Harnessing Omics Approaches on Advanced Preclinical Models to Discovery Novel Therapeutic Targets for the Treatment of Metastatic Colorectal Cancer

 ,
,  ,
, 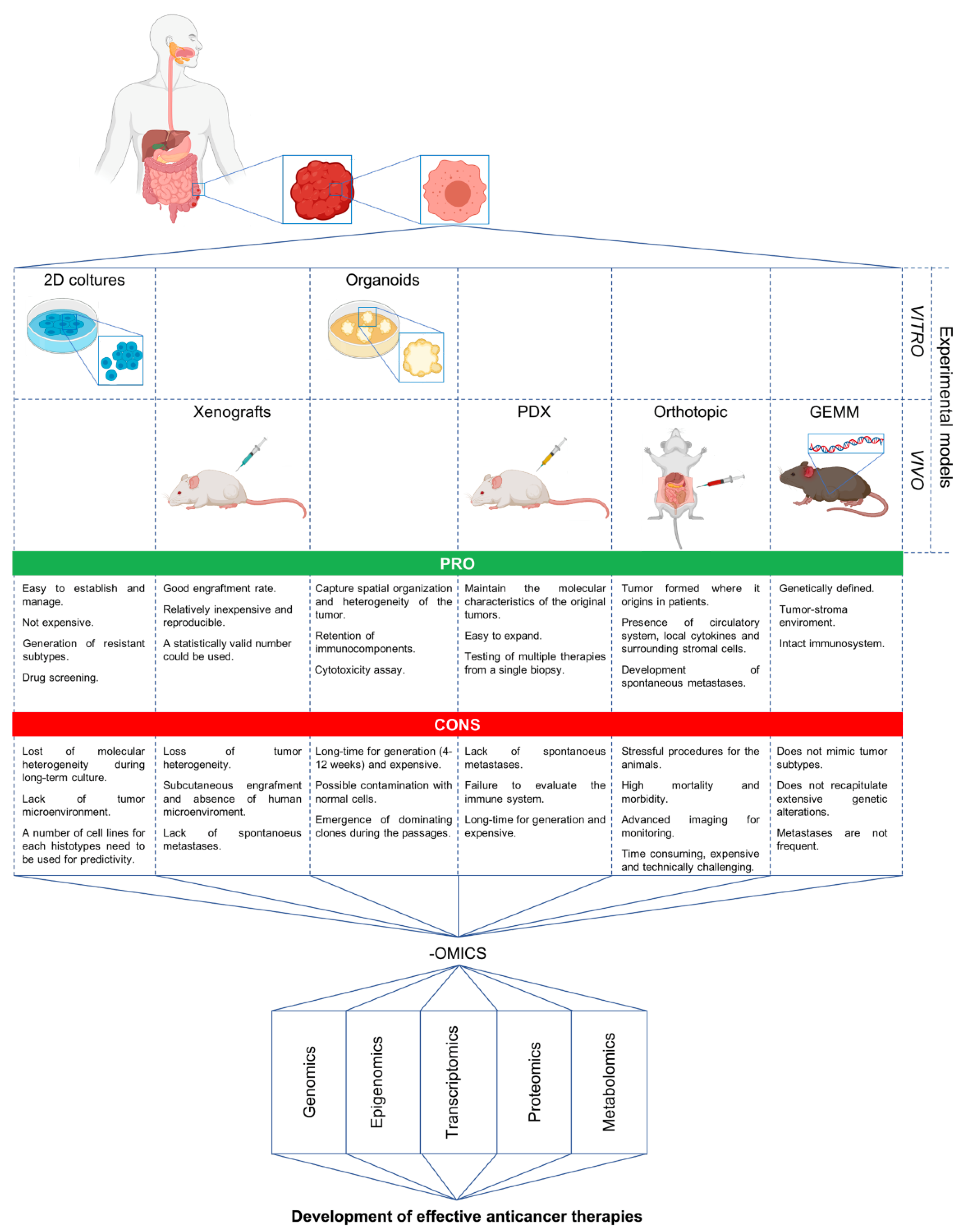{kind=link}
{kind=link}
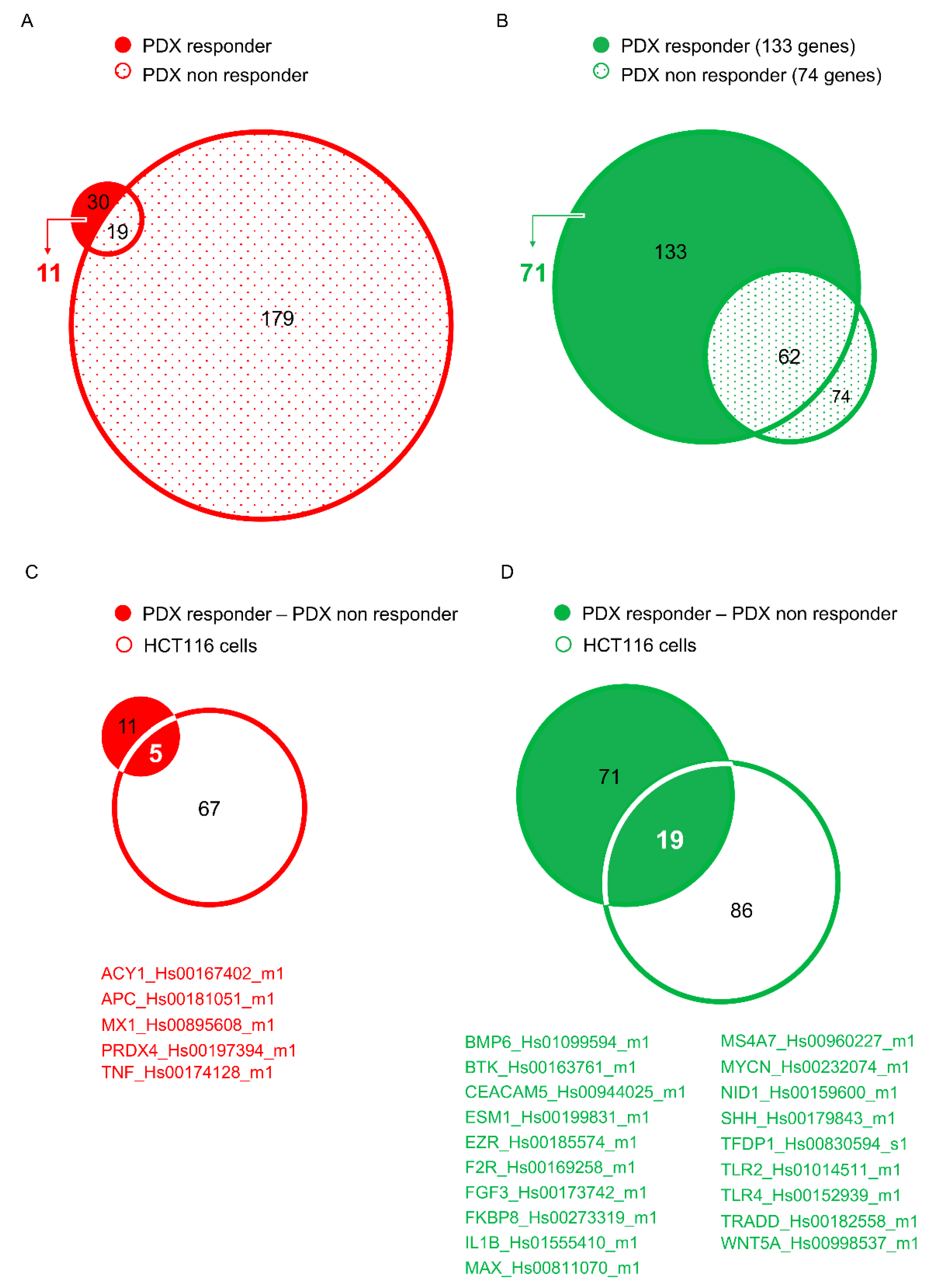{kind=link}
Abstract
:1. Introduction
2. Validated Biomarkers of Response to Anti-EGFR mAbs Treatment
3. Emerging Biomarkers of Response to Anti-EGFR mAbs Treatment
4. Secondary Resistance to Anti-EGFR Treatment
5. Advanced Preclinical Models and Omics in the Discovery of New Strategies against mCRC
6. Development of Novel Therapeutics in Targeting mCRC: The Experience of Our Laboratory
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: Globocan Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; van Krieken, J.H.; Aderka, D.; Aguilar, E.A.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. Esmo Consensus Guidelines for the Management of Patients with Metastatic Colorectal Cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.B. Epidermal Growth Factor Receptor as a Therapeutic Target in Colorectal Cancer. Clin. Colorectal Cancer 2003, 2, 246–251. [Google Scholar] [CrossRef]
- Galizia, G.; Lieto, E.; de Vita, F.; Orditura, M.; Castellano, P.; Troiani, T.; Imperatore, V.; Ciardiello, F. Cetuximab, a Chimeric Human Mouse Anti-Epidermal Growth Factor Receptor Monoclonal Antibody, in the Treatment of Human Colorectal Cancer. Oncogene 2007, 26, 3654–3660. [Google Scholar] [CrossRef] [Green Version]
- Benvenuti, S.; Sartore-Bianchi, A.; di Nicolantonio, F.; Zanon, C.; Moroni, M.; Veronese, S.; Siena, S.; Bardelli, A. Oncogenic Activation of the Ras/Raf Signaling Pathway Impairs the Response of Metastatic Colorectal Cancers to Anti-Epidermal Growth Factor Receptor Antibody Therapies. Cancer Res. 2007, 67, 2643–2648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lievre, A.; Bachet, J.B.; le Corre, D.; Boige, V.; Landi, B.; Emile, J.F.; Cote, J.F.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. Kras Mutation Status Is Predictive of Response to Cetuximab Therapy in Colorectal Cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baselga, J. The Egfr as a Target for Anticancer Therapy--Focus on Cetuximab. Eur. J. Cancer 2001, 37 (Suppl. 4), S16–S22. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the Braf Gene in Human Cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Di Nicolantonio, F.; Martini, M.; Molinari, F.; Sartore-Bianchi, A.; Arena, S.; Saletti, P.; de Dosso, S.; Mazzucchelli, L.; Frattini, M.; Siena, S.; et al. Wild-Type Braf Is Required for Response to Panitumumab or Cetuximab in Metastatic Colorectal Cancer. J. Clin. Oncol. 2008, 26, 5705–5712. [Google Scholar] [CrossRef]
- van Brummelen, E.M.J.; de Boer, A.; Beijnen, J.H.; Schellens, J.H.M. Braf Mutations as Predictive Biomarker for Response to Anti-Egfr Monoclonal Antibodies. Oncologist 2017, 22, 864–872. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas, Network. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertotti, A.; Migliardi, G.; Galimi, F.; Sassi, F.; Torti, D.; Isella, C.; Cora, D.; di Nicolantonio, F.; Buscarino, M.; Petti, C.; et al. A Molecularly Annotated Platform of Patient-Derived Xenografts (“Xenopatients”) Identifies Her2 as an Effective Therapeutic Target in Cetuximab-Resistant Colorectal Cancer. Cancer Discov. 2011, 1, 508–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavuri, S.M.; Jain, N.; Galimi, F.; Cottino, F.; Leto, S.M.; Migliardi, G.; Searleman, A.; Shen, W.; Monsey, J.; Trusolino, L.; et al. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov. 2015, 5, 832–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bregni, G.; Sciallero, S.; Sobrero, A. HER2 Amplification and Anti-EGFR Sensitivity in Advanced Colorectal Cancer. JAMA Oncol. 2019, 5, 605–606. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Comoglio, P.M. Scatter-factor and semaphorin receptors: Cell signalling for invasive growth. Nat. Rev. Cancer 2002, 2, 289–300. [Google Scholar] [CrossRef]
- Lee, H.E.; Kim, M.A.; Lee, H.S.; Jung, E.-J.; Yang, H.-K.; Lee, B.L.; Bang, Y.-J.; Kim, W.H. MET in gastric carcinomas: Comparison between protein expression and gene copy number and impact on clinical outcome. Br. J. Cancer 2012, 107, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.M.; Grandis, J.R. Pharmacokinetic and pharmacodynamic properties of EGFR inhibitors under clinical investigation. Cancer Treat. Rev. 2004, 30, 255–268. [Google Scholar] [CrossRef]
- Frattini, M.; Saletti, P.; Romagnani, E.; Martin, V.; Molinari, F.; Ghisletta, M.; Camponovo, A.; Etienne, L.L.; Cavalli, F.; Mazzucchelli, L. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br. J. Cancer 2007, 97, 1139–1145. [Google Scholar] [CrossRef] [Green Version]
- Karapetis, C.; Jonker, D.; Daneshmand, M.; Hanson, J.E.; O’Callaghan, C.J.; Marginean, C.; Zalcberg, J.R.; Simes, J.; Moore, M.J.; Tebbutt, N.C.; et al. PIK3CA, BRAF, and PTEN Status and Benefit from Cetuximab in the Treatment of Advanced Colorectal Cancer—Results from NCIC CTG/AGITG CO.17. Clin. Cancer Res. 2013, 20, 744–753. [Google Scholar] [CrossRef] [Green Version]
- Samuels, Y. High Frequency of Mutations of the PIK3CA Gene in Human Cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligresti, G.; Militello, L.; Steelman, L.S.; Cavallaro, A.; Basile, F.; Nicoletti, F.; Stivala, F.; McCubrey, J.A.; Libra, M. PIK3CA mutations in human solid tumors. Cell Cycle 2009, 8, 1352–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartore-Bianchi, A.; Martini, M.; Molinari, F.; Veronese, S.M.; Nichelatti, M.; Artale, S.; Di Nicolantonio, F.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; et al. PIK3CA Mutations in Colorectal Cancer Are Associated with Clinical Resistance to EGFR-Targeted Monoclonal Antibodies. Cancer Res. 2009, 69, 1851–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prenen, H.; De Schutter, J.; Jacobs, B.; De Roock, W.; Biesmans, B.; Claes, B.; Lambrechts, D.; Van Cutsem, E.; Tejpar, S. PIK3CA Mutations Are Not a Major Determinant of Resistance to the Epidermal Growth Factor Receptor Inhibitor Cetuximab in Metastatic Colorectal Cancer. Clin. Cancer Res. 2009, 15, 3184–3188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.Y.; Wu, X.Y.; Huang, Y.F.; Di, M.Y.; Zheng, D.Y.; Chen, J.Z.; Ding, H.; Mao, C.; Tang, J.L. Promising Biomarkers for Predicting the Outcomes of Patients with Kras Wild-Type Metastatic Colorectal Cancer Treated with Anti-Epidermal Growth Factor Receptor Monoclonal Antibodies: A Systematic Review with Meta-Analysis. Int. J. Cancer 2013, 133, 1914–1925. [Google Scholar] [CrossRef]
- Normanno, N.; Rachiglio, A.M.; Lambiase, M.; Martinelli, E.; Fenizia, F.; Esposito, C.; Roma, C.; Troiani, T.; Rizzi, D.; Tatangelo, F.; et al. Heterogeneity of KRAS, NRAS, BRAF and PIK3CA mutations in metastatic colorectal cancer and potential effects on therapy in the CAPRI GOIM trial. Ann. Oncol. 2015, 26, 1710–1714. [Google Scholar] [CrossRef]
- Misale, S.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Siena, S.; Bardelli, A. Resistance to Anti-EGFR Therapy in Colorectal Cancer: From Heterogeneity to Convergent Evolution. Cancer Discov. 2014, 4, 1269–1280. [Google Scholar] [CrossRef] [Green Version]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- De Mattos-Arruda, L.; Weigelt, B.; Cortés, J.; Won, H.H.; Ng, C.K.; Nuciforo, P.; Bidard, F.-C.; Aura, C.; Peg, V.; Piscuoglio, S.; et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: A proof-of-principle. Ann. Oncol. 2014, 25, 1729–1735. [Google Scholar] [CrossRef]
- Siravegna, G.; Lazzari, L.; Crisafulli, G.; Sartore-Bianchi, A.; Mussolin, B.; Cassingena, A.; Martino, C.; Lanman, R.B.; Nagy, R.J.; Fairclough, S.; et al. Radiologic and Genomic Evolution of Individual Metastases during HER2 Blockade in Colorectal Cancer. Cancer Cell 2018, 34, 148–162.e7. [Google Scholar] [CrossRef] [Green Version]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravergna, G.; et al. P2.08 Emergence of Kras Mutations and Acquired Resistance to Anti Egfr Therapy in Colorectal Cancer. Ann. Oncol. 2012, 23, v24. [Google Scholar] [CrossRef]
- Diaz, L.A., Jr.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef] [Green Version]
- Pietrantonio, F.; Vernieri, C.; Siravegna, G.; Mennitto, A.; Berenato, R.; Perrone, F.; Gloghini, A.; Tamborini, E.; Lonardi, S.; Morano, F.; et al. Heterogeneity of acquired resistance to anti-EGFR monoclonal antibodies in patients with metastatic colorectal cancer. Clin. Cancer Res. 2016, 23, 2414–2422. [Google Scholar] [CrossRef] [Green Version]
- Schmiegel, W.H.; Scott, R.J.; Dooley, S.; Lewis, W.; Meldrum, C.J.; Pockney, P.G.; Draganic, B.; Smith, S.; Hewitt, C.; Philimore, H.; et al. Blood-based detection ofRASmutations to guide anti-EGFR therapy in colorectal cancer patients: Concordance of results from circulating tumor DNA and tissue-based Ras testing. Mol. Oncol. 2017, 11, 208–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, J.; Muinelo, L.; Dalmases, A.; Jones, F.; Edelstein, D.; Iglesias, M.; Orrillo, M.; Abalo, A.; Rodríguez, C.; Brozos, E.; et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann. Oncol. 2017, 28, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, J.; Elez, E.; Caratù, G.; Matito, J.; Santos, C.; Macarulla, T.; Vidal, J.; Garcia, M.; Viéitez, J.; Paéz, D.; et al. Concordance of blood- and tumor-based detection of RAS mutations to guide anti-EGFR therapy in metastatic colorectal cancer. Ann. Oncol. 2017, 28, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; Cervantes, A.; Ciardiello, F.; De Luca, A.; Pinto, C. The liquid biopsy in the management of colorectal cancer patients: Current applications and future scenarios. Cancer Treat. Rev. 2018, 70, 1–8. [Google Scholar] [CrossRef]
- Siravegna, G.; Mussolin, B.; Venesio, T.; Marsoni, S.; Seoane, J.; Dive, C.; Papadopoulos, N.; Kopetz, S.; Corcoran, R.; Siu, L.; et al. How liquid biopsies can change clinical practice in oncology. Ann. Oncol. 2019, 30, 1580–1590. [Google Scholar] [CrossRef] [Green Version]
- Hutchinson, L.; Kirk, R. High drug attrition rates—Where are we going wrong? Nat. Rev. Clin. Oncol. 2011, 8, 189–190. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia Enables Predictive Modelling of Anticancer Drug Sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Medico, E.; Russo, M.; Picco, G.; Cancelliere, C.; Valtorta, E.; Corti, G.; Buscarino, M.; Isella, C.; Lamba, S.; Martinoglio, B.; et al. The molecular landscape of colorectal cancer cell lines unveils clinically actionable kinase targets. Nat. Commun. 2015, 6, 7002. [Google Scholar] [CrossRef]
- Marisa, L.; De Reynies, A.; Duval, A.; Selves, J.; Gaub, M.P.; Vescovo, L.; Etienne-Grimaldi, M.-C.; Schiappa, R.; Guenot, D.; Ayadi, M.; et al. Gene Expression Classification of Colon Cancer into Molecular Subtypes: Characterization, Validation, and Prognostic Value. PLoS Med. 2013, 10, e1001453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazzari, L.; Corti, G.; Picco, G.; Isella, C.; Montone, M.; Arcella, P.; Durinikova, E.; Zanella, E.R.; Novara, L.; Barbosa, F.; et al. Patient-Derived Xenografts and Matched Cell Lines Identify Pharmacogenomic Vulnerabilities in Colorectal Cancer. Clin. Cancer Res. 2019, 25, 6243–6259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Riedl, A.; Schlederer, M.; Pudelko, K.; Stadler, M.; Walter, S.; Unterleuthner, D.; Unger, C.; Kramer, N.; Hengstschläger, M.; Kenner, L.; et al. Comparison of cancer cells in 2D vs 3D culture reveals differences in AKT–mTOR–S6K signaling and drug responses. J. Cell Sci. 2016, 130, 203–218. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, D.; Andrieux, G.; Boehnke, K.; Keil, M.; Silvestri, A.; Silvestrov, M.; Keilholz, U.; Haybaeck, J.; Erdmann, G.; Sachse, C.; et al. Heterogeneous pathway activation and drug response modelled in colorectal-tumor-derived 3D cultures. PLoS Genet. 2019, 15, e1008076. [Google Scholar] [CrossRef]
- Schutte, M.; Risch, T.; Abdavi-Azar, N.; Boehnke, K.; Schumacher, D.; Keil, M.; Yildiriman, R.; Jandrasits, C.; Borodina, T.; Amstislavskiy, V.; et al. Molecular Dissection of Colorectal Cancer in Pre-Clinical Models Identifies Biomarkers Predicting Sensitivity to Egfr Inhibitors. Nat. Commun. 2017, 8, 14262. [Google Scholar] [CrossRef]
- Hollandsworth, H.M.; Amirfakhri, S.; Filemoni, F.; Schmitt, V.; Wennemuth, G.; Schmidt, A.; Hoffman, R.M.; Singer, B.B.; Bouvet, M. Anti-carcinoembryonic antigen-related cell adhesion molecule antibody for fluorescence visualization of primary colon cancer and metastases in patient-derived orthotopic xenograft mouse models. Oncotarget 2020, 11, 429–439. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.N.; Park, J.H.; Lwin, T.M.; Miyake, K.; Singh, S.R.; Hoffman, R.M.; Bouvet, M. Tumor-sealing Surgical Orthotopic Implantation of Human Colon Cancer in Nude Mice Induces Clinically-relevant Metastases Without Early Peritoneal Carcinomatosis. Anticancer. Res. 2019, 39, 4065–4071. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Murakami, T.; Suetsugu, A.; Kiyuna, T.; Igarashi, K.; Hiroshima, Y.; Zhao, M.; Zhang, Y.; Bouvet, M.; Clary, B.; et al. High-efficacy targeting of colon-cancer liver metastasis with Salmonella typhimurium A1-R via intra-portal-vein injection in orthotopic nude-mouse models. Oncotarget 2016, 8, 19065–19073. [Google Scholar] [CrossRef]
- Fumagalli, A.; Drost, J.; Suijkerbuijk, S.J.E.; Van Boxtel, R.; De Ligt, J.; Offerhaus, G.J.; Begthel, H.; Beerling, E.; Tan, E.H.; Sansom, O.J.; et al. Genetic dissection of colorectal cancer progression by orthotopic transplantation of engineered cancer organoids. Proc. Natl. Acad. Sci. USA 2017, 114, E2357–E2364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fumagalli, A.; Suijkerbuijk, S.J.E.; Begthel, H.; Beerling, E.; Oost, K.C.; Snippert, H.J.; Van Rheenen, J.; Drost, J. A surgical orthotopic organoid transplantation approach in mice to visualize and study colorectal cancer progression. Nat. Protoc. 2018, 13, 235–247. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, K.P.; Loizou, E.; Livshits, G.; Schatoff, E.M.; Baslan, T.; Manchado, E.; Simon, J.; Romesser, P.B.; Leach, B.; Han, T.; et al. Transplantation of engineered organoids enables rapid generation of metastatic mouse models of colorectal cancer. Nat. Biotechnol. 2017, 35, 577–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Kondo, J.; Inoue, M. Application of Cancer Organoid Model for Drug Screening and Personalized Therapy. Cells 2019, 8, 470. [Google Scholar] [CrossRef] [Green Version]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-quadruplexes in gene promoters: A novel anticancer strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef] [Green Version]
- Neidle, S. Quadruplex Nucleic Acids as Novel Therapeutic Targets. J. Med. Chem. 2016, 59, 5987–6011. [Google Scholar] [CrossRef]
- Lavrado, J.; Brito, H.; Borralho, P.; Ohnmacht, S.A.; Kim, N.-S.; Leitão, C.; Pisco, S.; Gunaratnam, M.; Rodrigues, C.M.P.; Moreira, R.; et al. KRAS oncogene repression in colon cancer cell lines by G-quadruplex binding indolo[3,2-c]quinolines. Sci. Rep. 2015, 5, 9696. [Google Scholar] [CrossRef] [Green Version]
- Biroccio, A.; Porru, M.; Rizzo, A.; Salvati, E.; D’Angelo, C.; Orlandi, A.; Passeri, D.; Franceschin, M.; Stevens, M.F.; Gilson, E.; et al. DNA Damage Persistence as Determinant of Tumor Sensitivity to the Combination of Topo I Inhibitors and Telomere-Targeting Agents. Clin. Cancer Res. 2011, 17, 2227–2236. [Google Scholar] [CrossRef] [Green Version]
- Porru, M.; Zizza, P.; Franceschin, M.; Leonetti, C.; Biroccio, A. EMICORON: A multi-targeting G4 ligand with a promising preclinical profile. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1362–1370. [Google Scholar] [CrossRef]
- Porru, M.; Artuso, S.; Salvati, E.; Bianco, A.; Franceschin, M.; Diodoro, M.G.; Passeri, D.; Orlandi, A.; Savorani, F.; D’Incalci, M.; et al. Targeting G-Quadruplex DNA Structures by EMICORON Has a Strong Antitumor Efficacy against Advanced Models of Human Colon Cancer. Mol. Cancer Ther. 2015, 14, 2541–2551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pompili, L.; Porru, M.; Caruso, C.; Biroccio, A.; Leonetti, C. Patient-derived xenografts: A relevant preclinical model for drug development. J. Exp. Clin. Cancer Res. 2016, 35, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, A.J.; Yu, L.; Bäckesjö, C.-M.; Vargas, L.; Faryal, R.; Aints, A.; Christensson, B.; Berglof, A.; Vihinen, M.; Nore, B.F.; et al. Bruton’s tyrosine kinase (Btk): Function, regulation, and transformation with special emphasis on the PH domain. Immunol. Rev. 2009, 228, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Novero, A.; Ravella, P.M.; Chen, Y.; Dous, G.; Liu, D. Ibrutinib for B cell malignancies. Exp. Hematol. Oncol. 2014, 3, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younes, A.; Sehn, L.H.; Johnson, P.; Zinzani, P.L.; Hong, X.; Zhu, J.; Patti, C.; Belada, D.; Samoilova, O.; Suh, C.; et al. Randomized Phase Iii Trial of Ibrutinib and Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone in Non-Germinal Center B-Cell Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2019, 37, 1285–1295. [Google Scholar] [CrossRef]
- Molina-Cerrillo, J.; Alonso-Gordoa, T.; Gajate, P.; Grande, E. Bruton’s tyrosine kinase (BTK) as a promising target in solid tumors. Cancer Treat. Rev. 2017, 58, 41–50. [Google Scholar] [CrossRef]
- Grassilli, E.; Pisano, F.; Cialdella, A.; Bonomo, S.; Missaglia, C.; Cerrito, M.G.; Masiero, L.; Ianzano, L.; Giordano, F.; Cicirelli, V.; et al. A novel oncogenic BTK isoform is overexpressed in colon cancers and required for RAS-mediated transformation. Oncogene 2016, 35, 4368–4378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavitrano, M.; Ianzano, L.; Bonomo, S.; Cialdella, A.; Cerrito, M.G.; Pisano, F.; Missaglia, C.; Giovannoni, R.; Romano, G.; McLean, C.M.; et al. BTK inhibitors synergise with 5-FU to treat drug-resistant TP53-null colon cancers. J. Pathol. 2019, 250, 134–147. [Google Scholar] [CrossRef]
- Michie, K.; Bermeister, A.; Robertson, N.O.; Goodchild, S.C.; Curmi, P.M.G. Two Sides of the Coin: Ezrin/Radixin/Moesin and Merlin Control Membrane Structure and Contact Inhibition. Int. J. Mol. Sci. 2019, 20, 1996. [Google Scholar] [CrossRef] [Green Version]
- Leiphrakpam, P.D.; Rajput, A.; Mathiesen, M.; Agarwal, E.; Lazenby, A.J.; Are, C.; Brattain, M.G.; Chowdhury, S. Ezrin expression and cell survival regulation in colorectal cancer. Cell. Signal. 2014, 26, 868–879. [Google Scholar] [CrossRef] [Green Version]
- Mori, K.; Toiyama, Y.; Otake, K.; Ide, S.; Imaoka, H.; Okigami, M.; Okugawa, Y.; Fujikawa, H.; Saigusa, S.; Hiro, J.; et al. Successful identification of a predictive biomarker for lymph node metastasis in colorectal cancer using a proteomic approach. Oncotarget 2017, 8, 106935–106947. [Google Scholar] [CrossRef] [Green Version]
- Anastas, J.N.; Moon, R.T. Wnt Signalling Pathways as Therapeutic Targets in Cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.L.; Silver, A. The opposing roles of Wnt-5a in cancer. Br. J. Cancer 2009, 101, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Weeraratna, A.T.; Jiang, Y.; Hostetter, G.; Rosenblatt, K.; Duray, P.; Bittner, M.; Trent, J.M. Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell 2002, 1, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Mikels, A.J.; Nusse, R. Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLoS Biol. 2006, 4, e115. [Google Scholar] [CrossRef] [PubMed]
- Bakker, E.R.; Das, A.M.; Helvensteijn, W.; Franken, P.F.; Swagemakers, S.; Van Der Valk, M.A.; Hagen, T.T.; Kuipers, E.J.; Van Veelen, W.; Smits, R. Wnt5a promotes human colon cancer cell migration and invasion but does not augment intestinal tumorigenesis in Apc 1638N mice. Carcinogenesis 2013, 34, 2629–2638. [Google Scholar] [CrossRef] [Green Version]
- Bergenfelz, C.; Medrek, C.; Ekström, E.; Jirström, K.; Janols, H.; Wullt, M.; Bredberg, A.; Leandersson, K. Wnt5a Induces a Tolerogenic Phenotype of Macrophages in Sepsis and Breast Cancer Patients. J. Immunol. 2012, 188, 5448–5458. [Google Scholar] [CrossRef] [Green Version]
- Mehmeti, M.; Bergenfelz, C.; Kallberg, E.; Millrud, C.R.; Bjork, P.; Ivars, F.; Johansson-Lindbom, B.; Kjellstrom, S.; Andre, I.; Leandersson, K. Wnt5a Is a Tlr2/4-Ligand That Induces Tolerance in Human Myeloid Cells. Commun. Biol. 2019, 2, 176. [Google Scholar] [CrossRef] [Green Version]
- Kelly, M.G.; Alvero, A.; Chen, R.; Silasi, D.-A.; Abrahams, V.M.; Chan, S.; Visintin, I.; Rutherford, T.; Mor, G. TLR-4 Signaling Promotes Tumor Growth and Paclitaxel Chemoresistance in Ovarian Cancer. Cancer Res. 2006, 66, 3859–3868. [Google Scholar] [CrossRef] [Green Version]
- Nicolussi, A.; D’Inzeo, S.; Capalbo, C.; Giannini, G.; Coppa, A. The role of peroxiredoxins in cancer. Mol. Clin. Oncol. 2017, 6, 139–153. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, S.; Pfeuffer, S.; Arnold, P.; Treitz, C.; Aden, K.; Ebsen, H.; Falk-Paulsen, M.; Gisch, N.; Fazio, A.; Kuiper, J.; et al. Prdx4 limits caspase-1 activation and restricts inflammasome-mediated signaling by extracellular vesicles. EMBO J. 2019, 38, e101266. [Google Scholar] [CrossRef] [PubMed]
- Yi, N.; Xiao, M.B.; Ni, W.K.; Jiang, F.; Lu, C.H.; Ni, R.-Z. High expression of peroxiredoxin 4 affects the survival time of colorectal cancer patients, but is not an independent unfavorable prognostic factor. Mol. Clin. Oncol. 2014, 2, 767–772. [Google Scholar] [CrossRef]
- Zhu, D.-J.; Chen, X.-W.; Wang, J.-Z.; Ju, Y.-L.; Yang, M.-Z.O.; Zhang, W.-J. Proteomic analysis identifies proteins associated with curcumin-enhancing efficacy of irinotecan-induced apoptosis of colorectal cancer LOVO cell. Int. J. Clin. Exp. Pathol. 2013, 7, 1–15. [Google Scholar]
- Shi, H.; Hayes, M.; Kirana, C.; Miller, R.J.; Keating, J.P.; Stubbs, R.S. Overexpression of aminoacylase 1 is associated with colorectal cancer progression. Hum. Pathol. 2013, 44, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Li, J.; Xie, H.; Ling, Q.; Wang, J.; Lu, D.; Zhou, L.; Xu, X.; Zheng, S. Proteomics-based identification of the tumor suppressor role of aminoacylase 1 in hepatocellular carcinoma. Cancer Lett. 2014, 351, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Liu, X.; Cao, X.; Zhang, M.; Chang, H. Study of the expression and function of ACY1 in patients with colorectal cancer. Oncol. Lett. 2017, 13, 2459–2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porru, M.; Zizza, P.; Panera, N.; Alisi, A.; Biroccio, A.; Leonetti, C. Harnessing Omics Approaches on Advanced Preclinical Models to Discovery Novel Therapeutic Targets for the Treatment of Metastatic Colorectal Cancer. Cancers 2020, 12, 1830. https://doi.org/10.3390/cancers12071830
Porru M, Zizza P, Panera N, Alisi A, Biroccio A, Leonetti C. Harnessing Omics Approaches on Advanced Preclinical Models to Discovery Novel Therapeutic Targets for the Treatment of Metastatic Colorectal Cancer. Cancers. 2020; 12(7):1830. https://doi.org/10.3390/cancers12071830
Chicago/Turabian StylePorru, Manuela, Pasquale Zizza, Nadia Panera, Anna Alisi, Annamaria Biroccio, and Carlo Leonetti. 2020. "Harnessing Omics Approaches on Advanced Preclinical Models to Discovery Novel Therapeutic Targets for the Treatment of Metastatic Colorectal Cancer" Cancers 12, no. 7: 1830. https://doi.org/10.3390/cancers12071830
APA StylePorru, M., Zizza, P., Panera, N., Alisi, A., Biroccio, A., & Leonetti, C. (2020). Harnessing Omics Approaches on Advanced Preclinical Models to Discovery Novel Therapeutic Targets for the Treatment of Metastatic Colorectal Cancer. Cancers, 12(7), 1830. https://doi.org/10.3390/cancers12071830