Monoclonal Gammopathies of Renal Significance: Renal Biopsy and Beyond

 , and
, and
Abstract
:1. Introduction
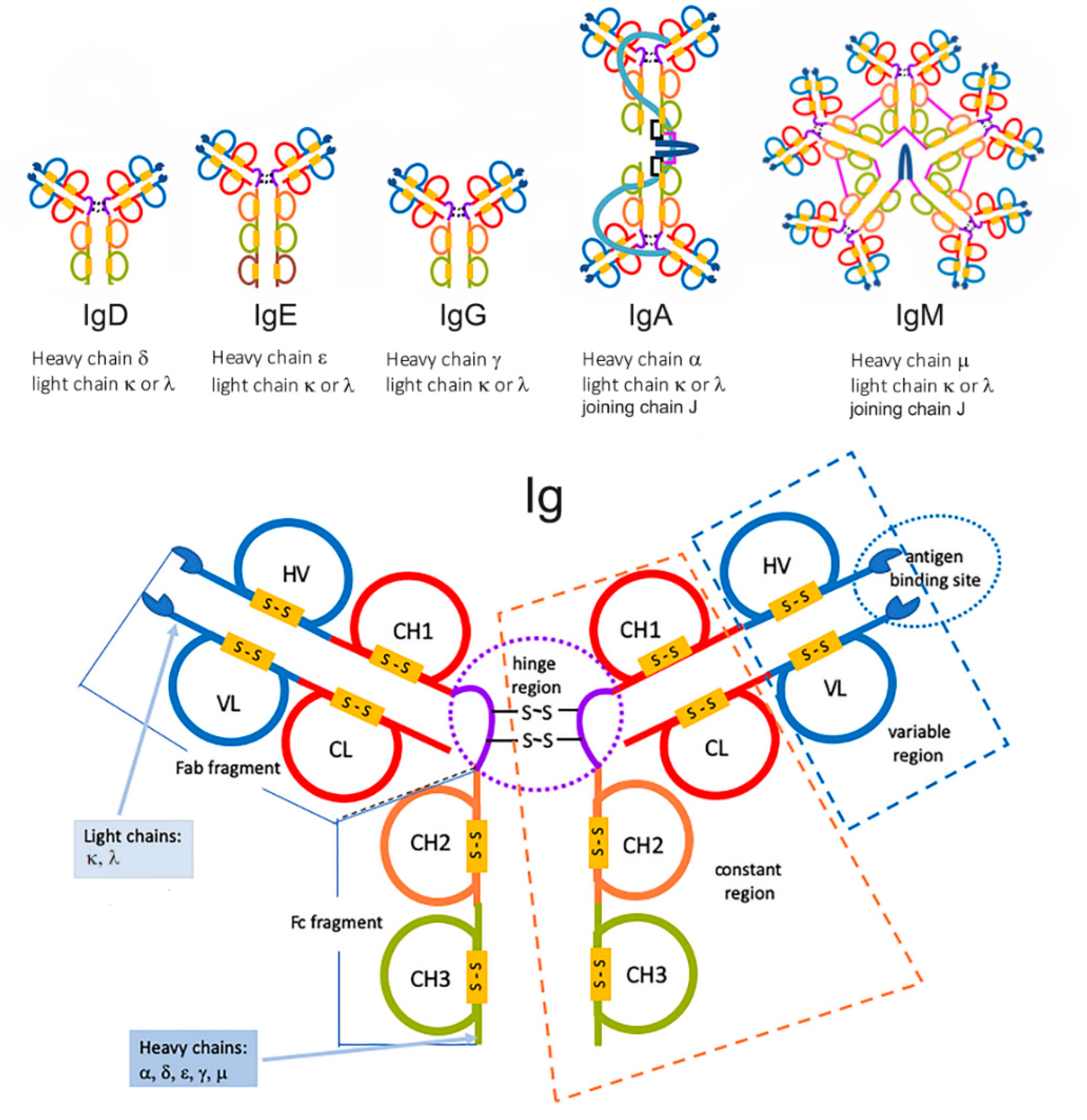
2. Biology of Immunoglobulin LC and Significance of MC Components
3. The Burden of Age: Epidemiology of MGRS
4. Directions: Hematologist to Nephrologist or Vice Versa?
5. Renal Biopsy in Persistent Urinary Abnormalities
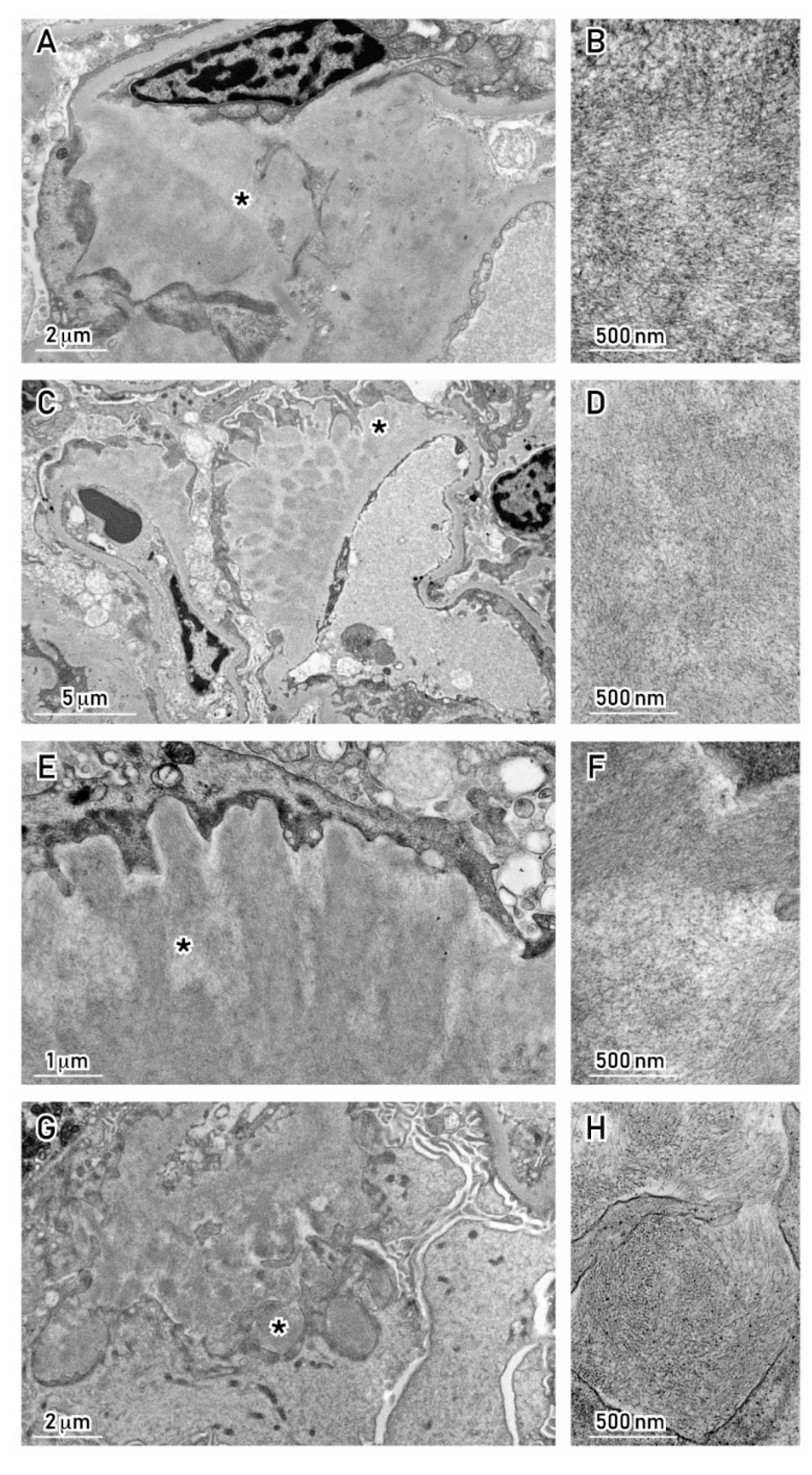
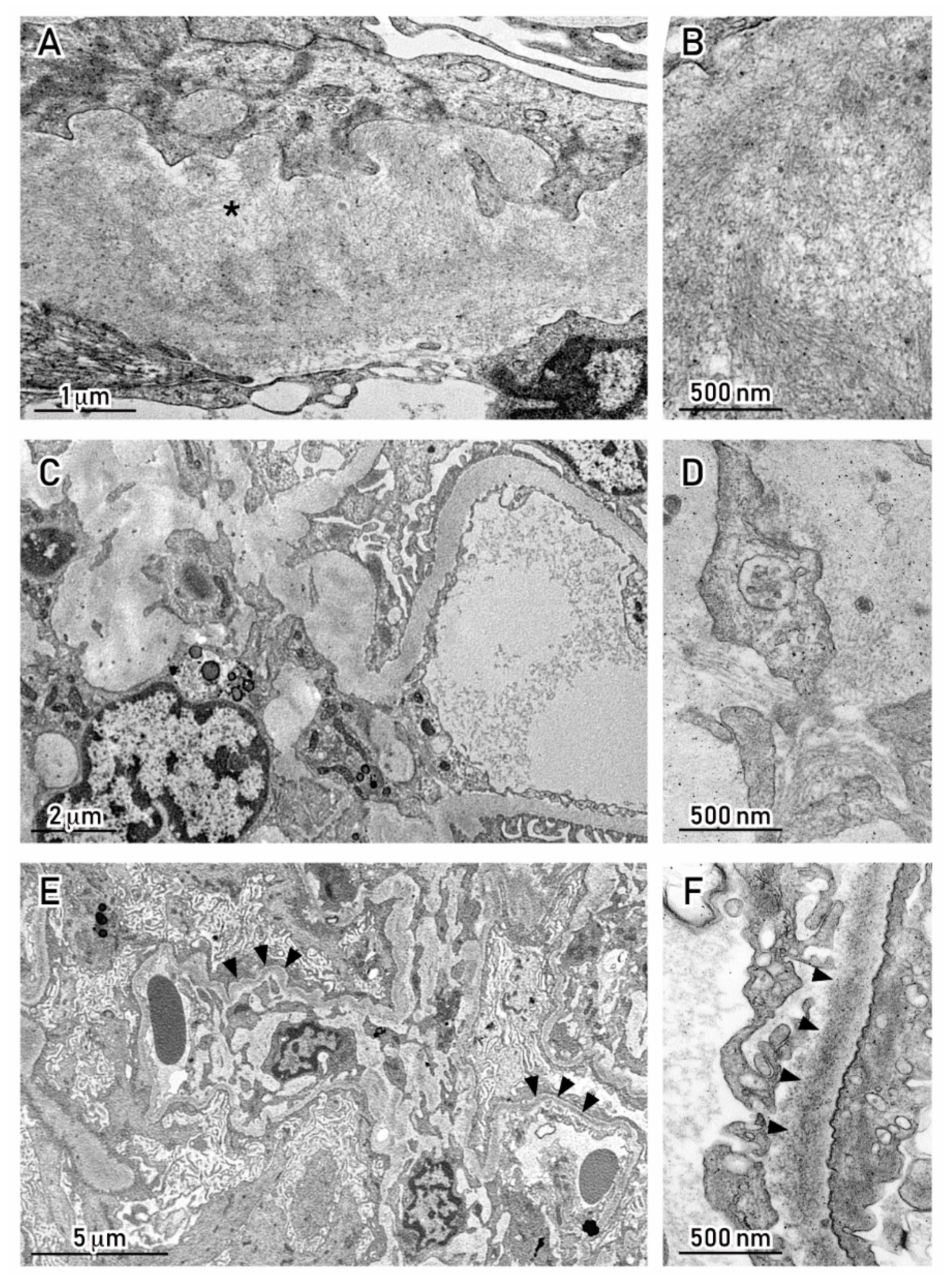
6. Mechanisms by Which MC Proteins Damage the Kidney
7. How to Assess the Link between MC Proteins and Renal Injury: The Role of the Renal Biopsy
8. When and How to Treat MGRS
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- van Nieuwenhuijzen, N.; Spaan, I.; Raymakers, R.; Peperzak, V. From MGUS to multiple myeloma, a paradigm for clonal evolution of premalignant cells. Cancer Res. 2018, 78, 2449–2456. [Google Scholar] [CrossRef] [Green Version]
- Mouhieddine, T.H.; Weeks, L.D.; Ghobrial, I.M. Monoclonal gammopathy of undetermined significance. Blood 2019, 133, 2484–2494. [Google Scholar] [CrossRef] [PubMed]
- Dispenzieri, A.; Katzmann, J.A.; Kyle, R.A.; Larson, D.R.; Melton, L.J., 3rd; Colby, C.L.; Therneau, T.M.; Clark, R.; Kumar, S.K.; Bradwell, A.; et al. Prevalence and risk of progression of light-chain monoclonal gammopathy of undetermined significance: A retrospective population-based cohort study. Lancet 2010, 375, 1721–1728. [Google Scholar] [CrossRef] [Green Version]
- Murray, D.; Kumar, S.K.; Kyle, R.A.; Dispenzieri, A.; Dasari, S.; Larson, D.R.; Vachon, C.; Cerhan, J.R.; Rajkumar, S.V. Detection and prevalence of monoclonal gammopathy of undetermined significance: A study utilizing mass spectrometry-based monoclonal immunoglobulin rapid accurate mass measurement. Blood Cancer J. 2019, 9, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkin, C.; Reddy-Kolanu, V.; Drayson, M.T.; Sapey, E.; Richter, A.G. The prevalence and significance of monoclonal gammopathy of undetermined significance in acute medical admissions. Br. J. Haematol. 2020, 189, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Leung, N.; Bridoux, F.; Hutchison, C.A.; Nasr, S.H.; Cockwell, P.; Fermand, J.P.; Dispenzieri, A.; Song, K.W.; Kyle, R.A. Monoclonal gammopathy of renal significance: When MGUS is no longer undetermined or insignificant. Blood 2012, 120, 4292–4295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motwani, S.S.; Herlitz, L.; Monga, D.; Jhaveri, K.D.; Lam, A.Q. Paraprotein-Related Kidney Disease: Glomerular Diseases Associated with Paraproteinemias. Clin. J. Am. Soc. Nephrol. 2016, 11, 2260–2272. [Google Scholar] [CrossRef]
- Sethi, S.; Rajkumar, S.V.; D’Agati, V.D. The Complexity and Heterogeneity of Monoclonal Immunoglobulin–Associated Renal Diseases. J. Am. Soc. Nephrol. 2018, 29, 1810–1823. [Google Scholar] [CrossRef] [Green Version]
- Doshi, M.; Lahot, A.; Danesh, F.R.; Batuman, V.; Sanders, P.W. Paraprotein-Related Kidney Disease: Kidney Injury from Paraproteins-What Determines the Site of Injury? Clin. J. Am. Soc. Nephrol. 2016, 11, 2288–2294. [Google Scholar] [CrossRef]
- Sogn, J.A.; Kindt, T.J. Immunoglobulin structure and function. Curr. Opin. Immunol. 1988, 1, 73–76. [Google Scholar] [CrossRef]
- Edmundson, A.B.; Ely, K.R.; Abola, E.E. Conformational flexibility in immunoglobulins. Contemp. Top. Mol. Immunol. 1978, 7, 95–118. [Google Scholar] [PubMed]
- Nezlin, R. Dynamic Aspects of the Immunoglobulin Structure. Immunol. Invest. 2019, 48, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Blancas-Mejia, L.M.; Misra, P.; Dick, C.J.; Cooper, S.A.; Redhage, K.R.; Bergman, M.R.; Jordan, T.L.; Maar, K.; Ramirez-Alvarado, M. Immunoglobulin light chain amyloid aggregation. Chem. Commun. (Camb). 2018, 54, 10664–10674. [Google Scholar] [CrossRef]
- Paladini, G.; Sala, P.G. Anion gap in multiple myeloma. Acta. Haematol. 1979, 62, 148–152. [Google Scholar] [CrossRef]
- Mansoor, S.; Siddiqui, I.; Adil, S.; Nabi Kakepoto, G.; Fatmi, Z.; Ghani, F. Anion gap among patients of multiple myeloma and normal individuals. Clin. Biochem. 2007, 40, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Kraut, J.A.; Madias, N.E. Serum anion gap: Its uses and limitations in clinical medicine. Clin. J. Am. Soc. Nephrol. 2007, 2, 162–174. [Google Scholar] [CrossRef] [Green Version]
- Grabar, P.; Williams, C.A. Method permitting the combined study of the electrophoretic and the immunochemical properties of protein mixtures; application to blood serum. Biochim. Biophys Acta. 1953, 10, 193–194. [Google Scholar] [CrossRef]
- Edelman, G.M.; Gally, J.A. The nature of Bence-Jones proteins. Chemical similarities to polypetide chains of myeloma globulins and normal gamma-globulins. J. Exp. Med. 1962, 116, 207–227. [Google Scholar] [CrossRef] [Green Version]
- Raju, T.N. The Nobel chronicles. 1972: Gerald M Edelman (b 1929) and Rodney R Porter (1917–85). Lancet 1999, 354, 1040. [Google Scholar] [CrossRef]
- Stevens, F.J.; Solomon, A.; Schiffer, M. Bence Jones proteins: A powerful tool for the fundamental study of protein chemistry and pathophysiology. Biochemistry 1991, 30, 6803–6805. [Google Scholar] [CrossRef] [Green Version]
- Pisitkun, T.; Shen, R.F.; Knepper, M.A. Identification and proteomic profiling of exosomes in human urine. Proc. Natl. Acad. Sci. USA 2004, 101, 13368–13373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez-Alvarado, M.; Ward, C.J.; Huang, B.Q.; Gong, X.; Hogan, M.C.; Madden, B.J.; Charlesworth, M.C.; Leung, N. Differences in immunoglobulin light chain species found in urinary exosomes in light chain amyloidosis (Al). PLoS ONE 2012, 7, e38061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez-Alvarado, M.; Barnidge, D.R.; Murray, D.L.; Dispenzieri, A.; Marin-Argany, M.; Dick, C.J.; Cooper, S.A.; Nasr, S.H.; Ward, C.J.; Dasari, S.; et al. Assessment of renal response with urinary exosomes in patients with AL amyloidosis: A proof of concept. Am. J. Hematol. 2017, 92, 536–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, A.; Weiss, D.T.; Kattine, A.A. Nephrotoxic potential of Bence Jones proteins. N. Engl. J. Med. 1991, 324, 1845–1851. [Google Scholar] [CrossRef]
- Luciani, A.; Sirac, C.; Terryn, S.; Javaugue, V.; Prange, J.A.; Bender, S.; Bonaud, A.; Cogné, M.; Aucouturier, P.; Ronco, P.; et al. Impaired Lysosomal Function Underlies Monoclonal Light Chain-Associated Renal Fanconi Syndrome. J. Am. Soc. Nephrol. 2016, 27, 2049–2061. [Google Scholar] [CrossRef]
- Ronco, P.; Plaisier, E.; Mougenot, B.; Aucouturier, P. Immunoglobulin light (heavy)-chain deposition disease: From molecular medicine to pathophysiology-driven therapy. Clin. J. Am. Soc. Nephrol. 2006, 1, 1342–1350. [Google Scholar] [CrossRef] [Green Version]
- Rane, S.; Rana, S.; Mudrabettu, C.; Jha, V.; Joshi, K. Heavy-chain deposition disease: A morphological, immunofluorescence and ultrastructural assessment. Clin. Kidney. J. 2012, 5, 383–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barwick, B.G.; Gupta, V.A.; Vertino, P.M.; Boisem, L.H. Cell of origin and genetic alterations in the pathogenesis of multiple myeloma. Front. Immunol. 2019, 10, 1121. [Google Scholar] [CrossRef] [Green Version]
- Bulati, M.; Caruso, C.; Colonna-Romano, G. From lymphopoiesis to plasma cells differentiation, the age-related modifications of B cell compartment are influenced by “inflamm-ageing”. Ageing Res. Rev. 2017, 36, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2018, 8, 1960. [Google Scholar] [CrossRef] [Green Version]
- Royal, V.; Leung, N.; Troyanov, S.; Nasr, S.H.; Écotière, L.; LeBlanc, R.; Adam, B.A.; Angioi, A.; Alexander, M.P.; Asunis, A.M.; et al. Clinicopathologic predictors of renal outcomes in light chain cast nephropathy: A multicenter retrospective study. Blood 2020, 135, 1833–1846. [Google Scholar] [CrossRef]
- Yadav, P.; Sathick, I.J.; Leung, N.; Brown, E.E.; Cook, M.; Sanders, P.W.; Cockwell, P. Serum free light chain level at diagnosis in myeloma cast nephropathy-a multicentre study. Blood Cancer J 2020, 10, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchison, C.A.; Cockwell, P.; Moroz, V.; Bradwell, A.R.; Fifer, L.; Gillmore, J.D.; Jesky, M.D.; Storr, M.; Wessels, J.; Winearls, C.G.; et al. High cutoff versus high-flux haemodialysis for myeloma cast nephropathy in patients receiving bortezomib-based chemotherapy (EuLITE): A phase 2 randomised controlled trial. Lancet. Haematol. 2019, 6, e217–e228. [Google Scholar] [CrossRef] [Green Version]
- Menè, P.; Giammarioli, E.; Fofi, C.; Antolino, G.; La Verde, G.; Tafuri, A.; Punzo, G.; Festuccia, F. Serum free light chains removal by HFR in patients with multiple myeloma and acute kidney injury: A case series. Kidney Blood Press. Res. 2018, 43, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Merlini, G.; Dispenzieri, A.; Sanchorawala, V.; Schönland, S.O.; Palladini, G.; Hawkins, P.N.; Gertz, M.A. Systemic immunoglobulin light chain amyloidosis. Nat. Rev. Dis. Primers. 2018, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Li, Q.; Kang, J.; Li, C.; Zhou, J. Non-secreting multiple myeloma switches to IgD of lambda type: A case report and review of literature. Int. J. Clin. Exp. Med. 2015, 8, 16984–16990. [Google Scholar]
- Selene, I.I.; Jose, J.A.; Khalil, M.J.; Faisal, M.S.; Malik, M.N. Presentation patterns, diagnostic markers, management strategies, and outcomes of IgD multiple myeloma: A systematic review of literature. Cureus 2019, 11, e4011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, G.A.; Joseph, L.; Gu, X.; Hough, A.; Barlogie, B. Renal pathologic spectrum in an autopsy series of patients with plasma cell dyscrasia. Arch. Patho.l Lab. Med. 2004, 128, 875–879. [Google Scholar]
- Joh, K. Pathology of glomerular deposition diseases. Pathol. Int. 2007, 57, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Picken, M.M. Amyloidosis-where are we now and where are we heading? Arch. Pathol. Lab. Med. 2010, 134, 545–551. [Google Scholar] [CrossRef]
- Herrera, G.A.; Picken, M.M. Renal Diseases Associated with Plasma Cell Dyscrasias, Amyloidoses, and Waldenström Macroglobulinemia; Jennette, J.C., Olson, J.L., Silva, F.G., D’Agati, V.D., Eds.; Heptinstall’s Pathology of the Kidney 7th. Chapter 22; Wolters Kluwer: Philadelphia, PA, USA, 2014; pp. 951–1014. [Google Scholar]
- Picken, M.M.; Herrera, G.A.; Dogan, A. Amyloid and Related Disorders; Humana Press: Totowa, NJ, USA, 2015. [Google Scholar]
- Picken, M.M. Proteomics and mass spectrometry in the diagnosis of renal amyloidosis. Clin. Kidney J. 2015, 8, 665–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridoux, F.; Leung, N.; Hutchison, C.A.; Touchard, G.; Sethi, S.; Fermand, J.P.; Picken, M.M.; Herrera, G.A.; Kastritis, E.; Merlini, G.; et al. Diagnosis of monoclonal gammopathy of renal significance. Kidney Int. 2015, 87, 698–711. [Google Scholar] [CrossRef] [Green Version]
- Herrera, G.A. The value of ultrastructural evaluation in medical renal diseases. Ultrastruct. Pathol. 2019, 43, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, J.L.; Markowitz, G.S.; Valeri, A.M.; Sacchi, G.; Appel, G.B.; D’Agati, V.D. Fibrillary and immunotactoid glomerulonephritis: Distinct entities with different clinical and pathologic features. Kidney Int. 2003, 63, 1450–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, N.; Drosou, M.E.; Nasr, S.H. Dysproteinemias and glomerular disease. Clin. J. Am. Soc. Nephrol. 2018, 13, 128–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herlitz, L.C.; D’Agati, V.D.; Markowitz, G.S. Crystalline nephropathies. Arch. Pathol. Lab. Med. 2012, 136, 713–720. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.J.; Zhou, X.J.; Wang, S.X.; Zhou, F.D.; Zhao, M.H. Monoclonal light chain crystalline podocytopathy and tubulopathy associated with monoclonal gammopathy of renal significance: A case report and literature review. BMC Nephrol. 2018, 12, 322. [Google Scholar] [CrossRef]
- Leboulleux, M.; Lelongt, B.; Mougenot, B.; Touchard, G.; Makdassi, R.; Rocca, A.; Noel, L.H.; Ronco, P.M.; Aucouturier, P. Protease resistance and binding of Ig light chains in myeloma-associated tubulopathies. Kidney Int. 1995, 48, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, V.; ElTers, M.; Kashani, K.; Leung, N.; Nasr, S.H. Crystalglobulin-induced nephropathy. J. Am. Soc. Nephrol. 2015, 26, 525–529. [Google Scholar] [CrossRef] [Green Version]
- Batuman, V. Proximal tubular injury in myeloma. Contrib. Nephrol. 2007, 153, 87–104. [Google Scholar]
- Herrera, G.A. Proximal tubulopathies associated with monoclonal light chains: The spectrum of clinicopathologic manifestations and molecular pathogenesis. Arch. Pathol. Lab. Med. 2014, 138, 1365–1380. [Google Scholar] [CrossRef] [PubMed]
- Stokes, M.B.; Valeri, A.M.; Herlitz, L.; Khan, A.M.; Siegel, D.S.; Markowitz, G.S.; D’Agati, V.D. Light chain proximal tubulopathy: Clinical and pathologic characteristics in the modern treatment era. J. Am. Soc. Nephrol. 2016, 27, 1555–1565. [Google Scholar] [CrossRef] [Green Version]
- Messiaen, T.; Deret, S.; Mougenot, B.; Bridoux, F.; Dequiedt, P.; Dion, J.J.; Makdassi, R.; Meeus, F.; Pourrat, J.; Touchard, G.; et al. Adult Fanconi syndrome secondary to light chain gammopathy. Clinicopathologic heterogeneity and unusual features in 11 patients. Med. (Baltim.) 2000, 79, 135–154. [Google Scholar] [CrossRef] [PubMed]
- Sanders, P.W.; Booker, B.B.; Bishop, J.B.; Cheung, H.C. Mechanisms of intranephronal proteinaceous cast formation by low molecular weight proteins. J. Clin. Invest. 1990, 85, 570–576. [Google Scholar] [CrossRef] [Green Version]
- Sanders, P.W.; Booker, B.B. Pathobiology of cast nephropathy from human Bence Jones proteins. J. Clin. Invest. 1992, 89, 630–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasr, S.H.; Satoskar, A.; Markowitz, G.S.; Valeri, A.M.; Appel, G.B.; Stokes, M.B.; Nadasdy, T.; D’Agati, V.D. Proliferative glomerulonephritis with monoclonal IgG deposits. J. Am. Soc. Nephrol. 2009, 20, 2055–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guiard, E.; Karras, A.; Plaisier, E.; Duong Van Huyen, J.P.; Fakhouri, F.; Rougier, J.P.; Noel, L.H.; Callard, P.; Delahousse, M.; Ronco, P. Patterns of noncryoglobulinemic glomerulonephritis with monoclonal Ig deposits: Correlation with IgG subclass and response to rituximab. Clin. J. Am. Soc. Nephrol. 2011, 6, 1609–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Servais, A.; Frémeaux-Bacchi, V.; Lequintrec, M.; Salomon, R.; Blouin, J.; Knebelmann, B.; Grünfeld, J.P.; Lesavre, P.; Noël, L.H.; Fakhouri, F. Primary glomerulonephritis with isolated C3 deposits: A new entity which shares common genetic risk factors with haemolytic uraemic syndrome. J. Med. Genet. 2007, 44, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Pickering, M.C.; D’Agati, V.D.; Nester, C.M.; Smith, R.J.; Haas, M.; Appel, G.B.; Alpers, C.E.; Bajema, I.M.; Bedrosian, C.; Braun, M.; et al. C3 glomerulopathy: Consensus report. Kidney Int. 2013, 84, 1079–1089. [Google Scholar] [CrossRef] [Green Version]
- Pirozzi, N.; Stoppacciaro, A.; Menè, P. Dominant c3 glomerulopathy: New roles for an old actor in renal pathology. J. Nephrol. 2018, 30, 503–510. [Google Scholar] [CrossRef]
- Thurman, J.M.; Nester, C.M. All things complement. Clin. J. Am. Soc. Nephrol. 2016, 11, 1856–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, C.P.; Messias, N.C.; Walker, P.D.; Fidler, M.E.; Cornell, L.D.; Hernandez, L.H.; Alexander, M.P.; Sethi, S.; Nasr, S.H. Membranoproliferative glomerulonephritis with masked monotypic immunoglobulin deposits. Kidney Int. 2015, 88, 867–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milani, P.; Merlini, G.; Palladini, G. Novel therapies in light chain amyloidosis. Kidney Int. Rep. 2017, 28, 530–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, N.; Bridoux, F.; Batuman, V.; Chaidos, A.; Cockwell, P.; D’Agati, V.D.; Dispenzieri, A.; Fervenza, F.C.; Fermand, J.P.; Gibbs, S.; et al. The evaluation of monoclonal gammopathy of renal significance: A consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat. Rev. Nephrol. 2019, 15, 45–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, A.C. Emerging options for combination therapy in multiple myeloma. Clin. Adv. Hematol. Oncol 2018, 16, 192–194. [Google Scholar]
- Rajkumar, S.V. Multiple myeloma: Every year a new standard? Hematol. Oncol. 2019, 37, 62–65. [Google Scholar] [CrossRef] [Green Version]
- Kazandjian, D.; Landgren, O. Delaying the use of high-dose melphalan with stem cell rescue in multiple myeloma is ready for prime time. Clin. Adv. Hematol. Oncol. 2019, 17, 559–568. [Google Scholar]
- Lin, Q.; Zhao, J.; Song, Y.; Liu, D. Recent updates on CAR T clinical trials for multiple myeloma. Mol. Cancer 2019, 18, 154. [Google Scholar] [CrossRef] [Green Version]
- Pinto, V.; Bergantim, R.; Caires, H.R.; Seca, H.; Guimarães, J.E.; Vasconcelos, M.H. Multiple myeloma: Available therapies and causes of drug resistance. Cancers (Basel) 2020, 12, 407. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Significance (MGRS) | |||||
|---|---|---|---|---|---|
| Patient | Age | M/F | eGFR, mL/min | uProt g/day | Diagnosis |
| T.A. | 64 | M | 52.2 | 11.0 | AL Amyloidosis, IgA λ mMM |
| M.M.B. | 72 | F | 10.6 | 5.0 | AL Amyloidosis, IgG λ |
| L.B.N. | 51 | F | 36.6 | 1.9 | AL Amyloidosis, IgG λ mMM |
| G.C. | 65 | M | 33.1 | 5.2 | LCDD, IgM λ |
| G.C. | 54 | M | 108.5 | 5.0 | AL Amyloidosis + FibGNF, IgM λ |
| R.C. | 68 | M | 100.0 | 4.8 | AL Amyloidosis, IgA λ |
| E.D.B. | 72 | M | 30.4 | 1.1 | LCDD, IgG λ |
| R.D.C. | 72 | F | 9.5 | 7.5 | AL Amyloidosis, IgG λ |
| I.D.T. | 71 | F | 50.7 | 13.2 | LCDD, IgG λ |
| L.F. | 66 | F | 25.1 | 8.8 | LCDD, IgG κ |
| R.G. | 54 | F | 160.2 | 3.1 | AL Amyloidosis, IgA λ MM |
| G.G. | 60 | F | 32.3 | 4.1 | LCDD, IgG κ |
| M.L. | 59 | M | 94.0 | 5.3 | AL Amyloidosis, IgG λ |
| E.L.C. | 70 | M | 84.3 | 4.6 | AL Amyl., B-cell lymphoma, IgG κ |
| M.L. | 45 | M | 96.7 | 8.8 | LCDD, IgG κ |
| P.L. | 70 | F | 90.1 | 5.2 | AL Amyloidosis, IgG λ |
| B.M. | 43 | F | 11.9 | 5.0 | Cast nephropathy, IgG κ MM |
| G.M.M. | 42 | F | 107.1 | 2.2 | LCDD, IgG κ |
| E.P. | 54 | F | 120.5 | 4.4 | AL Amyloidosis, IgG λ |
| A.P. | 76 | M | 12.4 | 10.0 | Cast nephropathy, AL Amyl., IgG λ |
| F.P. | 66 | M | 39.4 | 7.2 | AL Amyloidosis, IgG λ MM |
| M.C.S. | 52 | F | 65.3 | 5.8 | LCDD, IgG κ MM |
| D.S. | 64 | M | 45.8 | 8.5 | LCDD, IgG κ MM |
| V.T. | 62 | M | 26.3 | 1.2 | AL Amyloidosis, IgA λ |
| Data are expressed as mean ± SD | 62.58 ± 9.16 | 60.00 ± 40.54 | 5.73 ± 2.94 | ||
| Major Known Clinical/Pathological Presentations |
|---|
| Light chain deposition disease (LCDD, glomerular/tubular) |
| Heavy chain deposition disease (HCDD) |
| Tubulointerstitial LCDD with Fanconi syndrome |
| Tubular obstructive “cast nephropathy” |
| AL amyloidosis |
| Proliferative glomerulonephritis with monoclonal immune deposits (PGNMID) |
| “Dominant C3” glomerulonephritis |
| Fibrillary glomerulonephritis |
| Immunotactoid glomerulonephritis |
| Cryoglobulinemic membranoproliferative glomerulonephritis (“non-infectious”) |
| Nephritis with crystalline inclusions (“crystalline podocytopathy, tubulopathy”) |
| Major Regimens/Associations |
|---|
| Melphalan + prednisone (MP) ± thalidomide (MPT) * |
| Thalidomide + dexamethasone (TD) |
| Lenalidomide + dexamethasone (RD) |
| Bortezomib + dexamethasone (VD) |
| Bortezomib + melphalan + prednisone (VMP) |
| Bortezomib + thalidomide + dexamethasone (VTD) |
| Bortezomib + cyclophosphamide + dexamethasone (CyBorD, VCD) * |
| Bortezomib + lenalidomide + dexamethasone (VRD) |
| Carfilzomib + cyclophosphamide + dexamethasone (CCyD) * |
| Carfilzomib + lenalidomide + dexamethasone (KRD) |
| Pomalidomide + dexamethasone (Pom/Dex) |
| Carfilzomib + pomalidomide + dexamethasone (KPD) |
| Autologous Stem Cell Transplantation (ASCT) * |
| CD38-targeting immunotherapy: monoclonal Abs Daratumumab * |
| Daratumumab * |
| Elotuzumab |
| Anthracyclines, tetracyclines |
| Doxorubicin * |
| Doxycicline * |
| Aggresome inhibitors |
| Panobinostat |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menè, P.; De Alexandris, L.; Moioli, A.; Raffa, S.; Stoppacciaro, A. Monoclonal Gammopathies of Renal Significance: Renal Biopsy and Beyond. Cancers 2020, 12, 1741. https://doi.org/10.3390/cancers12071741
Menè P, De Alexandris L, Moioli A, Raffa S, Stoppacciaro A. Monoclonal Gammopathies of Renal Significance: Renal Biopsy and Beyond. Cancers. 2020; 12(7):1741. https://doi.org/10.3390/cancers12071741
Chicago/Turabian StyleMenè, Paolo, Lorenzo De Alexandris, Alessandra Moioli, Salvatore Raffa, and Antonella Stoppacciaro. 2020. "Monoclonal Gammopathies of Renal Significance: Renal Biopsy and Beyond" Cancers 12, no. 7: 1741. https://doi.org/10.3390/cancers12071741
APA StyleMenè, P., De Alexandris, L., Moioli, A., Raffa, S., & Stoppacciaro, A. (2020). Monoclonal Gammopathies of Renal Significance: Renal Biopsy and Beyond. Cancers, 12(7), 1741. https://doi.org/10.3390/cancers12071741