Study of Ras Mutations’ Prognostic Value in Metastatic Colorectal Cancer: STORIA Analysis

 ,
,  , , , ,
, , , ,  , ,
, ,  ,
,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Clinico-Pathological Characteristics of Studied Cohort
2.2. Patients’ Treatments
2.3. Overall Survival According to Specific KRAS Mutations
3. Discussion
4. Materials and Methods
4.1. Patient Management and Follow-Up
4.2. DNA Extraction and Sequencing
4.3. Statistical Analyses, Study Design, and Data Presentation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [Green Version]
- Vatandoust, S.; Price, T.J.; Karapetis, C.S. Colorectal cancer: Metastases to a single organ. World J. Gastroenterol. 2015, 21, 11767–11776. [Google Scholar] [CrossRef] [PubMed]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS proteins and their regulators in human disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Normanno, N.; Tejpar, S.; Morgillo, F.; De Luca, A.; Van Cutsem, E.; Ciardiello, F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat. Rev. Clin. Oncol. 2009, 6, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Jinesh, G.G.; Sambandam, V.; Vijayaraghavan, S.; Balaji, K.; Mukherjee, S. Molecular genetics and cellular events of K-Ras-driven Tumorigenesis. Oncogene 2018, 37, 839–846. [Google Scholar] [CrossRef] [Green Version]
- Waring, P.; Tie, J.; Maru, D.; Karapetis, C.S. RAS Mutations as Predictive Biomarkers in Clinical Management of Metastatic Colorectal Cancer. Clin. Colorectal Cancer 2016, 15, 95–103. [Google Scholar] [CrossRef]
- Petrelli, F.; Coinu, A.; Cabiddu, M.; Ghilardi, M.; Barni, S. KRAS as Prognostic Biomarker in Metastatic Colorectal Cancer Patients Treated with Bevacizumab: A Pooled Analysis of 12 Published Trials. Med. Oncol. 2013, 30, 650. [Google Scholar] [CrossRef]
- Smith, J.C.; Brooks, L.; Hoff, P.M.; McWalter, G.; Dearden, S.; Morgan, S.R.; Wilson, D.; Robertson, J.D.; Jürgensmeier, J.M. KRAS Mutations Are Associated with Inferior Clinical Outcome in Patients with Metastatic Colorectal Cancer, but Are Not Predictive for Benefit with Cediranib. Eur. J. Cancer 2013, 49, 2424–2432. [Google Scholar] [CrossRef]
- Koike, J.; Ushigome, M.; Funahashi, K.; Shiokawa, H.; Kaneko, T.; Arai, K.; Matsuda, S.; Kagami, S.; Suzuki, T.; Kurihara, A.; et al. Significance of KRAS Mutation in Patients Receiving mFOLFOX6 with or without Bevacizumab for Metastatic Colorectal Cancer. Hepatogastroenterology 2014, 61, 2222–2226. [Google Scholar]
- Petrelli, F.; Coinu, A.; Cabiddu, M.; Borgonovo, K.; Lonati, V.; Ghilardi, M.; Barni, S. Prognostic factors for survival with bevacizumab-based therapy in colorectal cancer patients: A systematic review and pooled analysis of 11,585 patients. Med. Oncol. 2015, 32, 456. [Google Scholar] [CrossRef]
- Modest, D.P.; Ricard, I.; Heinemann, V.; Hegewisch-Becker, S.; Schmiegel, W.; Porschen, R.; Stintzing, S.; Graeven, U.; Arnold, D.; von Weikersthal, L.F. Outcome according to KRAS-, NRAS- and BRAF-mutation as well as KRAS mutation variants: Pooled analysis of five randomized trials in metastatic colorectal cancer by the AIO colorectal cancer study group. Ann Oncol. 2016, 27, 1746–1753. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Balmain, A.; Counter, C.M. A model for RAS mutation patterns in cancers: Finding the sweet spot. Nat. Rev. Cancer 2018, 18, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining Glutaminolysis Bolsters Chemotherapy while NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res. 2020, 80, 1630–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef] [PubMed]
- Ottaiano, A.; Circelli, L.; Lombardi, A.; Scala, S.; Martucci, N.; Galon, J.; Buonanno, M.; Scognamiglio, G.; Botti, G.; Hermitte, F.; et al. Genetic trajectory and immune microenvironment of lung-specific oligometastatic colorectal cancer. Cell Death Dis. 2020, 11, 275. [Google Scholar] [CrossRef]
- Visscher, M.; Arkin, M.R.; Dansen, T.B. Covalent targeting of acquired cysteines in cancer. Curr. Opin. Chem. Biol. 2016, 30, 61–67. [Google Scholar] [CrossRef]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 2018, 172, 578–589. [Google Scholar] [CrossRef] [Green Version]
- Patricelli, M.P.; Janes, M.R.; Li, L.S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef] [Green Version]
- Govindan, R.; Fakih, M.G.; Price, T.J.; Falchook, G.S.; Desai, J.; Kuo, J.C.; Strickler, J.H.; Krauss, J.C.; Li, B.T.; Denlinger, C.S.; et al. Phase 1 study of AMG 510, a novel molecule targeting KRAS G12C mutant solid tumors. Ann. Oncol. 2019, 30, v163–v164. [Google Scholar] [CrossRef]
- Pantsar, T. The current understanding of KRAS protein structure and dynamics. Comput. Struct. Biotechnol. J. 2019, 18, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Jang, H.; Nussinov, R. The structural basis for Ras activation of PI3Kα lipid kinase. Phys. Chem. Chem. Phys. 2019, 21, 12021–12028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Jang, H.; Nussinov, R. The mechanism of PI3Kα activation at the atomic level. Chem. Sci. 2019, 10, 3671–3680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Jang, H.; Muratcioglu, S.; Gursoy, A.; Keskin, O.; Nussinov, R.; Zhang, J. Ras conformational ensembles, allostery, and signalling. Chem. Rev. 2016, 116, 6607–6665. [Google Scholar] [CrossRef]
- Lu, S.; Jang, H.; Gu, S.; Zhang, J.; Nussinov, R. Nussinov Drugging Ras GTPase: A comprehensive mechanistic and signaling structural view. Chem. Soc. Rev. 2016, 45, 4929–4952. [Google Scholar] [CrossRef] [Green Version]
- Mehaffey, M.R.; Schardon, C.L.; Novelli, E.T.; Cammarata, M.B.; Webb, L.J.; Fast, W.; Brodbelt, J.S. Investigation of GTP-dependent dimerization of G12X K-Ras variants using ultraviolet photodissociation mass spectrometry. Chem. Sci. 2019, 10, 8025–8034. [Google Scholar] [CrossRef]
- Misale, S.; Fatherree, J.P.; Cortez, E.; Li, C.; Bilton, S.; Timonina, D.; Myers, D.T.; Lee, D.; Gomez-Caraballo, M.; Greenberg, M.; et al. KRAS G12C NSCLC Models Are Sensitive to Direct Targeting of KRAS in Combination with PI3K Inhibition. Clin. Cancer Res. 2019, 25, 796–807. [Google Scholar] [CrossRef] [Green Version]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.C.; Er, T.K.; Liu, Y.Y.; Hwang, J.K.; Barrio, M.J.; Rodrigo, M.; Garcia-Toro, E.; Herreros-Villanueva, M. Computational analysis of KRAS mutations: Implications for different effects on the KRAS p.G12D and p.G13D mutations. PLoS ONE 2013, 8, e55793. [Google Scholar] [CrossRef]
- Tejpar, S.; Celik, I.; Schlichting, M.; Sartorius, U.; Bokemeyer, C.; Van Cutsem, E. Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J. Clin. Oncol. 2012, 30, 3570–3577. [Google Scholar] [CrossRef]
- Messner, I.; Cadeddu, G.; Huckenbeck, W.; Knowles, H.J.; Gabbert, H.E.; Baldus, S.E.; Schaefer, K.L. KRAS p.G13D mutations are associated with sensitivity to anti-EGFR antibody treatment in colorectal cancer cell lines. J. Cancer Res. Clin. Oncol. 2013, 139, 201–209. [Google Scholar] [CrossRef] [PubMed]
- De Roock, W.; Jonker, D.J.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Tu, D.; Siena, S.; Lamba, S.; Arena, S.; Frattini, M.; Piessevaux, H.; et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 2010, 304, 1812–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pupo, E.; Avanzato, D.; Middonti, E.; Bussolino, F.; Lanzetti, L. KRAS-Driven Metabolic Rewiring Reveals Novel Actionable Targets in Cancer. Front. Oncol. 2019, 9, 848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Normanno, N.; Rachiglio, A.M.; Lambiase, M.; Martinelli, E.; Fenizia, F.; Esposito, C.; Roma, C.; Troiani, T.; Rizzi, D.; Tatangelo, F.; et al. Heterogeneity of KRAS, NRAS, BRAF and PIK3CA mutations in metastatic colorectal cancer and potential effects on therapy in the CAPRI GOIM trial. Ann. Oncol. 2015, 26, 1710–1714. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene | Mutation (Proteic Change) | Age | Gender | Grading | Side of Primary Tumor | pT * | Lymph Node Involvement (pN) * | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <65 | ≥65 | p | M | F | p | G1/G2 | G3 | p | Left | Right | p | T1/T2 | T3 | T4 | p | 0 | 1–3 | >3 | p | ||
| KRAS | |||||||||||||||||||||
| p.G12D | 22 | 35 | 27 | 30 | 10 | 47 | 26 | 31 | 12 | 30 | 10 | 1 | 6 | 45 | |||||||
| p.G12V | 17 | 21 | 20 | 18 | 6 | 32 | 14 | 24 | 11 | 15 | 9 | 1 | 9 | 25 | |||||||
| p.G13D | 15 | 20 | 18 | 17 | 4 | 31 | 12 | 23 | 7 | 16 | 6 | 0 | 5 | 24 | |||||||
| p.G12A | 6 | 8 | 11 | 3 | 4 | 10 | 4 | 10 | 2 | 5 | 4 | 1 | 3 | 7 | |||||||
| p.G12C | 5 | 8 | 5 | 8 | 3 | 10 | 2 | 11 | 4 | 5 | 3 | 0 | 2 | 10 | |||||||
| p.G12S | 5 | 7 | 0.99 | 4 | 8 | 0.22 | 2 | 10 | 0.78 | 3 | 9 | 0.33 | 2 | 5 | 3 | 0.94 | 0 | 1 | 9 | 0.59 | |
| p.A146T | 2 | 6 | 5 | 3 | 4 | 4 | 4 | 4 | 1 | 2 | 3 | 2 | 1 | 3 | |||||||
| p.A146V | 1 | 2 | 2 | 1 | 0 | 3 | 1 | 2 | 1 | 1 | 0 | 1 | 0 | 1 | |||||||
| p.K117N | 2 | 1 | 0 | 3 | 0 | 3 | 0 | 3 | 0 | 1 | 2 | 0 | 1 | 2 | |||||||
| p.G13C | 0 | 3 | 3 | 0 | 1 | 2 | 1 | 2 | 0 | 1 | 1 | 0 | 0 | 2 | |||||||
| p.G13R | 1 | 1 | 0 | 2 | 1 | 1 | 1 | 1 | 2 | 0 | 0 | 1 | 0 | 1 | |||||||
| p.G12_G13insG | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | |||||||
| p.G12F | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | |||||||
| p.G13D; pG12D | 1 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | |||||||
| p.A59E | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | |||||||
| NRAS | |||||||||||||||||||||
| p.G12C | 2 | 2 | 3 | 1 | 1 | 3 | 1 | 3 | 0 | 3 | 0 | 0 | 1 | 2 | |||||||
| p.Q61H | 1 | 3 | 2 | 2 | 1 | 3 | 0 | 4 | 0 | 3 | 0 | 0 | 0 | 3 | |||||||
| p.G12R | 1 | 1 | 2 | 0 | 0 | 2 | 0 | 2 | 1 | 0 | 1 | 0 | 0 | 2 | |||||||
| p.Q61E | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | |||||||
| p.Q61L | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | |||||||
| p.Q61R | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | |||||||
| p.V14I | 1 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | |||||||
| Gene | Mutation (Proteic Change) | Metastatic Involvement | Type of First-Line CT | Best Response to First-Line CT | No. of Chemotherapy Lines | Time on Therapy (Months) * | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| One Site | Two or More Sites | Multiple Sites Including Peritoneum | p | CT | CT/AA | p | CR, PR or SD | PD | NA | p | 1 | 2 | >2 | p | Median (Range) | ||
| KRAS | |||||||||||||||||
| p.G12D | 12 | 33 | 12 | 9 | 48 | 27 | 22 | 8 | 57 | 31 | 20 | 19.2 (11.7–23.7) | |||||
| p.G12V | 11 | 18 | 9 | 5 | 33 | 19 | 12 | 7 | 38 | 18 | 13 | 18.8 (8.5–23.2) | |||||
| p.G13D | 7 | 22 | 6 | 6 | 29 | 19 | 11 | 5 | 35 | 20 | 13 | 19.1 (8.8–25.0) | |||||
| p.G12A | 2 | 8 | 4 | 3 | 11 | 7 | 7 | 0 | 14 | 9 | 7 | 18.2 (11.1–23.6) | |||||
| p.G12C | 5 | 5 | 3 | 2 | 11 | 4 | 9 | 0 | 13 | 6 | 2 | 10.7 (4.3–23.1) | |||||
| p.G12S | 2 | 7 | 3 | 0.87 | 4 | 8 | 0.71 | 2 | 8 | 2 | 0.14 | 12 | 4 | 0 | 0.76 | 5.9 (3.9–13.1) | |
| p.A146T | 3 | 2 | 3 | 2 | 6 | 5 | 3 | 0 | 8 | 6 | 6 | 16.9 (13.3–21.8) | |||||
| p.A146V | 2 | 1 | 0 | 2 | 1 | 2 | 0 | 1 | 3 | 3 | 2 | 16.3, 18.6, 18.9 | |||||
| p.K117N | 0 | 1 | 2 | 0 | 3 | 1 | 2 | 0 | 3 | 2 | 2 | 17.4, 19.0, 19.2 | |||||
| p.G13C | 1 | 1 | 1 | 1 | 2 | 2 | 1 | 0 | 3 | 3 | 2 | 17.2, 21.6, 22.4 | |||||
| p.G13R | 2 | 0 | 0 | 0 | 2 | 2 | 0 | 0 | 2 | 2 | 0 | 10.4, 19.5 | |||||
| p.G12_G13insG | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 1 | 12.2 | |||||
| p.G12F | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 1 | 20.6 | |||||
| p.G13D; pG12D | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 22.7 | |||||
| p.A59E | 0 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 25.2 | |||||
| NRAS | |||||||||||||||||
| p.G12C | 0 | 4 | 0 | 1 | 3 | 3 | 1 | 0 | 4 | 2 | 0 | 15.5 (6.6–18.0) | |||||
| p.Q61H | 0 | 4 | 0 | 0 | 4 | 1 | 1 | 2 | 4 | 3 | 1 | 11.6 (8.4–19.3) | |||||
| p.G12R | 1 | 0 | 1 | 1 | 1 | 2 | 0 | 0 | 2 | 0 | 0 | 15.3, 19.2 | |||||
| p.Q61E | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 1 | 11.4 | |||||
| p.Q61L | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | 0 | 23.2 | |||||
| p.Q61R | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 0 | 21.1 | |||||
| p.V14I | 0 | 0 | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 18.7 | |||||
| Gene | Proteic Change | No. of Events/Patients | Mutation Incidence (%) | ClinVar ID | Median Survival (Months) | 95% CI | p at Log Rank Test |
|---|---|---|---|---|---|---|---|
| KRAS | |||||||
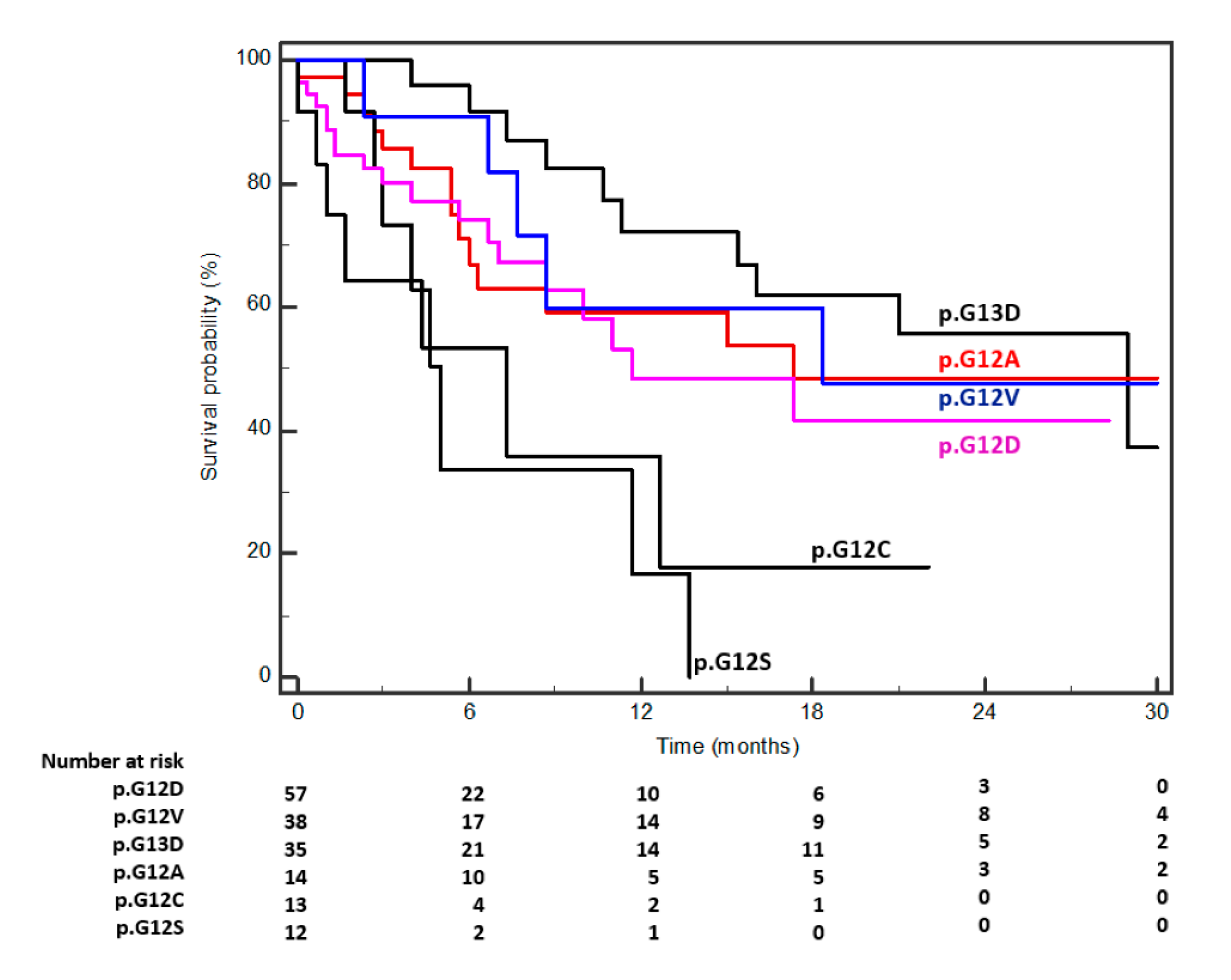
| p.G12D | 19/57 | 27.7 | 27261 | 11.6 | 8.6–17.3 | ||
| p.G12V | 15/38 | 18.4 | 27622 | 17.3 | 6.0–31.6 | ||
| p.G13D | 10/35 | 17.0 | 27619 | 27.0 | 15.3–29.0 | ||
| p.G12A | 5/14 | 6.8 | 54289 | 16.3 | 7.6–18.3 | ||
| p.G12C | 7/13 | 6.3 | 27617 | 7.3 | 1.6–12.6 | ||
| p.G12S | 8/12 | 5.8 | 12584 | 5.0 | 3.0–11.6 | p = 0.0006 | |
| p.A146T | 6/8 | 3.9 | 194404 | 19.3 | 6.4–21.4 | ||
| p.A146V | 2/3 | 1.4 | 362841 | 2.0 | 0.6–2.0 | ||
| p.K117N | 2/3 | 1.4 | 362843 | 4.6 | 2.3–6.1 | ||
| p.G13C | 1/3 | 1.4 | 54290 | 29.8 | 5.4–36.3 | ||
| p.G13R | 1/2 | 0.9 | 27632 | 0.6 and 6.3 | NA | ||
| p.G12_G13insG | 0/1 | 0.5 | NR | 19.6 | NA | ||
| p.G12F | 1/1 | 0.5 | NR | 1.4 | NA | ||
| p.G13D; pG12D | 0/1 | 0.5 | NR | 19.6 | NA | ||
| p.A59E | 0/1 | 0.5 | NR | 10.0 | NA | ||
| NRAS | |||||||
| p.G12C | 3/4 | 1.9 | 48938 | 4.8 | 2.9–7.9 | ||
| p.Q61H | 2/4 | 1.9 | 359197 | 9.0 | 8.6–9.9 | ||
| p.G12R | 1/2 | 0.9 | 48939 | 1.6 and 4.6 | NA | ||
| p.Q61E | 1/1 | 0.5 | 362754 | 6.2 | NA | ||
| p.Q61L | 0/1 | 0.5 | 362753 | 20.3 | NA | ||
| p.Q61R | 1/1 | 0.5 | 28939 | 5.5 | NA | ||
| p.V14I | 1/1 | 0.5 | 27628 | 9.6 | NA |
| Co-Variate | Dichotomization | Median Survivals | No. of Events/Patients | p at Univariate | HR | 95% CI | p at Multivariate |
|---|---|---|---|---|---|---|---|
| Age | <65 year vs. ≥65 year | 17.3 vs. 12.6 | 40/82 vs. 46/124 | 0.4088 | 0.82 | 0.52–1.29 | 0.548 |
| Gender | M vs. F | 13.6 vs. 15.0 | 35/107 vs. 51/99 | 0.8902 | 0.96 | 0.62–1.50 | 0.872 |
| Side | L vs. R | 15.3 vs. 13.6 | 32/73 vs. 54/133 | 0.6084 | 0.88 | 0.56–1.39 | 0.309 |
| Metastatic involvement | 1 site vs. >1 | NR vs. 9.6 | 12/49 vs. 74/157 | <0.0001 | 0.36 | 0.22–0.58 | 0.001 |
| Response to first-line CT | DC vs. not DC | 29.0 vs. 9.0 | 37/98 vs. 30/80 | 0.0032 | 0.46 | 0.28–0.77 | 0.073 |
| KRAS mutations * | p.G12D vs. other mut | 11.6 vs. 15.3 | 19/57 vs. 45/112 | 0.6510 | 0.87 | 0.50–1.53 | 0.669 |
| p.G12V vs. other mut | 17.3 vs. 13.6 | 15/38 vs. 49/131 | 0.5684 | 0.84 | 0.48–1.48 | 0.580 | |
| p.G13D vs. other mut | 27.0 vs. 11.6 | 10/35 vs. 54/134 | 0.0384 | 0.55 | 0.31–0.96 | 0.165 | |
| p.G12A vs. other mut | 16.3 vs. 15.0 | 5/14 vs. 59/155 | 0.4104 | 0.71 | 0.32–1.58 | 0.634 | |
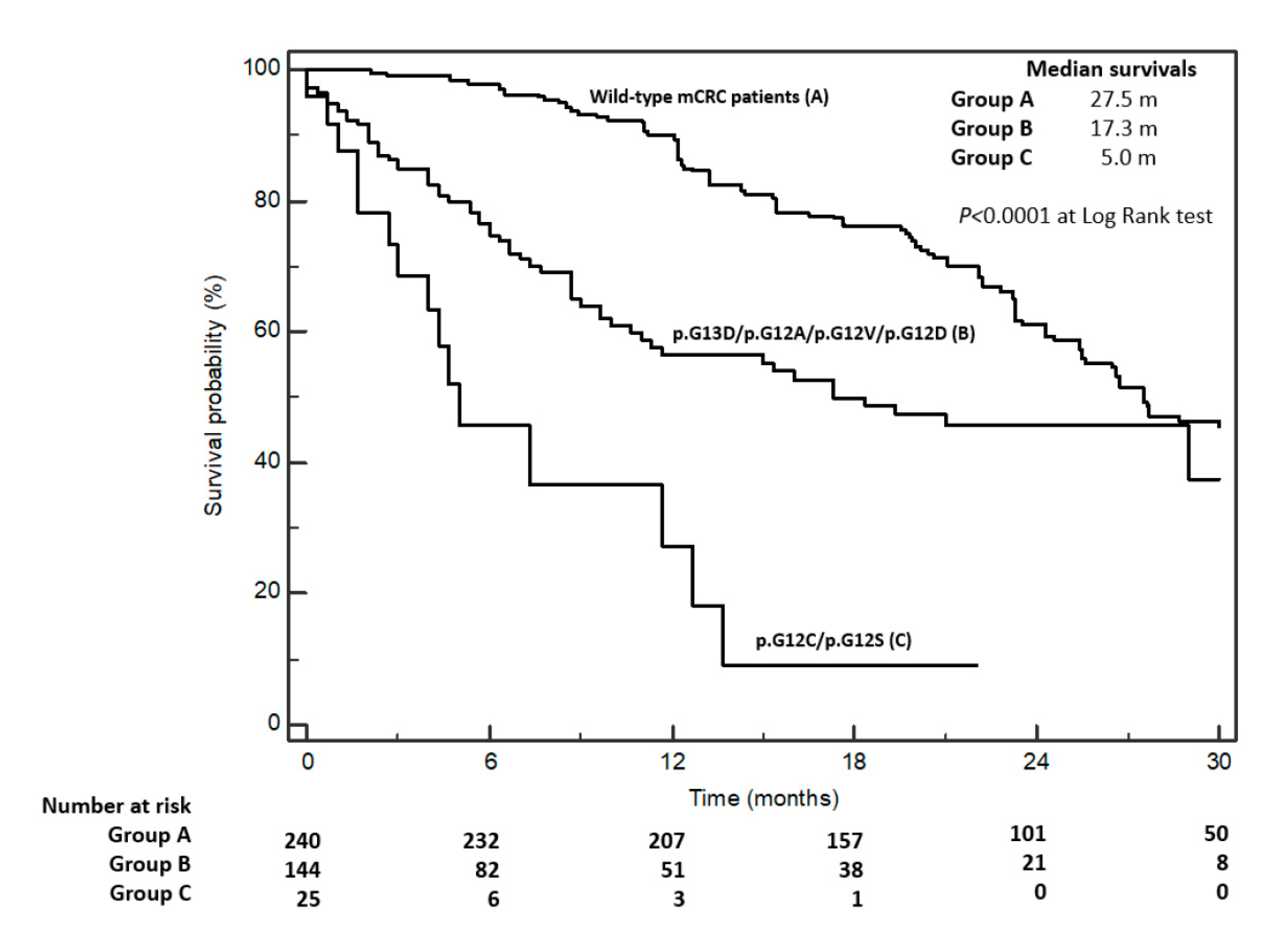
| p.G12C/p.G12S vs. other mut | 5.0 vs. 18.3 | 15/25 vs. 49/144 | 0.0002 | 4.99 | 2.15–11.5 | 0.002 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ottaiano, A.; Normanno, N.; Facchini, S.; Cassata, A.; Nappi, A.; Romano, C.; Silvestro, L.; De Stefano, A.; Rachiglio, A.M.; Roma, C.; et al. Study of Ras Mutations’ Prognostic Value in Metastatic Colorectal Cancer: STORIA Analysis. Cancers 2020, 12, 1919. https://doi.org/10.3390/cancers12071919
Ottaiano A, Normanno N, Facchini S, Cassata A, Nappi A, Romano C, Silvestro L, De Stefano A, Rachiglio AM, Roma C, et al. Study of Ras Mutations’ Prognostic Value in Metastatic Colorectal Cancer: STORIA Analysis. Cancers. 2020; 12(7):1919. https://doi.org/10.3390/cancers12071919
Chicago/Turabian StyleOttaiano, Alessandro, Nicola Normanno, Sergio Facchini, Antonino Cassata, Anna Nappi, Carmela Romano, Lucrezia Silvestro, Alfonso De Stefano, Anna Maria Rachiglio, Cristin Roma, and et al. 2020. "Study of Ras Mutations’ Prognostic Value in Metastatic Colorectal Cancer: STORIA Analysis" Cancers 12, no. 7: 1919. https://doi.org/10.3390/cancers12071919
APA StyleOttaiano, A., Normanno, N., Facchini, S., Cassata, A., Nappi, A., Romano, C., Silvestro, L., De Stefano, A., Rachiglio, A. M., Roma, C., Maiello, M. R., Scala, S., Delrio, P., Tatangelo, F., Di Mauro, A., Botti, G., Avallone, A., & Nasti, G. (2020). Study of Ras Mutations’ Prognostic Value in Metastatic Colorectal Cancer: STORIA Analysis. Cancers, 12(7), 1919. https://doi.org/10.3390/cancers12071919