Mechanisms of Androgen Receptor Agonist- and Antagonist-Mediated Cellular Senescence in Prostate Cancer



Abstract
:1. Introduction
1.1. Prostate Cancer (PCa)
1.2. The Androgen Receptor (AR) and Adaptive Response of PCa
1.3. PCa Cell Response to Androgen Levels
1.4. Cellular Senescence in PCa
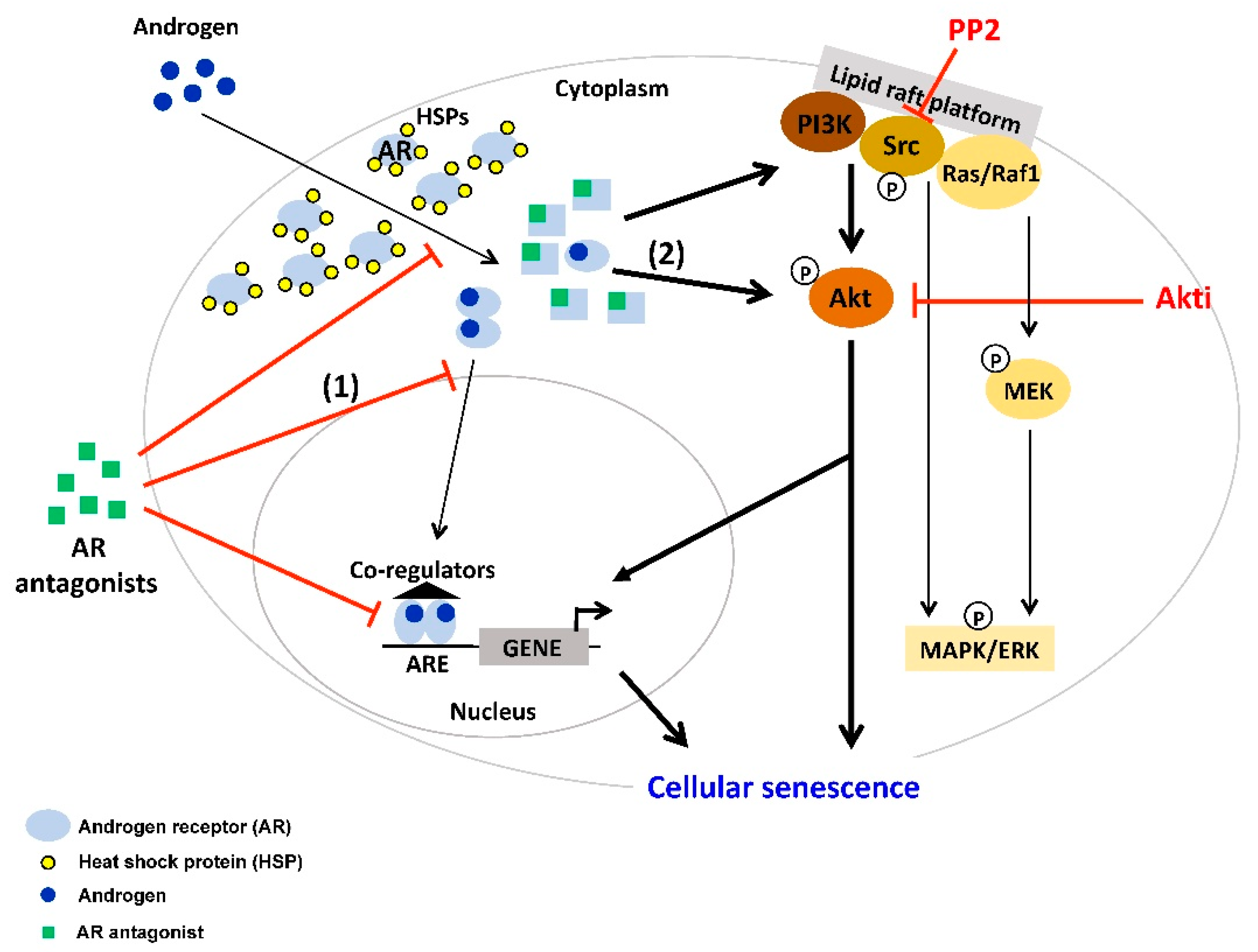
2. AR Antagonist-Induced Cellular Senescence
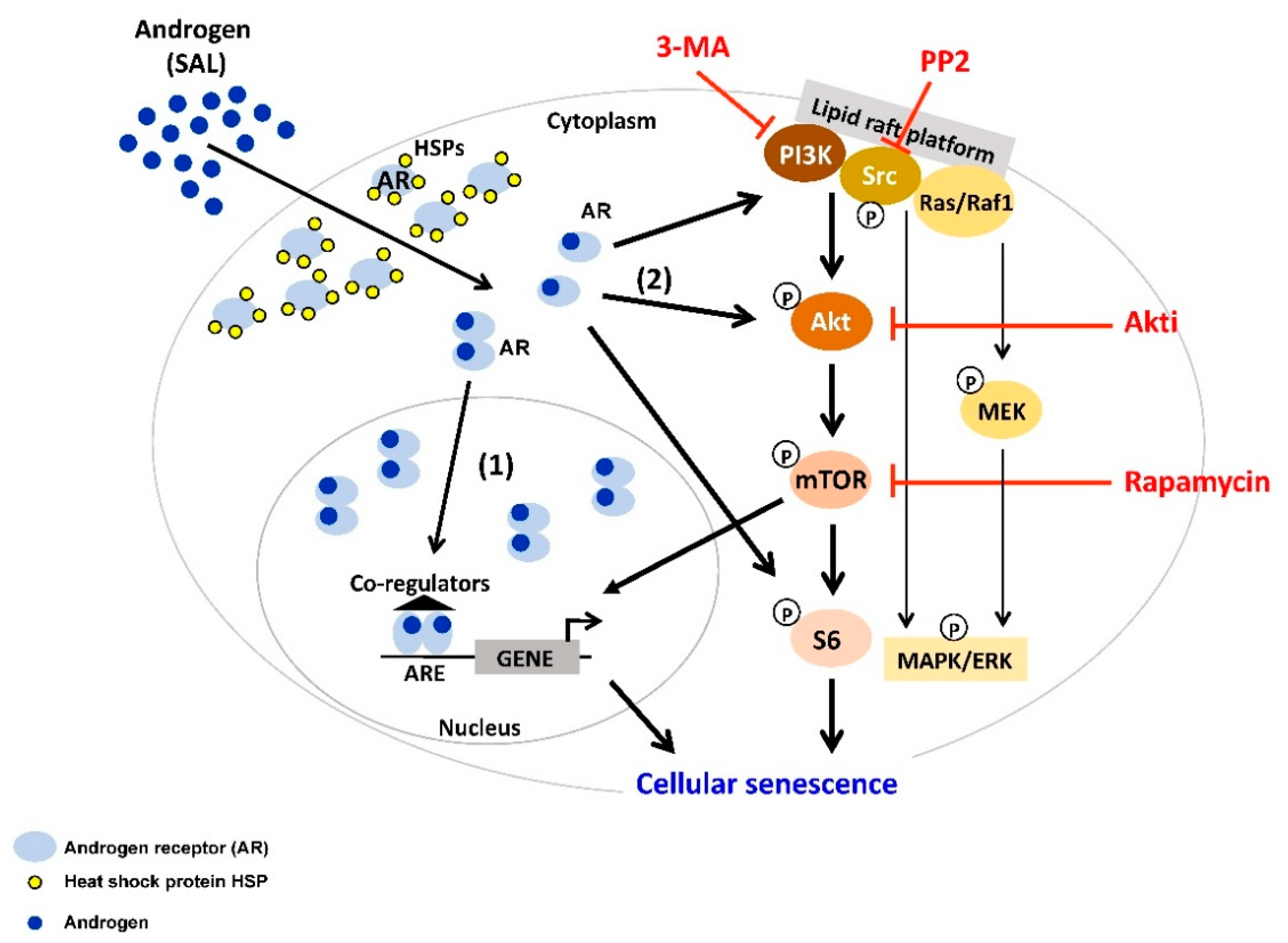
3. Supraphysiological Levels of Androgens Induce Cellular Senescence
4. Interplay between AR-Signaling and other Cellular Signaling Pathways in Senescent PCa
5. Targeting AR Ligand-Induced Cellular Senescent PCa Cells with Senolytic Compounds
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AA | atraric acid |
| ADT | androgen deprivation therapy |
| AR | androgen receptor |
| Apa | Apalutamide |
| BAT | bipolar androgen therapy |
| Bic | bicalutamide |
| CRPC | castration-resistant PCa |
| CSPC | castration-sensitive PCa |
| CDK | cyclin-dependent kinase |
| DHT | dihydrotestosterone |
| Enz | enzalutamide |
| LAL | low androgen level |
| LBD | ligand binding domain |
| nmCRPC | non-metastatic CRPC |
| mCRPC | metastatic CRPC |
| mTORC | mammalian target of rapamycin complex |
| PSA | prostate-specific antigen |
| PCa | prostate cancer |
| ROS | reactive oxygen species |
| SA-β-Gal | senescence-associated β-galactosidase |
| SAHF | senescence-associated heterochromatin foci |
| SAL | supraphysiological androgen level |
| SASP | senescence-associated secretory phenotype |
| TKs | tyrosine kinases |
| VDEC | vas deferens epithelial cells |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Mearini, L.; Zucchi, A.; Nunzi, E.; Villirillo, T.; Bini, V.; Porena, M. Low serum testosterone levels are predictive of prostate cancer. World J. Urol. 2013, 31, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Pastuszak, A.W.; Rodriguez, K.M.; Nguyen, T.M.; Khera, M. Testosterone therapy and prostate cancer. Transl. Androl. Urol. 2016, 5, 909–920. [Google Scholar] [CrossRef] [Green Version]
- Bousset, L.; Rambur, A.; Fouache, A.; Bunay, J.; Morel, L.; Lobaccaro, J.A.; Baron, S.; Trousson, A.; de Joussineau, C. New Insights in Prostate Cancer Development and Tumor Therapy: Modulation of Nuclear Receptors and Specific Role of Liver X Receptors. Int. J. Mol. Sci. 2018, 19, 2545. [Google Scholar] [CrossRef] [Green Version]
- Alpajaro, S.I.R.; Harris, J.A.K.; Evans, C.P. Non-metastatic castration resistant prostate cancer: A review of current and emerging medical therapies. Prostate Cancer Prostatic Dis. 2019, 22, 16–23. [Google Scholar] [CrossRef]
- Lin, T.T.; Chen, Y.H.; Wu, Y.P.; Chen, S.Z.; Li, X.D.; Lin, Y.Z.; Chen, S.H.; Zheng, Q.S.; Wei, Y.; Xu, N.; et al. Risk factors for progression to castration-resistant prostate cancer in metastatic prostate cancer patients. J. Cancer 2019, 10, 5608–5613. [Google Scholar] [CrossRef]
- Helsen, C.; Van den Broeck, T.; Voet, A.; Prekovic, S.; Van Poppel, H.; Joniau, S.; Claessens, F. Androgen receptor antagonists for prostate cancer therapy. Endocr. Relat. Cancer 2014, 21, T105–T118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, C.; Chen, S.; Ng, P.; Bubley, G.J.; Nelson, P.S.; Mostaghel, E.A.; March, B.; Matsumoto, A.M.; Simon, N.I.; Wang, H.; et al. Intratumoral De Novo Steroid Synthesis Activates Androgen Receptor in Castration Resistant Prostate Cancer and is Upregulated by Treatment with CYP17A1 Inhibitors. Cancer Res. 2011, 71, 6503–6513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Fang, H.; Chen, C.; Wu, Y.; Wang, Y.; Ge, H.; Wang, L.; Wan, Y.; He, H. Metastatic castration-resistant prostate cancer: Academic insights and perspectives through bibliometric analyses. Medicine 2020, 99, e19760. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.A.; Malhotra, S.V.; Stoyanova, T. Second-Generation Antiandrogens: From Discovery to Standard of Care in Castration Resistant Prostate Cancer. Front. Oncol. 2019, 9, 801. [Google Scholar] [CrossRef]
- Nguyen, M.M.; Dincer, Z.; Wade, J.R.; Alur, M.; Michalak, M.; Defranco, D.B.; Wang, Z. Cytoplasmic localization of the androgen receptor is independent of calreticulin. Mol. Cell. Endocrinol. 2009, 302, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Lakshmana, G.; Baniahmad, A. Interference with the androgen receptor protein stability in therapy-resistant prostate cancer. Int. J. Cancer 2019, 144, 1775–1779. [Google Scholar] [CrossRef]
- Gil, D.; Zarzycka, M.; Dulinska-Litewka, J.; Ciolczyk-Wierzbicka, D.; Lekka, M.; Laidler, P. Dihydrotestosterone increases the risk of bladder cancer in men. Hum. Cell 2019, 32, 379–389. [Google Scholar] [CrossRef] [Green Version]
- Zboray, L.; Pluciennik, A.; Curtis, D.; Liu, Y.; Berman-Booty, L.D.; Orr, C.; Kesler, C.T.; Berger, T.; Gioeli, D.; Paschal, B.M.; et al. Preventing the Androgen Receptor N/C Interaction Delays Disease Onset in a Mouse Model of SBMA. Cell Rep. 2015, 13, 2312–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hankey, W.; Chen, Z.; Wang, Q. Shaping Chromatin States in Prostate Cancer by Pioneer Transcription Factors. Cancer Res. 2020, 80, 2427–2436. [Google Scholar] [CrossRef] [Green Version]
- De Silva, D.; Zhang, Z.; Liu, Y.; Parker, J.S.; Xu, C.; Cai, L.; Wang, G.G.; Earp, H.S.; Whang, Y.E. Interaction between androgen receptor and coregulator SLIRP is regulated by Ack1 tyrosine kinase and androgen. Sci. Rep. 2019, 9, 18637. [Google Scholar] [CrossRef] [Green Version]
- Regufe da Mota, S.; Bailey, S.; Strivens, R.A.; Hayden, A.L.; Douglas, L.R.; Duriez, P.J.; Borrello, M.T.; Benelkebir, H.; Ganesan, A.; Packham, G.; et al. LSD1 inhibition attenuates androgen receptor V7 splice variant activation in castration resistant prostate cancer models. Cancer Cell Int. 2018, 18, 71. [Google Scholar] [CrossRef]
- Van de Wijngaart, D.J.; Dubbink, H.J.; van Royen, M.E.; Trapman, J.; Jenster, G. Androgen receptor coregulators: Recruitment via the coactivator binding groove. Mol. Cell. Endocrinol. 2012, 352, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Heemers, H.V.; Tindall, D.J. Androgen Receptor (AR) Coregulators: A Diversity of Functions Converging on and Regulating the AR Transcriptional Complex. Endocr. Rev. 2007, 28, 778–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Q.; Zhang, Z.; Wu, Y.; Yu, W.Y.; Zhang, J.; Jiang, Z.M.; Zhang, Y.; Liang, H.; Gui, Y.T. Non-Genomic Action of Androgens is Mediated by Rapid Phosphorylation and Regulation of Androgen Receptor Trafficking. Cell. Physiol. Biochem. 2017, 43, 223–236. [Google Scholar] [CrossRef] [Green Version]
- Castoria, G.; Giovannelli., P.; Di Donato, M.; Ciociola, A.; Hayashi, R.; Bernal, F.; Appella, E.; Auricchio, F.; Migliaccio, A. Role of non-genomic androgen signaling in suppressing proliferation of fibroblasts and fibrosarcoma cells. Cell Death Dis. 2014, 5, e1548. [Google Scholar] [CrossRef] [Green Version]
- Decker, K.F.; Zheng, D.; Yuhong, H.; Bowman, T.; Edwards, J.R.; Jia, L. Persistent androgen receptor-mediated transcription in castration-resistant prostate cancer under androgen-deprived conditions. Nucleic Acids Res. 2012, 40, 10765–10779. [Google Scholar] [CrossRef] [PubMed]
- Mills, I.G. Maintaining and reprogramming genomic androgen receptor activity in prostate cancer. Nat. Rev. Cancer 2014, 14, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Perner, S.; Cronauer, M.V.; Schrader, A.J.; Klocker, H.; Culig, Z.; Baniahmad, A. Adaptive responses of androgen receptor signaling in castration-resistant prostate cancer. Oncotarget 2015, 6, 35542–35555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasekar, T.; Yang., J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar]
- Dutt, S.S.; Gao, A.C. Molecular mechanisms of castration-resistant prostate cancer progression. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.J. Enzalutamide: A Review in Castration-Resistant Prostate Cancer. Drugs 2018, 78, 1913–1924. [Google Scholar] [CrossRef]
- Brave, M.; Weinstock, C.; Brewer, J.R.; Chi, D.C.; Suzman, D.L.; Cheng, J.; Zhang, L.; Sridhara, R.; Ibrahim, A.; Kluetz, P.G.; et al. An FDA Review of Drug Development in Non-Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef] [Green Version]
- Chi, K.N.; Agarwal, N.; Bjartell, A.; Chung, B.H.; Pereira de Santana Gomes, A.J.; Given, R.; Soto, A.J.; Merseburger, A.S.; Özgüroglu, M.; Uemura, H.; et al. Apalutamide for Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2019, 381, 13–24. [Google Scholar] [CrossRef]
- Sonnenschein, C.; Olea, N.; Pasanen, M.E.; Soto, A.M. Negative controls of cell proliferation: Human prostate cancer cells and androgens. Cancer Res. 1989, 49, 3474–3481. [Google Scholar]
- Mirochnik, Y.; Veliceasa, D.; Williams, L.; Maxwell, K.; Yemelyanov, A.; Budunova, I.; Volpert, O.V. Androgen receptor drives cellular senescence. PLoS ONE 2012, 7, e31052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roediger, J.; Hessenkemper, W.; Bartsch, S.; Manvelyan, M.; Huettner, S.S.; Liehr, T.; Esmaeili, M.; Foller, S.; Petersen, I.; Grimm, M.O.; et al. Supraphysiological androgen levels induce cellular senescence in human prostate cancer through the Src-Akt pahtway. Mol. Cancer 2014, 13, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaacs, J.T.; D’Antonio, J.M.; Chen, S.; Antony, L.; Dalrymple, S.P.; Ndikuyeze, G.H.; Luo, J.; Denmeade, S.R. Adaptive auto-regulation of androgen receptor provides a paradigm shifting rationale for bipolar androgen therapy (BAT) for castrate resistant human prostate cancer. Prostate 2012, 72, 1491–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, Y.; Altuwaijri, S.; Wu, C.T.; Ricke, W.A.; Messing, E.M.; Yao, J.; Yeh, S.; Chang, C. Androgen receptor is a tumor suppressor and proliferator in prostate cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 12182–12187. [Google Scholar] [CrossRef] [Green Version]
- Teply, B.A.; Wang, H.; Luber, B.; Sullivan, R.; Rifkind, I.; Bruns, A.; Spitz, A.; DeCarli, M.; Sinibaldi, V.; Pratz, C.F.; et al. Bipolar androgen therapy in men with metastatic castration-resistant prostate cancer after progression on enzalutamide: An open-label, phase 2, multicohort study. Lancet Oncol. 2018, 1, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Denmeade, S.R. Bipolar Androgen Therapy in the Treatment of Prostate Cancer. Clin. Adv. Hematol. Oncol. 2018, 6, 408–411. [Google Scholar]
- Hessenkemper, W.; Roediger, J.; Bartscht, S.; Houtsmuller, A.B.; van Royen, M.E.; Petersen, I.; Grimm, M.O.; Baniahmad, A. A natural androgen receptor antagonist induces cellular senescence in prostate cancer cells. Mol. Endocrinol. 2014, 28, 1831–1840. [Google Scholar] [CrossRef] [Green Version]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguria, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Senescence in premalignant tumours. Nature 2005, 436, 642. [Google Scholar] [CrossRef]
- Alimonti, A.; Nardella, C.; Chen, Z.; Clohessy, J.G.; Carracedo, A.; Trotman, L.C.; Cheng, K.; Varmeh, S.; Kozma, S.C.; Thomas, G.; et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J. Clin. Invest. 2010, 120, 681–693. [Google Scholar] [CrossRef]
- Qin, S.; Schulte, B.A.; Wang, G.W. Role of senescence induction in cancer treatment. World J. Clin. Oncol. 2018, 9, 180–187. [Google Scholar] [CrossRef]
- Fridlyanskaya, I.; Alekseenko, L.; Nikolsky, N. Senescence as a general cellular response to stress: A mini-review. Exp. Gerontol. 2015, 72, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Liu, F.; Yang, P.; Xiong, F.; Yu, Q.; Li, J.; Zhou, Z.; Zhang, S.; Wang, C.Y. Aging and stress induced β cell senescence and its implication in diabetes development. Aging 2019, 11, 9947–9959. [Google Scholar] [CrossRef]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 99, 118. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J.; Robert., L. Cell senescence: Role in aging and age-related diseases. Interdiscip. Top. Gerontol. 2014, 39, 45–61. [Google Scholar] [PubMed] [Green Version]
- Muñoz-Espin, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indovina, P.; Marcelli, E.; Casini, N.; Rizzo, V.; Giordano, A. Emerging roles of RB family: New defense mechanisms against tumor progression. J. Cell. Physiol. 2013, 228, 525–535. [Google Scholar] [CrossRef]
- Kotolloshi, R.; Mirzakhani, K.; Ahlburg, J.; Kraft, F.; Pungsrinont, T.; Baniahmad, A. Thyroid hormone induces cellular senescence in prostate cancer cells through induction of DEC1. J. Steroid Biochem. Mol. Biol. 2020, 201, 105689. [Google Scholar] [CrossRef]
- Esmaeili, M.; Hennej, S.; Ludwig, S.; Klitzsch, A.; Kraft, F.; Melle, C.; Baniahmad, A. The tumor suppressor ING1b is a novel corepressor for the androgen receptor and induces cellular senescence in prostate cancer cells. J. Mol. Cell. Biol. 2016, 8, 207–220. [Google Scholar] [CrossRef]
- Esmaeili, M.; Pungsrinont, T.; Schaefer, A.; Baniahmad, A. A novel crosstalk between the tumor suppressors ING1 and ING2 regulates androgen receptor signaling. J. Mol. Med. 2016, 94, 1167–1179. [Google Scholar] [CrossRef]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavaré, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Future Oncol. 2009, 5, 1403–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, H.Y.; Han, M.W.; Chang, H.W.; Kim, S.Y.; Kim, S.W. Prolonged autophagy by MTOR inhibitor leads radioresistant cancer cells into senescence. Autophagy 2013, 9, 1631–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.B.; Kanno, A.; Ozawa, T.; Tao, H.; Umezawa, Y. Nongenomic Activity of Ligands in the Association of Androgen Receptor with Src. ACS Chem. Bio. 2007, 7, 484–492. [Google Scholar] [CrossRef]
- Yuan, X.; Cai, C.; Chen, S.; Chen, S.; Yu, Z.; Balk, S.P. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene 2014, 33, 2815–2825. [Google Scholar] [CrossRef] [Green Version]
- Sugawara, T.; Baumgart, S.J.; Nevedomskaya., E.; Reichert, K.; Streuber, H.; Lejeune, P.; Mumberg, D.; Haendler, B. Darolutamide is a potent androgen receptor antagonist with strong efficacy in prostate cancer models. Int. J. Cancer 2019, 145, 1382–1394. [Google Scholar] [CrossRef] [PubMed]
- Obst, J.K.; Wang, J.; Jian, K.; Williams, D.E.; Tien, A.H.; Mawji, N.; Tam, T.; Yang, Y.C.; Andersen, R.J.; Chi, K.N.; et al. Revealing Metabolic Liabilities of Ralaniten To Enhance Novel Androgen Receptor Targeted Therapies. ACS Pharm. Transl. Sci. 2019, 2, 453–467. [Google Scholar] [CrossRef]
- Burton, D.G.A.; Giribaldi, M.G.; Munoz, A.; Halvorsen, K.; Patel, A.; Jorda, M.; Perez-Stable, C.; Rai, P. Androgen Deprivation-Induced Senescence Promotes Outgrowth of Androgen-Refractory Prostate Cancer Cells. PLoS ONE 2013, 8, e68003. [Google Scholar] [CrossRef] [PubMed]
- McCormick, J.R.; Blute, M.L.; Yang, B.; Damaschke, N.; Jarrad, D.F. Synthetic lethal metabolic targeting of cellular senescence in prostate cancer with the repurposed drug metformin. J. Urol. 2018, 195, e673–e674. [Google Scholar] [CrossRef]
- Carpenter, V.J.; Saleh, T.; Gewirtz, D.A. The antiandrogen bicalutamide induces senescence in LNCaP cells and quiescence in Myc CaP cells. Cancer Res. 2019, 79. [Google Scholar] [CrossRef]
- Pungsrinont, T.; Sutter, M.F.; Ertingshausen, M.C.C.M.; Lakshmana, G.; Kokal, M.; Khan, A.S.; Baniahmad, A. Senolytic compounds control a distinct fate of androgen receptor agonist- and antagonist-induced cellular senescent LNCaP prostate cancer cells. Cell Biosci. 2020, 10, 59. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Pungsrinont, T.; Ženata, O.; Neubert, L.; Vrzal, R.; Baniahmad, A. Interleukin-23 represses the level of cell senescence induced by the androgen receptor antagonists enzalutamide and darolutamide in castration-resistant prostate cancer cells. Horm. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Fousteris, M.A.; Schubert, U.; Roell, D.; Roediger, J.; Bailis, N.; Nokolaropoulos, S.S.; Baniahmad, A.; Giannis, A. 20-Aminosteroids as a novel class of selective and complete androgen receptor antagonists and inhibitors of prostate cancer cell growth. Bioorg. Med. Chem. 2010, 18, 6960–6969. [Google Scholar] [CrossRef]
- Roell, D.; Rösler, T.W.; Hessenkemper, W.; Kraft, F.; Hauschild, M.; Bartsch, S.; Abraham, T.E.; Houtsmuller, M.; Matusch, R.; van Royen, M.E.; et al. Halogen-substituted anthranilic acid derivatives provide a novel chemical platform for androgen receptor antagonists. J. Steroid Biochem. Mol. Biol. 2019, 188, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, R.; Coss, C.C.; Yepuru, M.; Kearby, J.D.; Miller, D.D.; Dalton, J.T. Steroidal Androgens and Nonsteroidal, Tissue-Selective Androgen Receptor Modulator, S-22, Regulate Androgen Receptor Function through Distinct Genomic and Nongenomic Signaling Pathways. Mol. Endocrinol. 2008, 22, 2448–2465. [Google Scholar] [CrossRef] [Green Version]
- Dotzlaw, H.; Papaioannou, M.; Moehren, U.; Claessens, F.; Baniahmad, A. Agonist-antagonist induced coactivator and corepressor interplay on the human androgen receptor. Mol. Cell. Endocrinol. 2003, 213, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Bitzer, J.; Römer, T.; Lopes da Silva Filho, A. The use of cyproterone acetate/ethinyl estradiol in hyperandrogenic skin symptoms—A review. Eur. J. Contracep. Repr. 2017, 22, 172–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farooqi, A.A.; Sarkar, F.H.; Yuhong, H. Overview on the complexity of androgen receptor-targeted therapy for prostate cancer. Cancer Cell Int. 2015, 15, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klokk, T.I.; Kurys, P.; Elbi, C.; Nagaich, A.K.; Hendarwanto, A.; Slagsvold, T.; Chang, C.Y.; Hager, G.L.; Saatcioglu, F. Ligand-specific dynamics of the androgen receptor at its response element in living cells. Mol. Cell. Biol. 2007, 27, 1823–1843. [Google Scholar] [CrossRef] [Green Version]
- Masiello, D.; Cheng, S.; Bubley, G.J.; Lu, M.L.; Balk, S.P. Bicalutamide functions as an androgen receptor antagonist by assembly of a transcriptionally inactive receptor. J. Biol. Chem. 2002, 277, 26321–26326. [Google Scholar] [CrossRef] [Green Version]
- Farla, P.; Hersmus, R.; Trapman, J.; Houtsmuller, A.B. Antiandrogens prevent stable DNA-binding of the androgen receptor. J. Cell Sci. 2005, 118, 4187–4198. [Google Scholar] [CrossRef] [Green Version]
- McLeod, D.G.; Iversen, P.; See, W.A.; Morris, T.; Armstrong, J.; Wirth, M.P. Bicalutamide 150 mg plus standard care vs standard care alone for early prostate cancer. BJU Int. 2006, 97, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Wirth, M.; Tyrell, C.; Delaere, K.; Sánchez-Chapado, M.; Ramon, J.; Wallace, D.M.; Hetherington, J.; Pina, F.; Heyns, C.F.; Navani, S.; et al. Bicalutamide (Casodex) 150 mg plus standard care in early non-metastatic prostate cancer. Results from Early Prostate Cancer Trial 24 at a median 7 years’ follow-up. Prostate Cancer Prostatic Dis. 2007, 10, 87–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Vida, A.; Galazi, M.; Rudman, S.; Chowdhury, S.; Sternber, C.N. Enzalutamide for the treatment of metastatic castration-resistant prostate cancer. Drug Des. Dev. Ther. 2015, 9, 3325–3339. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.E.; Ouk, S.; Yoo, D.; Sawyers, C.L.; Chen, C.; Tran, C.; Wongvipat, J. Structure-activity relationship for thiohydantoin androgen receptor antagonists for castration-resistant prostate cancer (CRPC). J. Med. Chem. 2010, 53, 2779–2796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merseburger, A.S.; Haas, G.P.; Klot, C.A. An update on enzalutamide in the treatment of prostate cancer. Ther. Adv. Urol. 2015, 7, 9–21. [Google Scholar] [CrossRef]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef] [Green Version]
- Niraula, S.; Chi, K.; Joshua, A.M. Beyond castration-defining future directions in the hormonal treatment of prostate cancer. Horm. Cancer 2012, 3, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann-Censits, J.; Kelly, K.W. Enzalutamide. A novel antiandrogen for patients with castrate-resistant prostate cancer. Clin. Cancer Res. 2013, 19, 1335–1339. [Google Scholar] [CrossRef] [Green Version]
- Ghashghaei, M.; Muanza, T.; Paliouras, M.; Niazi, T.M. Effect of enzalutamide on sensitivity in prostate cancer cells to radiation by inhibition of DNA double strand break repair. J. Clin. Oncol. 2017, 35, 208. [Google Scholar] [CrossRef]
- Al-Salama, Z.T. Apalutamide: A Review in Non-Metastatic Castration-Resistant Prostate Cancer. Drugs 2019, 79, 1591–1598. [Google Scholar] [CrossRef]
- Rathkopf, D.A.; Scher, H.I. Apalutamide for the treatment of prostate cancer. Expert Rev. Anticancer Ther. 2019, 18, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Clegg, N.J.; Wangvipat, J.; Joseph, J.D.; Tran, C.; Ouk, S.; Dilhas, A.; Chen, Y.; Grillot, K.; Bischoff, E.D.; Cai, L.; et al. ARN-509: A Novel Antiandrogen for Prostate Cancer Treatment. Cancer Res. 2012, 72, 1494–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borno, H.T.; Small, E.J. Apalutamide and its use in the treatment of prostate cancer. Future Oncol. 2019, 15, 591–599. [Google Scholar] [CrossRef]
- Fujiza, K.; Nonomura, N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Mens. Health 2019, 37, 288–295. [Google Scholar]
- Bastos, D.A.; Antonarakis, E.S. Darolutamide For Castration-Resistant Prostate Cancer. Onco. Targets Ther. 2019, 12, 8769–8777. [Google Scholar] [CrossRef] [Green Version]
- Fizazi, K.; Massard, C.; Bono, P.; Jones, R.; Kataja, V.; James, N.; Garcia, J.A.; Protheroe, A.; Tammela, T.L.; Elliott, T.; et al. Activity and safety of ODM-201 in patients with progressive metastatic castration-resistant prostate cancer (ARADES). An open-label 1 dose-escalation and randomized phase 2 dose expansion trial. Lancet Oncol. 2014, 15, 975–985. [Google Scholar] [CrossRef]
- Fizazi, K.; Albiges, L.; Loriot, J.; Massard, C. ODM-201: A new-generation androgen receptor inhibitor in castration-resistant prostate cancer. Expert Rev. Anticancer Ther. 2015, 15, 1007–1017. [Google Scholar] [CrossRef]
- Schleich, S.; Papaioannou, M.; Baniahmad, A.; Matusch, R. Extracts from Pygeum africanum and other ethnobotanical species with antiandrogenic activity. Planta Med. 2006, 72, 807–813. [Google Scholar] [CrossRef]
- Papaioannou, M.; Söderholm, A.A.; Hong, W.; Roediger, J.; Roell, D.; Thiele, M.; Nyrönen, T.H.; Baniahmad, A. Computational and functional analysis of the androgen receptor antagonist atraric acid and its derivatives. Anticancer Agents Med. Chem. 2013, 13, 801–810. [Google Scholar] [CrossRef]
- Roell, D.; Baniahmad, A. The natural compounds atraric acid and N-butylbenzene-sulfonamide as antagonists of the human androgen receptor and inhibitors of prostate cancer cell growth. Mol. Cell. Endocrinol. 2011, 322, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaioannou, M.; Schleich, S.; Prade, I.; Degen, S.; Roell, D.; Schubert, U.; Tanner, T.; Claessens, F.; Matusch, R.; Baniahmad, A. The natural compound atraric acid is an antagonist of the human androgen receptor inhibiting cellular invasiveness and prostate cancer cell growth. J. Cell. Mol. Med. 2009, 13, 2210–2223. [Google Scholar] [CrossRef] [PubMed]
- Fenner, A. A new class of AR antagonists? Nat. Rev. Endocrinol. 2019, 15, 128. [Google Scholar] [CrossRef]
- Schweizer, M.T.; Antonarakis, E.S.; Wang, H.; Ajiboye, A.S.; Spitz, A.; Cao, H.; Luo, J.; Haffner, M.C.; Yegnasubramanian, S.; Carducci, M.A.; et al. Effect of bipolar androgen therapy for asymptomatic men with castration-resistant prostate cancer: Results from a pilot clinical study. Sci. Transl. Med. 2015, 7, 269ra2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaacs, J.T.; Brennen, W.N.; Denmeade, S.R. Rationale for bipolar androgen therapy (BAT) for metastatic prostate cancer. Cell Cycle 2017, 16, 1639–1640. [Google Scholar] [CrossRef] [Green Version]
- Lam, H.M.; Corey, E. Supraphysiological Testosterone Therapy as Treatment for Castration-Resistant Prostate Cancer. Front. Oncol. 2018, 8, 167. [Google Scholar] [CrossRef] [Green Version]
- Kokontis, J.M.; Lin, H.P.; Jiang, S.S.; Lin, C.Y.; Fukuchi, J.; Hiipakka, R.A.; Chung, C.J.; Chan, T.M.; Liao, S.; Chang, C.H.; et al. Androgen suppresses the proliferation of androgen receptor-positive castration-resistant prostate cancer cells via inhibition of Cdk2, CyclinA, and Skp2. PLoS ONE 2014, 9, e109170. [Google Scholar] [CrossRef]
- Liao, R.S.; Shihong, M.; Lu, M.; Li, R.; Yin, Y.; Raj, G.V. Androgen receptor-mediated non-genomic regulation of prostate cancer cell proliferation. Transl. Androl. Urol. 2013, 2, 187–196. [Google Scholar]
- Cinar, B.; Mukhopadhyay, N.K.; Meng, G.; Freeman, M.R. Phosphoinositide 3-kinase-independent non-genomic signals transit from the androgen receptor to Akt1 in membrane raft microdomains. J. Biol. Chem. 2007, 282, 29584–29593. [Google Scholar] [CrossRef] [Green Version]
- Peterziel, H.; Mink, S.; Schonert, A.; Becker, M.; Klocker, H.; Cato, A.C. Rapid signaling by androgen receptor in prostate cancer cells. Oncogene 1999, 18, 6322–6329. [Google Scholar] [CrossRef] [Green Version]
- Vlaeminck-Guillem, V.; Gillet, G.; Rimokh, R. SRC: Marker or actor in prostate cancer aggressiveness. Front. Oncol. 2014, 4, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, J.K.; Sadar, M.D. Non-Genomic Actions of the Androgen Receptor in Prostate Cancer. Front. Endocrinol. 2017, 15, 273–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandel, A.; Larsson, P.; Sarwar, M.; Semenas, J.; Syed Khaja, A.S.; Persson, J.L. The interplay between AR, EGF receptor and MMP-9 signaling pathways in invasive prostate cancer. Mol. Med. 2018, 24, 34. [Google Scholar] [CrossRef] [PubMed]
- Saranyutanon, S.; Srivastava, S.K.; Pai, S.; Singh, S.; Singh, A.P. Therapies targeted to androgen receptor signaling axis in prostate cancer: Progress, challenges, and hope. Cancers 2019, 12, 51. [Google Scholar] [CrossRef] [Green Version]
- Tatarov, O.; Mitchell, T.J.; Seywright, M.; Leung, H.Y.; Brunton, V.G.; Edwards, J. SRC family kinase activity is up-regulated in hormone-refractory prostate cancer. Clin. Cancer Res. 2009, 15, 3540–3549. [Google Scholar] [CrossRef] [Green Version]
- Migliaccio, A.; Castoria, G.; Di Domenico, M.; de Falco, A.; Bilancio, A.; Lombardi, M.; Barone, M.V.; Ametrano, D.; Zannini, M.S.; Abbondanza, C.; et al. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J. 2000, 19, 5406–5417. [Google Scholar] [CrossRef] [Green Version]
- Zarif, J.C.; Lamb, L.E.; Schulz, V.V.; Nollet, E.A.; Miranti, C.K. Androgen receptor non-nuclear regulation of prostate cancer cell invasion mediated by Src and matriptase. Oncotarget 2015, 6, 6862–6876. [Google Scholar] [CrossRef] [Green Version]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.; Peter, H.G.; Jie, N.; Hao, J.; Bucci, J.; Cozzi, P.J.; Li, Y. Targeting PI3K/Akt/mTOR signaling pathway in the treatment of prostate cancer radioresistance. Crit. Rev. Oncol. Hematol. 2015, 96, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Lorente, D.; De Bono, J.S. Molecular alterations and emerging targets in castration resistant prostate cancer. Eur. J. Cancer 2014, 50, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edlind, M.P.; Hsieh, A.C. PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance. Asian J. Androl. 2014, 16, 378–386. [Google Scholar] [PubMed]
- Mulholland, D.J.; Tran, L.M.; Li, Y.; Cai, H.; Morim, A.; Wang, S.; Plaisier, S.; Garraway, I.P.; Huang, J.; Graeber, T.G.; et al. Cell Autonomous Role of PTEN in Regulating Castration-Resistant Prostate Cancer Growth. Cancer Cell 2011, 19, 792–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodon, J.; Dienstmann, R.; Serra, V.; Tabernero, J. Development of PI3K inhibitors: Lessons learned from early clinical trials. Nat. Rev. Clin. Oncol. 2013, 10, 143–153. [Google Scholar] [CrossRef]
- Statz, C.M.; Patterson, S.E.; Mockus, S.M. mTOR Inhibitors in Castration-Resistant Prostate Cancer: A Systematic Review. Target. Oncol. 2017, 12, 47–59. [Google Scholar] [CrossRef]
- Majumder, P.K.; Yeh, J.J.; George, D.J.; Febbo, P.G.; Kum, J.; Xue, Q.; Bikoff, R.; Ma, H.; Kantoff, P.W.; Golub, T.R.; et al. Prostate intraepithelial neoplasia induced by prostate restricted Akt activation: The MPAKT model. Proc. Natl. Acad. Sci. USA 2003, 100, 7841–7846. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- McCall, P.; Gemmell, L.K.; Mukherjee, R.; Bartlett, J.M.; Edwards, J. Phosphorylation of the androgen receptor is associated with reduced survival in hormone-refractory prostate cancer patients. Br. J. Cancer 2008, 98, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xie, S.; Jamaluddin, M.S.; Altuwaijri, S.; Ni, J.; Kim, E.; Chen, Y.T.; Hu, Y.C.; Wang, L.; Chuang, K.H.; et al. Induction of androgen receptor expression by phosphatidylinositol 3-kinase/Akt downstream substrate, FOXO3a, and their roles in apoptosis of LNCaP prostate cancer cells. J. Biol. Chem. 2005, 280, 33558–33565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Li, S.; Gan, L.; Kao, T.P.; Huang, H. A Transcription-Independent Function of FOXO1 in Inhibition of Androgen-Independent Activation of the Androgen Receptor in Prostate Cancer Cells. Cancer Res. 2008, 68, 10290–10299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.; Hu, M.C.; Makino, K.; Spohn, B.; Bartholomeusz, G.; Yan, D.H.; Hung, M.C. HER-2/neu promotes androgen-independent survival and growth of prostate cancer cells through the Akt pathway. Cancer Res. 2000, 60, 6841–6845. [Google Scholar]
- Singh, R.; Dhanyamraju, P.K.; Lauth, M. DYRK1B blocks canonical and promotes non-canonical Hedgehog signaling through activation of the mTOR/AKT pathway. Oncotarget 2017, 8, 833–845. [Google Scholar] [CrossRef] [Green Version]
- Malik, S.N.; Brattain, M.; Ghosh, P.M.; Toyer, D.A.; Prihoda, T.; Bedolla, R.; Kreisberg, J.I. Immunohistochemical demonstration of phospho-Akt in high Gleason grade prostate cancer. Clin. Cancer Res. 2002, 8, 1168–1171. [Google Scholar]
- Axanova, L.S.; Chen, Y.Q.; McCoy, T.; Sui, G.; Cramer, S.D. 1,25-dihydroxyvitamin D(3) and PI3K/AKT inhibitors synergistically inhibit growth and induce senescence in prostate cancer cells. Prostate 2010, 70, 1658–1671. [Google Scholar] [CrossRef] [Green Version]
- Nowicki, T.S.; Zhao, H.; Darzynkiewicz, Z.; Moscatello, A.; Shin, E.; Schantz, S.; Tiwari, R.K.; Geliebter, J. Downregulation of uPAR inhibits migration, invasion, proliferation, FAK/PI3K/Akt signaling and induces senescence in papillary thyroid carcinoma cells. Cell Cycle 2011, 10, 100–107. [Google Scholar] [CrossRef] [Green Version]
- Van der Steen, T.; Tindall, D.J.; Huang, H. Posttranslational Modification of the Androgen Receptor in Prostate Cancer. Int. J. Mol. Sci. 2013, 14, 14833–14859. [Google Scholar] [CrossRef] [Green Version]
- Elia, U.; Flescher, E. PI3K/Akt Pathway Activation Attenuates the Cytotoxic Effect of Methyl Jasmonate Toward Sarcoma Cells. Neoplasia 2008, 11, 1303–1313. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Qiao, L.; Wang, S.; Rong, S.B.; Meuillet, E.J.; Berggren, M.; Gallegos, A.; Powis, G.; Kozikowski, A.P. 3-(Hydroxymethyl)-bearing Phosphatidylinositol Ether Lipid Analogues and Carbonate Surrogates Block PI3-K, Akt, and Cancer Cell Growth. J. Med. Chem. 2000, 43, 3045–3051. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Stevens, D.M.; Saitoh, M.; Kinkel, S.; Crossby, K.; Sheen, J.H.; Mullholland, D.J.; Magnuson, M.A.; Wu, H.; Sabatini, D.M. The mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 2009, 15, 148–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, S.M.; Manin, C.; Beaudoin, L.; Leotoing, L.; Communai, Y.; Veyssiere, G.; Morel, L. Androgen receptor mediates non-genomic activation of phosphatidylinositol 3-OH kinase in androgen-sensitive epithelial cells. J. Biol. Chem. 2004, 279, 14579–14586. [Google Scholar] [CrossRef] [Green Version]
- Correia-Melo, C.; Birch, J.; Fielder, E.; Rahmatika, D.; Taylor, J.; Chapman, J.; Lagnado, A.; Carroll, B.M.; Miwa, S.; Richardson, G.; et al. Rapamycin improves healthspan but not inflammaging in nfκb1−/− mice. Aging Cell 2019, 18, e12882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, B.G.; Nelson, Y.; Rabanal-Ruiz, O.; Kucheryavenko, O.; Dunhill-Turner, N.A.; Chesterman, C.C.; Zahari, Q.; Zhang, T.; Conduit, S.E.; Mitchell, C.A.; et al. Persistent mTORC1 signaling in cell senescence results from defects in amino acid and growth factor sensing. J. Cell. Biol. 2017, 216, 1949–1957. [Google Scholar] [CrossRef]
- Carroll, B.V.; Korolchuk, I.; Sarkar, S. Amino acids and autophagy: Cross-talk and co-operation to control cellular homeostasis. Amino Acids 2015, 47, 2065–2088. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Young, A.R.; Arakawa, S.; Samarajiwa, S.A.; Nakashima, T.; Yoshida, S.; Hong, S.; Berry, L.S.; Reichelt, S.; Ferreira, M.; et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 2011, 332, 966–970. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.; Qudrat, A.; Al Mosabbir, A.; Truong, K. Advances in Senotherapies. In Molecular Basis and Emerging Strategies for Anti-Aging Interventions; Rizvi, S.I., Çakatay, U., Eds.; Springer: Singapore, 2018; pp. 67–82. [Google Scholar]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Denmaria, M. Unmasking transcriptional heterogeneity in senescent cells. Curr. Biol. 2017, 27, 2652–2660. [Google Scholar] [CrossRef] [Green Version]
- Jeon, Y.J.; Kim, I.K.; Hong, S.H.; Nan, H.; Kim, H.J.; Lee, H.J.; Masuda, E.S.; Meyuhas, O.; Oh, B.H.; Jung, Y.K. Ribosomal protein S6 is a selective mediator of TRAIL-apoptotic signaling. Oncogene 2008, 27, 4344–4352. [Google Scholar] [CrossRef] [Green Version]
- Meyuhas, O. Ribosomal protein S6 phosphorylation: Four decades of research. Int. Rev. Cell Mol. Biol. 2015, 320, 41–73. [Google Scholar]
- Wittenberg, A.D.; Azar, S.; Klochendler, A.; Stolovich-Rain, M.; Avraham, S.; Birnbaum, L.; Binder Gallimidi, A.; Katz, M.; Dor, Y.; Meyuhas, O. Phosphorylated ribosomal protein S6 is required for Akt-driven hyperplasia and malignant transformation, but not for hypertrophy, aneuploidy and hyperfunction of pancreatic β-cells. PLoS ONE 2016, 11, e0149995. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| AR Ligands | Detection of Cellular Senescence and Molecular Pathways | Cell Lines and PCa Tissues | References |
|---|---|---|---|
| AR antagonists | |||
| Bicalutamide | SA-β-gal, p16INK4A, p27KIP1 | LNCaP, PC3 AR, CWR22PC | [50,58,59,60] |
| Enzalutamide | SA-β-gal, p16INK4A | LNCaP, C4-2 | [61,62] |
| Darolutamide | SA-β-gal, p16INK4A | LNCaP, C4-2 | [62] |
| Atraric acid | SA-β-gal, p16INK4A, pRb, Src, Akt | LNCaP | [37] |
| Novel 20-aminosteroid (Compound 18) | SA-β-gal | LNCaP | [63] |
| Halogen-substituted anthranilic acid esters | SA-β-gal | LNCaP | [64] |
| AR agonists | |||
| Dihydrotestosterone | SA-β-gal, SAHF, p14ARF, p16INK4A, p21CIP1, Cyclin D1, pRb, p63, mTOR, ROS, PML | PC3 AR, LNCaP, C4-2, RWPE AR, PCa tissue ex vivo | [31,32,50] |
| Methyltrienolone | SA-β-gal, SAHF, p14ARF, p16INK4A, p21CIP1, p27KIP1, Cyclin D1, E2F1, pRb, Src, Akt | PC3 AR, LNCaP, C4-2, PCa tissue ex vivo | [32,50] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kokal, M.; Mirzakhani, K.; Pungsrinont, T.; Baniahmad, A. Mechanisms of Androgen Receptor Agonist- and Antagonist-Mediated Cellular Senescence in Prostate Cancer. Cancers 2020, 12, 1833. https://doi.org/10.3390/cancers12071833
Kokal M, Mirzakhani K, Pungsrinont T, Baniahmad A. Mechanisms of Androgen Receptor Agonist- and Antagonist-Mediated Cellular Senescence in Prostate Cancer. Cancers. 2020; 12(7):1833. https://doi.org/10.3390/cancers12071833
Chicago/Turabian StyleKokal, Miriam, Kimia Mirzakhani, Thanakorn Pungsrinont, and Aria Baniahmad. 2020. "Mechanisms of Androgen Receptor Agonist- and Antagonist-Mediated Cellular Senescence in Prostate Cancer" Cancers 12, no. 7: 1833. https://doi.org/10.3390/cancers12071833
APA StyleKokal, M., Mirzakhani, K., Pungsrinont, T., & Baniahmad, A. (2020). Mechanisms of Androgen Receptor Agonist- and Antagonist-Mediated Cellular Senescence in Prostate Cancer. Cancers, 12(7), 1833. https://doi.org/10.3390/cancers12071833