J.L.; Garcia-Murillas, I.; Fribbens, C.; Cutts, R.; Martelotto, L.G.; Ng, C.K.Y.; et al. Diverse BRCA1 and BRCA2 Reversion Mutations in Circulating Cell-Free DNA of Therapy-Resistant Breast or Ovarian Cancer. Clin. Cancer. Res. 2017, 23, 6708–6720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domchek, S.M. Reversion Mutations with Clinical Use of PARP Inhibitors: Many Genes, Many Versions. Cancer Discov. 2017, 7, 937–939. [Google Scholar] [CrossRef] [Green Version]
- Lheureux, S.; Bruce, J.P.; Burnier, J.V.; Karakasis, K.; Shaw, P.A.; Clarke, B.A.; Yang, S.Y.; Quevedo, R.; Li, T.; Dowar, M.; et al. Somatic BRCA1/2 Recovery as a Resistance Mechanism After Exceptional Response to Poly (ADP-ribose) Polymerase Inhibition. J. Clin. Oncol. 2017, 35, 1240–1249. [Google Scholar] [CrossRef]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [Green Version]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 9, 3970. [Google Scholar] [CrossRef] [Green Version]
- Johnson, N.; Li, Y.C.; Walton, Z.E.; Cheng, K.A.; Li, D.; Rodig, S.J.; Moreau, L.A.; Unitt, C.; Bronson, R.T.; Thomas, H.D.; et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat. Med. 2011, 17, 875–882. [Google Scholar] [CrossRef] [Green Version]
- Johnson, N.; Johnson, S.F.; Yao, W.; Li, Y.C.; Choi, Y.E.; Bernhardy, A.J.; Wang, Y.; Capelletti, M.; Sarosiek, K.A.; Moreau, L.A.; et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc. Natl. Acad. Sci. USA 2013, 110, 17041–17046. [Google Scholar] [CrossRef] [Green Version]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Alvarez-Quilon, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef]
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013, 3, 68–81. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Somyajit, K.; Narita, T.; Maskey, E.; Stanlie, A.; Kremer, M.; Typas, D.; Lammers, M.; Mailand, N.; Nussenzweig, A.; et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 2018, 173, 972–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dev, H.; Chiang, T.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018, 20, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunting, S.F.; Callen, E.; Kozak, M.L.; Kim, J.M.; Wong, N.; Lopez-Contreras, A.J.; Ludwig, T.; Baer, R.; Faryabi, R.B.; Malhowski, A.; et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol. Cell 2012, 46, 125–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Xu, X.; Bunting, S.F.; Liu, J.; Wang, R.H.; Cao, L.L.; Wu, J.J.; Peng, T.N.; Chen, J.; Nussenzweig, A.; et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol. Cell 2009, 35, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Bowden, N.A. Nucleotide excision repair: Why is it not used to predict response to platinum-based chemotherapy? Cancer Lett. 2014, 346, 163–171. [Google Scholar] [CrossRef]
- Ceccaldi, R.; O’Connor, K.W.; Mouw, K.W.; Li, A.Y.; Matulonis, U.A.; D’Andrea, A.D.; Konstantinopoulos, P.A. A unique subset of epithelial ovarian cancers with platinum sensitivity and PARP inhibitor resistance. Cancer Res. 2015, 75, 628–634. [Google Scholar] [CrossRef] [Green Version]
- Mesquita, K.A.; Alabdullah, M.; Griffin, M.; Toss, M.S.; Fatah, T.; Alblihy, A.; Moseley, P.; Chan, S.Y.T.; Rakha, E.A.; Madhusudan, S. ERCC1-XPF deficiency is a predictor of olaparib induced synthetic lethality and platinum sensitivity in epithelial ovarian cancers. Gynecol. Oncol. 2019, 153, 416–424. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, A.; Zhao, Y.; Xiang, J.; Yu, D.; Liang, Z.; Xu, C.; Zhang, Q.; Li, J.; Duan, P. The association of polymorphisms in nucleotide excision repair genes with ovarian cancer susceptibility. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Arora, S.; Kothandapani, A.; Tillison, K.; Kalman-Maltese, V.; Patrick, S.M. Downregulation of XPF-ERCC1 enhances cisplatin efficacy in cancer cells. DNA Repair 2010, 9, 745–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darcy, K.M.; Tian, C.; Reed, E. A Gynecologic Oncology Group study of platinum-DNA adducts and excision repair cross-complementation group 1 expression in optimal, stage III epithelial ovarian cancer treated with platinum-taxane chemotherapy. Cancer Res. 2007, 67, 4474–4481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colmegna, B.; Uboldi, S.; Frapolli, R.; Licandro, S.A.; Panini, N.; Galmarini, C.M.; Badri, N.; Spanswick, V.J.; Bingham, J.P.; Kiakos, K.; et al. Increased sensitivity to platinum drugs of cancer cells with acquired resistance to trabectedin. Br. J. Cancer 2015, 113, 1687–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Hu, P.; Cao, Y.; Wang, G.Y.; Wang, N.; Zhou, R.M. Predicting the outcome of platinum-based chemotherapies in epithelial ovarian cancer using the 8092C/A polymorphism of ERCC1: A meta-analysis. Biomark. Biochem. Indic. Expo. Response Susceptibility Chem. 2014, 19, 128–134. [Google Scholar]
- Rubatt, J.M.; Darcy, K.M.; Tian, C.; Muggia, F.; Dhir, R.; Armstrong, D.K.; Bookman, M.A.; Niedernhofer, L.J.; Deloia, J.; Birrer, M.; et al. Pre-treatment tumor expression of ERCC1 in women with advanced stage epithelial ovarian cancer is not predictive of clinical outcomes: A Gynecologic Oncology Group study. Gynecol. Oncol. 2012, 125, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Krivak, T.C.; Darcy, K.M.; Tian, C.; Armstrong, D.; Baysal, B.E.; Gallion, H.; Ambrosone, C.B.; DeLoia, J.A. Relationship between ERCC1 polymorphisms, disease progression, and survival in the Gynecologic Oncology Group Phase III Trial of intraperitoneal versus intravenous cisplatin and paclitaxel for stage III epithelial ovarian cancer. J. Clin. Oncol. 2008, 26, 3598–3606. [Google Scholar] [CrossRef] [Green Version]
- Steffensen, K.D.; Smoter, M.; Waldstrom, M.; Grala, B.; Bodnar, L.; Stec, R.; Szczylik, C.; Jakobsen, A. Resistance to first line platinum paclitaxel chemotherapy in serous epithelial ovarian cancer: The prediction value of ERCC1 and Tau expression. Int. J. Oncol. 2014, 44, 1736–1744. [Google Scholar] [CrossRef]
- Mouw, K.W.; D’Andrea, A.D.; Konstantinopoulos, P.A. Nucleotide excision repair (NER) alterations as evolving biomarkers and therapeutic targets in epithelial cancers. Oncoscience 2015, 2, 942–943. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef]
- Schlacher, K. PARPi focus the spotlight on replication fork protection in cancer. Nat. Cell Biol. 2017, 19, 1309–1310. [Google Scholar] [CrossRef]
- Rondinelli, B.; Gogola, E.; Yucel, H.; Duarte, A.A.; van de Ven, M.; van der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 2017, 68, 414–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Feng, Y.; Yu, G.K.; Ru, Y.; Tang, S.W.; Shen, Y.; Pommier, Y. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget 2016, 7, 76534–76550. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Tang, S.W.; Leo, E.; Baechler, S.A.; Redon, C.E.; Zhang, H.; Al Abo, M.; Rajapakse, V.N.; Nakamura, E.; Jenkins, L.M.M.; et al. SLFN11 Blocks Stressed Replication Forks Independently of ATR. Mol. Cell 2018, 69, 371–384. [Google Scholar] [CrossRef] [Green Version]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef]
- Li, T.; Peng, J.; Zeng, F.; Zhang, K.; Liu, J.; Li, X.; Ouyang, Q.; Wang, G.; Wang, L.; Liu, Z.; et al. Association between polymorphisms in CTR1, CTR2, ATP7A, and ATP7B and platinum resistance in epithelial ovarian cancer. Int. J. Clin. Pharmacol. Ther. 2017, 55, 774–780. [Google Scholar] [CrossRef]
- Li, Y.Q.; Yin, J.Y.; Liu, Z.Q.; Li, X.P. Copper efflux transporters ATP7A and ATP7B: Novel biomarkers for platinum drug resistance and targets for therapy. IUBMB Life 2018, 70, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Kilari, D.; Guancial, E.; Kim, E.S. Role of copper transporters in platinum resistance. World J. Clin. Oncol. 2016, 7, 106–113. [Google Scholar] [CrossRef] [Green Version]
- Briz, O.; Perez-Silva, L.; Al-Abdulla, R.; Abete, L.; Reviejo, M.; Romero, M.R.; Marin, J.J.G. What “The Cancer Genome Atlas”database tells us about the role of ATP-binding cassette (ABC) proteins in chemoresistance to anticancer drugs. Expert Opin. Drug Metab. Toxicol. 2019, 15, 577–593. [Google Scholar] [CrossRef]
- Tian, C.; Ambrosone, C.B.; Darcy, K.M.; Krivak, T.C.; Armstrong, D.K.; Bookman, M.A.; Davis, W.; Zhao, H.; Moysich, K.; Gallion, H.; et al. Common variants in ABCB1, ABCC2 and ABCG2 genes and clinical outcomes among women with advanced stage ovarian cancer treated with platinum and taxane-based chemotherapy: A Gynecologic Oncology Group study. Gynecol. Oncol. 2012, 124, 575–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaidyanathan, A.; Sawers, L.; Gannon, A.L.; Chakravarty, P.; Scott, A.L.; Bray, S.E.; Ferguson, M.J.; Smith, G. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br. J. Cancer 2016, 115, 431–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christie, E.L.; Pattnaik, S.; Beach, J.; Copeland, A.; Rashoo, N.; Fereday, S.; Hendley, J.; Alsop, K.; Brady, S.L.; Lamb, G.; et al. Multiple ABCB1 transcriptional fusions in drug resistant high-grade serous ovarian and breast cancer. Nat. Commun. 2019, 10, 1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawlor, D.; Martin, P.; Busschots, S.; Thery, J.; O’Leary, J.J.; Hennessy, B.T.; Stordal, B. PARP Inhibitors as P-glyoprotein Substrates. J. Pharm. Sci. 2014, 103, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.A.; Paulo, A. Small molecule inhibitors of multidrug resistance gene (MDR1) expression: Preclinical evaluation and mechanisms of action. Curr. Cancer Drug Targets 2013, 13, 814–828. [Google Scholar] [CrossRef]
- Kleih, M.; Bopple, K.; Dong, M.; Gaissler, A.; Heine, S.; Olayioye, M.A.; Aulitzky, W.E.; Essmann, F. Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell Death Dis. 2019, 10, 851. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, B.; Ramadoss, S.; Chaudhuri, G.; Janzen, C.; Sen, S.; Mascharak, P.K. Carbon monoxide sensitizes cisplatin-resistant ovarian cancer cell lines toward cisplatin via attenuation of levels of glutathione and nuclear metallothionein. J. Inorg. Biochem. 2019, 191, 29–39. [Google Scholar] [CrossRef]
- Jinawath, N.; Vasoontara, C.; Jinawath, A.; Fang, X.; Zhao, K.; Yap, K.-L.; Guo, T.; Lee, C.S.; Wang, W.; Balgley, B.M. Oncoproteomic analysis reveals co-upregulation of RELA and STAT5 in carboplatin resistant ovarian carcinoma. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- Ojalvo, L.S.; Thompson, E.D.; Wang, T.L.; Meeker, A.K.; Shih, I.M.; Fader, A.N.; Cimino-Mathews, A.; Emens, L.A. Tumor-associated macrophages and the tumor immune microenvironment of primary and recurrent epithelial ovarian cancer. Hum. Pathol. 2018, 74, 135–147. [Google Scholar] [CrossRef]
- Martin de la Fuente, L.; Westbom-Fremer, S.; Arildsen, N.S.; Hartman, L.; Malander, S.; Kannisto, P.; Masback, A.; Hedenfalk, I. PD-1/PD-L1 expression and tumor-infiltrating lymphocytes are prognostically favorable in advanced high-grade serous ovarian carcinoma. Virchows Arch. Int. J. Pathol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, W.T.; Adams, S.F.; Tahirovic, E.; Hagemann, I.S.; Coukos, G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2012, 124, 192–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, D.; Fiegl, H.; Zeimet, A.G.; Wieser, V.; Marth, C.; Sprung, S.; Sopper, S.; Hartmann, G.; Reimer, D.; Boesch, M. High RIG-I expression in ovarian cancer associates with an immune-escape signature and poor clinical outcome. Int. J. Cancer 2020, 146, 2007–2018. [Google Scholar] [CrossRef] [PubMed]
- Konecny, G.E.; Wang, C.; Hamidi, H.; Winterhoff, B.; Kalli, K.R.; Dering, J.; Ginther, C.; Chen, H.W.; Dowdy, S.; Cliby, W.; et al. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Foote, K.M.; Nissink, J.W.M.; McGuire, T.; Turner, P.; Guichard, S.; Yates, J.W.T.; Lau, A.; Blades, K.; Heathcote, D.; Odedra, R.; et al. Discovery and Characterization of AZD6738, a Potent Inhibitor of Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Kinase with Application as an Anticancer Agent. J. Med. Chem. 2018, 61, 9889–9907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradbury, A.; Hall, S.; Curtin, N.; Drew, Y. Targeting ATR as Cancer Therapy: A new era for synthetic lethality and synergistic combinations? Pharmacol. Ther. 2020, 207, 107450. [Google Scholar] [CrossRef]
- O’Sullivan Coyne, G.H.; Do, K.T.; Kummar, S.; Takebe, N.; Quinn, M.F.; Piha-Paul, S.A.; Bruns, A.; Juwara, L.; Sharon, E.; Piekarz, R. Phase I trial of the triplet M6620 (formerly VX970)+ veliparib+ cisplatin in patients with advanced solid tumors. Am. Soc. Clin. Oncol. 2018, 36 (Suppl. 15), 2549. [Google Scholar] [CrossRef]
- Esteller, M.; Silva, J.M.; Dominguez, G.; Bonilla, F.; Matias-Guiu, X.; Lerma, E.; Bussaglia, E.; Prat, J.; Harkes, I.C.; Repasky, E.A. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. JNCI J. Natl. Cancer Inst. 2000, 92, 564–569. [Google Scholar] [CrossRef]
- Li, M.; Balch, C.; Montgomery, J.S.; Jeong, M.; Chung, J.H.; Yan, P.; Huang, T.H.; Kim, S.; Nephew, K.P. Integrated analysis of DNA methylation and gene expression reveals specific signaling pathways associated with platinum resistance in ovarian cancer. BMC Med. Genom. 2009, 2, 34. [Google Scholar] [CrossRef] [Green Version]
- Glasspool, R.; Brown, R.; Gore, M.; Rustin, G.; McNeish, I.; Wilson, R.; Pledge, S.; Paul, J.; Mackean, M.; Hall, G. A randomised, phase II trial of the DNA-hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in combination with carboplatin vs carboplatin alone in patients with recurrent, partially platinum-sensitive ovarian cancer. Br. J. Cancer 2014, 110, 1923–1929. [Google Scholar] [CrossRef]
- Matei, D.; Shen, C.; Fang, F.; Schilder, J.; Li, M.; Arnold, A.; Zeng, Y.; Pilrose, J.; Kulesavage, C.; Balch, C. A phase II study of decitabine and carboplatin in recurrent platinum (Pt)-resistant ovarian cancer (OC). J. Clin. Oncol. 2011, 29 (Suppl. 15), 5011. [Google Scholar] [CrossRef]
- Fang, F.; Zuo, Q.; Pilrose, J.; Wang, Y.; Shen, C.; Li, M.; Wulfridge, P.; Matei, D.; Nephew, K.P. Decitabine reactivated pathways in platinum resistant ovarian cancer. Oncotarget 2014, 5, 3579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, S.; Hu, W.; Iyer, R.; Kavanagh, J.J.; Coleman, R.L.; Levenback, C.F.; Sood, A.K.; Wolf, J.K.; Gershenson, D.M.; Markman, M. Phase 1b-2a study to reverse platinum resistance through use of a hypomethylating agent, azacitidine, in patients with platinum-resistant or platinum-refractory epithelial ovarian cancer. Cancer 2011, 117, 1661–1669. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Matulonis, U.A.; Alvarez Secord, A.; Nemunaitis, J.; Roman, L.D.; Blagden, S.P.; Banerjee, S.; McGuire, W.P.; Ghamande, S.; Birrer, M.J.; et al. A Randomized Phase II Trial of Epigenetic Priming with Guadecitabine and Carboplatin in Platinum-resistant, Recurrent Ovarian Cancer. Clin. Cancer Res. 2020, 26, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Cardenas, H.; Huang, H.; Jiang, G.; Perkins, S.M.; Zhang, C.; Keer, H.N.; Liu, Y.; Nephew, K.P.; Matei, D. Genomic and epigenomic signatures in ovarian cancer associated with resensitization to platinum drugs. Cancer Res. 2018, 78, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Kogan, A.A.; Mclaughlin, L.J.; Topper, M.; Muvarak, N.; Stojanovic, L.; Creed, T.M.; Bentzen, S.; Civin, C.I.; Baer, M.R.; Kingsbury, T.J. DNA Demethylating Agents Generate a Brcaness Effect in Multiple Sporadic Tumor Types: Prediction for Sensitivity to PARP Inhibitors in AML. Blood 2017, 130 (Suppl. 1), 3347. [Google Scholar]
- Wiegmans, A.P.; Yap, P.-Y.; Ward, A.; Lim, Y.C.; Khanna, K.K. Differences in expression of key DNA damage repair genes after epigenetic-induced BRCAness dictate synthetic lethality with PARP1 inhibition. Mol. Cancer Ther. 2015, 14, 2321–2331. [Google Scholar] [CrossRef] [Green Version]
- Dizon, D.S.; Blessing, J.A.; Penson, R.T.; Drake, R.D.; Walker, J.L.; Johnston, C.M.; DiSilvestro, P.A.; Fader, A.N. A phase II evaluation of belinostat and carboplatin in the treatment of recurrent or persistent platinum-resistant ovarian, fallopian tube, or primary peritoneal carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2012, 125, 367–371. [Google Scholar] [CrossRef] [Green Version]
- Dizon, D.S.; Damstrup, L.; Finkler, N.J.; Lassen, U.; Celano, P.; Glasspool, R.; Crowley, E.; Lichenstein, H.S.; Knoblach, P.; Penson, R.T. Phase II activity of belinostat (PXD-101), carboplatin, and paclitaxel in women with previously treated ovarian cancer. Int. J. Gynecol. Cancer 2012, 22, 979–986. [Google Scholar] [CrossRef]
- Petermann, E.; Woodcock, M.; Helleday, T. Chk1 promotes replication fork progression by controlling replication initiation. Proc. Natl. Acad. Sci. USA 2010, 107, 16090–16095. [Google Scholar] [CrossRef] [Green Version]
- Buisson, R.; Boisvert, J.L.; Benes, C.H.; Zou, L. Distinct but concerted roles of ATR, DNA-PK, and Chk1 in countering replication stress during S phase. Mol. Cell 2015, 59, 1011–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.M.; Nair, J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Merino, M.J.; Swisher, E.M.; Harrell, M.I.; Trepel, J.B.; Lee, M.J.; et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: A first-in-class proof-of-concept phase 2 study. Lancet Oncol. 2018, 19, 207–215. [Google Scholar] [CrossRef]
- Do, K.; Doroshow, J.H.; Kummar, S. Wee1 kinase as a target for cancer therapy. Cell Cycle 2013, 12, 3159–3164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leijen, S.; van Geel, R.M.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients With TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lheureux, S.; Cabanero, M.; Cristea, M.C.; Mantia-Smaldone, G.; Olawaiye, A.; Ellard, S.; Weberpals, J.I.; Wahner Hendrickson, A.E.; Fleming, G.F.; Welch, S.; et al. A randomized double-blind placebo-controlled phase II trial comparing gemcitabine monotherapy to gemcitabine in combination with adavosertib in women with recurrent, platinum resistant epithelial ovarian cancer: A trial of the Princess Margaret, California, Chicago and Mayo Phase II Consortia. J. Clin. Oncol. 2019, 37 (Suppl. 15), 5518. [Google Scholar]
- Dawson, M.; Stein, E.M.; Huntly, B.J.P.; Karadimitris, A.; Kamdar, M.; Fernandez de Larrea, C.; Dickinson, M.J.; Yeh, P.S.-H.; Daver, N.; Chaidos, A.; et al. A Phase I Study of GSK525762, a Selective Bromodomain (BRD) and Extra Terminal Protein (BET) Inhibitor: Results from Part 1 of Phase I/II Open Label Single Agent Study in Patients with Acute Myeloid Leukemia (AML). Blood 2017, 130 (Suppl. 1), 1377. [Google Scholar]
- Karakashev, S.; Zhu, H.; Yokoyama, Y.; Zhao, B.; Fatkhutdinov, N.; Kossenkov, A.V.; Wilson, A.J.; Simpkins, F.; Speicher, D.; Khabele, D.; et al. BET Bromodomain Inhibition Synergizes with PARP Inhibitor in Epithelial Ovarian Cancer. Cell Rep. 2017, 21, 3398–3405. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.J.; Stubbs, M.; Liu, P.; Ruggeri, B.; Khabele, D. The BET inhibitor INCB054329 reduces homologous recombination efficiency and augments PARP inhibitor activity in ovarian cancer. Gynecol. Oncol. 2018, 149, 575–584. [Google Scholar] [CrossRef]
- Sun, C.; Yin, J.; Fang, Y.; Chen, J.; Jeong, K.J.; Chen, X.; Vellano, C.P.; Ju, Z.; Zhao, W.; Zhang, D.; et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell 2018, 33, 401–416. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, A.R.; Gueble, S.E.; Liu, Y.; Oeck, S.; Kim, H.; Yun, Z.; Glazer, P.M. Cediranib suppresses homology-directed DNA repair through down-regulation of BRCA1/2 and RAD51. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Barry, W.T.; Wenham, R.M.; Hendrickson, A.E.W.; Armstrong, D.K.; Chan, N.; Cohn, D.E.; Lee, J.-m.; Penson, R.T.; Cristea, M.C.; et al. A phase 2 biomarker trial of combination cediranib and olaparib in relapsed platinum (plat) sensitive and plat resistant ovarian cancer (ovca). J. Clin. Oncol. 2018, 36 (Suppl. 15), 5519. [Google Scholar] [CrossRef]
- Lheureux, S.; Oaknin, A.; Garg, S.; Bruce, J.; Dhani, N.C.; Madariaga, A.; Bonilla, L.; Lee, Y.C.; Colombo, I.; Bhat, G.; et al. Evolve: A post PARP inhibitor clinical translational phase II trial of cediranib-olaparib in ovarian cancer—A Princess Margaret Consortium—GCIG Phase II Trial. J. Clin. Oncol. 2019, 37 (Suppl. 15), 5521. [Google Scholar] [CrossRef]
- Todd, A.K.; Johnston, M.; Neidle, S. Highly prevalent putative quadruplex sequence motifs in human DNA. Nucleic Acids Res. 2005, 33, 2901–2907. [Google Scholar] [CrossRef] [Green Version]
- Chambers, V.S.; Marsico, G.; Boutell, J.M.; Di Antonio, M.; Smith, G.P.; Balasubramanian, S. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 2015, 33, 877–881. [Google Scholar] [CrossRef] [Green Version]
- Huppert, J.L.; Balasubramanian, S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005, 33, 2908–2916. [Google Scholar] [CrossRef] [Green Version]
- Youds, J.L.; O’Neil, N.J.; Rose, A.M. Homologous recombination is required for genome stability in the absence of DOG-1 in Caenorhabditis elegans. Genetics 2006, 173, 697–708. [Google Scholar] [CrossRef] [Green Version]
- Koole, W.; van Schendel, R.; Karambelas, A.E.; van Heteren, J.T.; Okihara, K.L.; Tijsterman, M. A Polymerase Theta-dependent repair pathway suppresses extensive genomic instability at endogenous G4 DNA sites. Nat. Commun. 2014, 5, 3216. [Google Scholar] [CrossRef] [Green Version]
- Betous, R.; Rey, L.; Wang, G.; Pillaire, M.J.; Puget, N.; Selves, J.; Biard, D.S.; Shin-ya, K.; Vasquez, K.M.; Cazaux, C.; et al. Role of TLS DNA polymerases eta and kappa in processing naturally occurring structured DNA in human cells. Mol. Carcinog. 2009, 48, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, R.; Miller, K.M.; Forment, J.V.; Bradshaw, C.R.; Nikan, M.; Britton, S.; Oelschlaegel, T.; Xhemalce, B.; Balasubramanian, S.; Jackson, S.P. Small-molecule–induced DNA damage identifies alternative DNA structures in human genes. Nat. Chem. Biol. 2012, 8, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Khot, A.; Brajanovski, N.; Cameron, D.P.; Hein, N.; Maclachlan, K.H.; Sanij, E.; Lim, J.; Soong, J.; Link, E.; Blombery, P.; et al. First-in-Human RNA Polymerase I Transcription Inhibitor CX-5461 in Patients with Advanced Hematologic Cancers: Results of a Phase I Dose-Escalation Study. Cancer Discov. 2019, 9, 1036–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, M.K.; Pujade-Lauraine, E.; Aoki, D.; Mirza, M.R.; Lorusso, D.; Oza, A.M.; du Bois, A.; Vergote, I.; Reuss, A.; Bacon, M.; et al. Fifth Ovarian Cancer Consensus Conference of the Gynecologic Cancer InterGroup: Recurrent disease. Ann. Oncol. 2017, 28, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Hanker, L.C.; Loibl, S.; Burchardi, N.; Pfisterer, J.; Meier, W.; Pujade-Lauraine, E.; Ray-Coquard, I.; Sehouli, J.; Harter, P.; du Bois, A.; et al. The impact of second to sixth line therapy on survival of relapsed ovarian cancer after primary taxane/platinum-based therapy. Ann. Oncol. 2012, 23, 2605–2612. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Study Name/NCT | Target | Study Treatment | Study Population | Study Phase |
|---|---|---|---|---|
| DUETTE NCT04239014 | ATR/PARP | Ceralasertib (AZD6738) + Olaparib, or Olaparib monotherapy, or Placebo | Relapsed platinum-sensitive OC, who have acquired resistance from prior PARPi treatment | II, RCT |
| NCT02723864 | ATR/PARP | VX-970 + Veliparib and Cisplatin | Advanced refractory solid tumours | I |
| NCT02901899 | DNMT/PD-1 | Guadecitabine + pembrolizumab | Recurrent Platinum Resistant OC | II, open-label |
| NCT03924245 | HDAC/PARP | Entinostat + Olaparib | Recurrent platinum refractory and resistant EOC | I/II |
| NCT02915523 | PD-L1/HDAC | Avelumab ± Entinostat | Advanced OC Which Has Progressed or Recurred After First-line Platinum-based Chemotherapy and at Least Two Subsequent Lines of Treatment | Ib/II, RCT |
| NCT02797977 | CHK1 | SRA737 + gemcitabine + cisplatin, or gemcitabine monotherapy | Advanced solid tumours, including HGSOC which is BRCA1 and BRCA2 wild type. | I/II, non-randomised |
| NCT03057145 | CHK1/PARP | (Prexasertib) LY2606368 + Olaparib | Advanced solid tumours | I |
| NCT03579316 | WEE-1/PARP | Adavosertib (AZD1775) + Olaparib, or adavosertib monotherapy | Recurrent OC with progression on prior PARPi therapy | II, RCT |
| NCT02502266 | Angiogenesis/PARP | Cediranib + Olaparib, or chemotherapy | Platinum resistant or Refractory OC | III, RCT |
| DNA Damage Repair Pathway | Key Pathway Functions | Key Genes | Effect of Pathway Alterations on Therapeutic Resistance |
|---|---|---|---|
| Homologous Recombination (HR) | Repair of DSBs or stalled replication forks during S and G2 phases of cell cycle | BRCA1, BRCA2, RAD51, HSP90 | Reactivation of HR pathway enables repair of DSBs and resolves replisome blocks, promoting cancer cell progression through the cell cycle despite the presence of cytotoxic DNA damage. |
| Non-Homologous End Joining (NHEJ) | Repair of DSBs during interphase | 53BP1 | Loss of 53BP1 re-wires NHEJ pathway, reactivating HR independent of BRCA1 |
| Base Excision Repair (BER) | Repair of SSBs and DNA base lesions | PARP-1, XRCC1, Pol β | Functional BER pathway leads to loss of synthetic lethality and PARPi resistance |
| Nucleotide Excision Repair (NER) | Removes “bulky lesions” which distort the DNA double helix, including intra-strand crosslinks formed by platinum adducts. | ERCC1, XPF | Upregulation of ERCC1 and XPF potentially restores NER function. NER pathway alteration potentially confers sensitivity to platinum, and not PARPi. |
| Fanconi Anemia (FA) | Removes intra-strand DNA crosslinks, coordinates DNA replication by fine-tuning mitotic checkpoints and replication fork stabilisation | FANCC, FANCD2, FANCA | Mutations in FA pathway genes may have a similar effect to BRCA1 and BRCA2 mutation, in promoting progression of cancer cell through the cell cycle, even in setting of DNA damage and replication stress |
| Mismatch Repair (MMR) Deficiency | Recognise, excise and resynthesise mismatched or unmatched DNA base pairs or insertion-deletion loops. | MLH1, MSH2 | MMR deficiency results in microsatellite instability, interfering with detection of cytotoxic DNA damage, allowing cancer cells to proliferate despite DNA damage. |
| Resistance Mechanism | Function |
|---|---|
| BRCA (or HR gene) reversion mutation | Restores open reading frame of gene, resulting in functional protein expression |
| Loss of BRCA1 promoter methylation | Restores BRCA1 function |
| Upregulated HSP90 | Promotes BRCA-independent RAD51 loading onto damaged DNA |
| BRCA1 C-terminal domain mutation | Upregulation of BRCA1, in absence of BRCA1 reversion mutation |
| Loss 53BP1 | Recruits Shieldin complex to inhibit DNA resection, initiating HR in a BRCA-independent manner. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McMullen, M.; Karakasis, K.; Madariaga, A.; Oza, A.M. Overcoming Platinum and PARP-Inhibitor Resistance in Ovarian Cancer. Cancers 2020, 12, 1607. https://doi.org/10.3390/cancers12061607
McMullen M, Karakasis K, Madariaga A, Oza AM. Overcoming Platinum and PARP-Inhibitor Resistance in Ovarian Cancer. Cancers. 2020; 12(6):1607. https://doi.org/10.3390/cancers12061607
Chicago/Turabian StyleMcMullen, Michelle, Katherine Karakasis, Ainhoa Madariaga, and Amit M. Oza. 2020. "Overcoming Platinum and PARP-Inhibitor Resistance in Ovarian Cancer" Cancers 12, no. 6: 1607. https://doi.org/10.3390/cancers12061607
APA StyleMcMullen, M., Karakasis, K., Madariaga, A., & Oza, A. M. (2020). Overcoming Platinum and PARP-Inhibitor Resistance in Ovarian Cancer. Cancers, 12(6), 1607. https://doi.org/10.3390/cancers12061607