Merkel Cell Polyomavirus T Antigens Induce Merkel Cell-Like Differentiation in GLI1-Expressing Epithelial Cells

 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Results
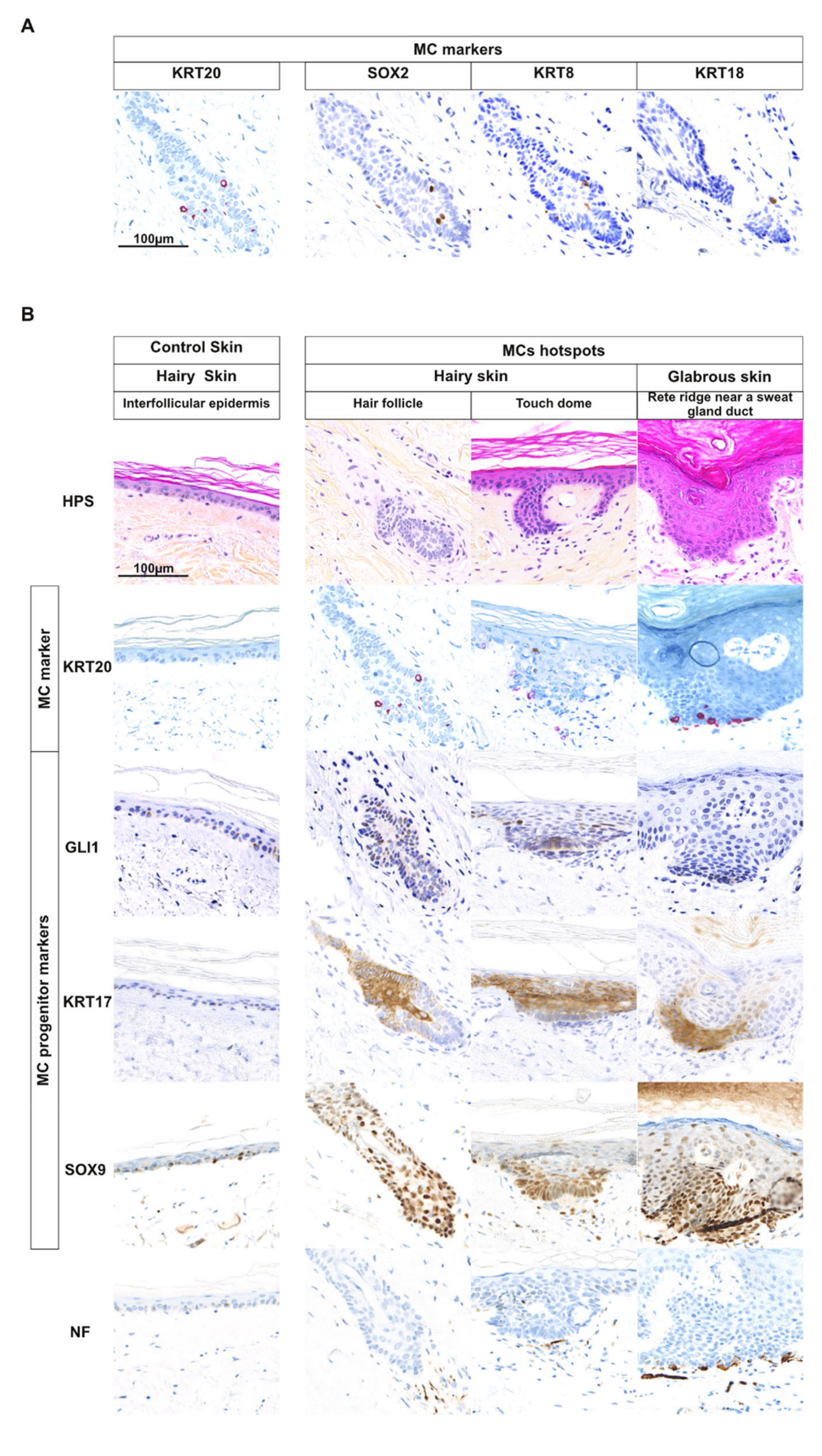
2.1. MCs Are Often Located in Appendage Structures in Human Skin
2.2. Cells with an MC Progenitor Phenotype Characterized by GLI1 Expression are Found in Close Proximity of MCs in Human Hairy Skin
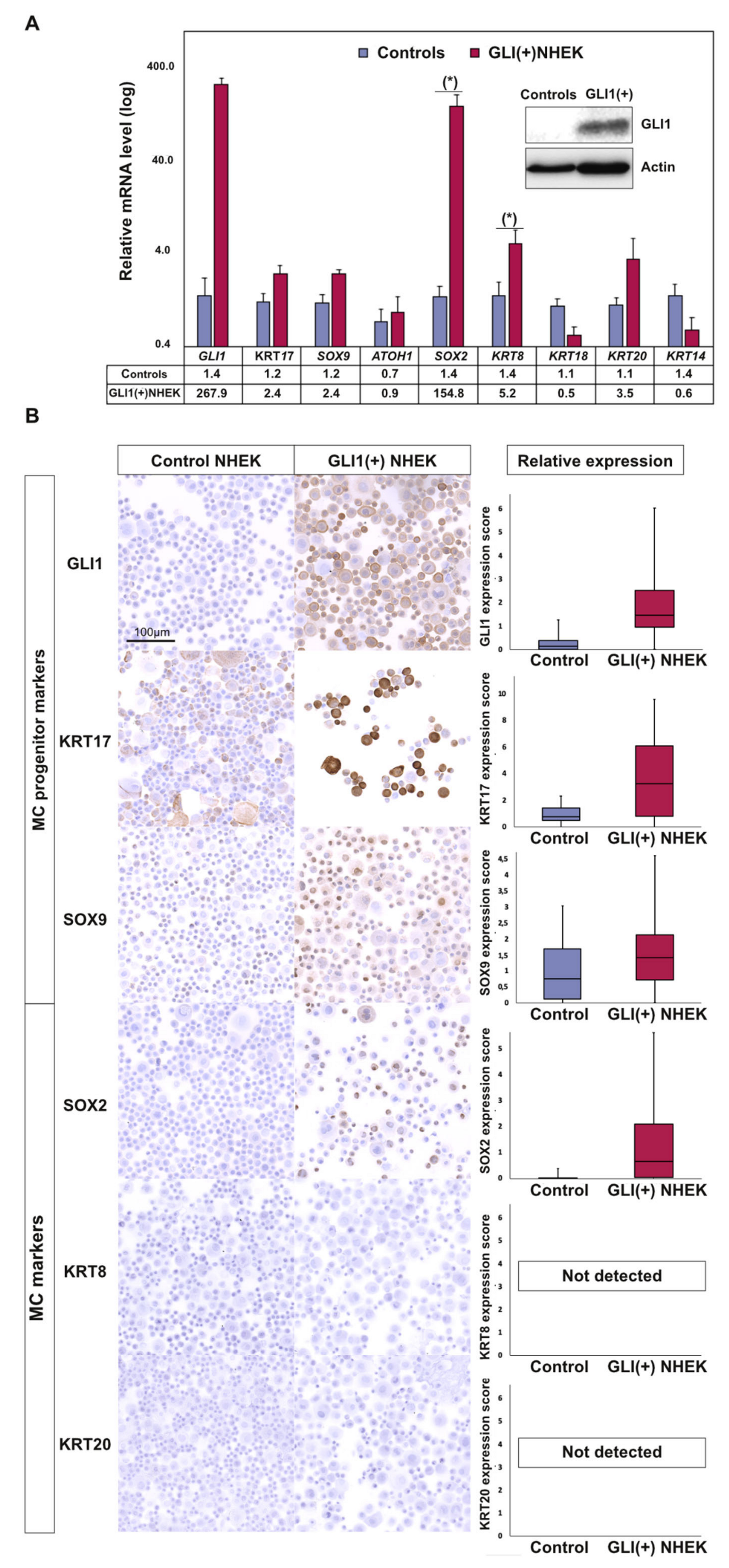
2.3. GLI1 Expression in Keratinocytes Induces MC Lineage Markers
2.4. MC-Progenitor and MC Markers Are Expressed in Trichoblastoma and Merkel Cell Carcinoma
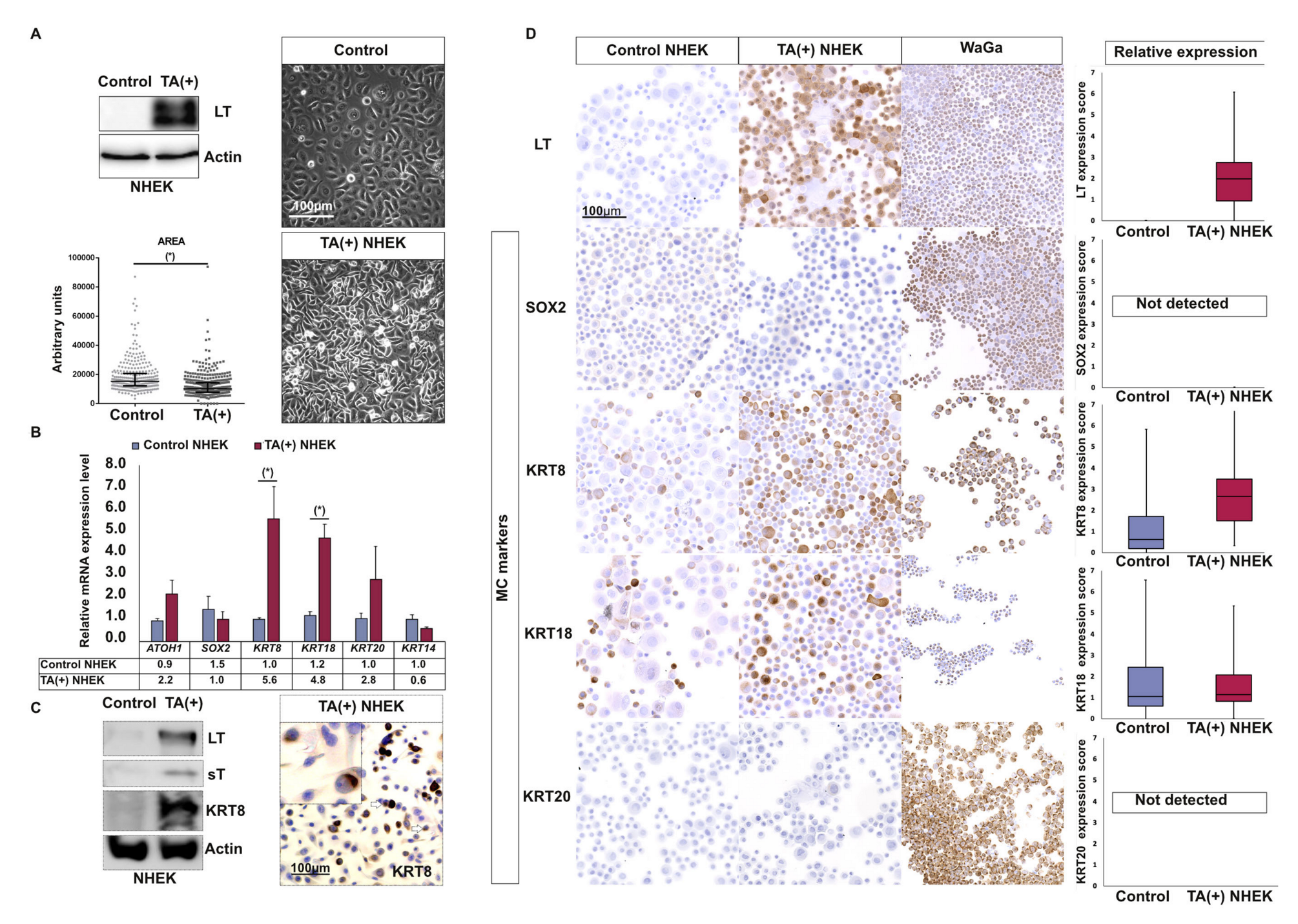
2.5. T Antigens Can Trigger Early MC Differentiation Marker Expression in Epidermal Cells
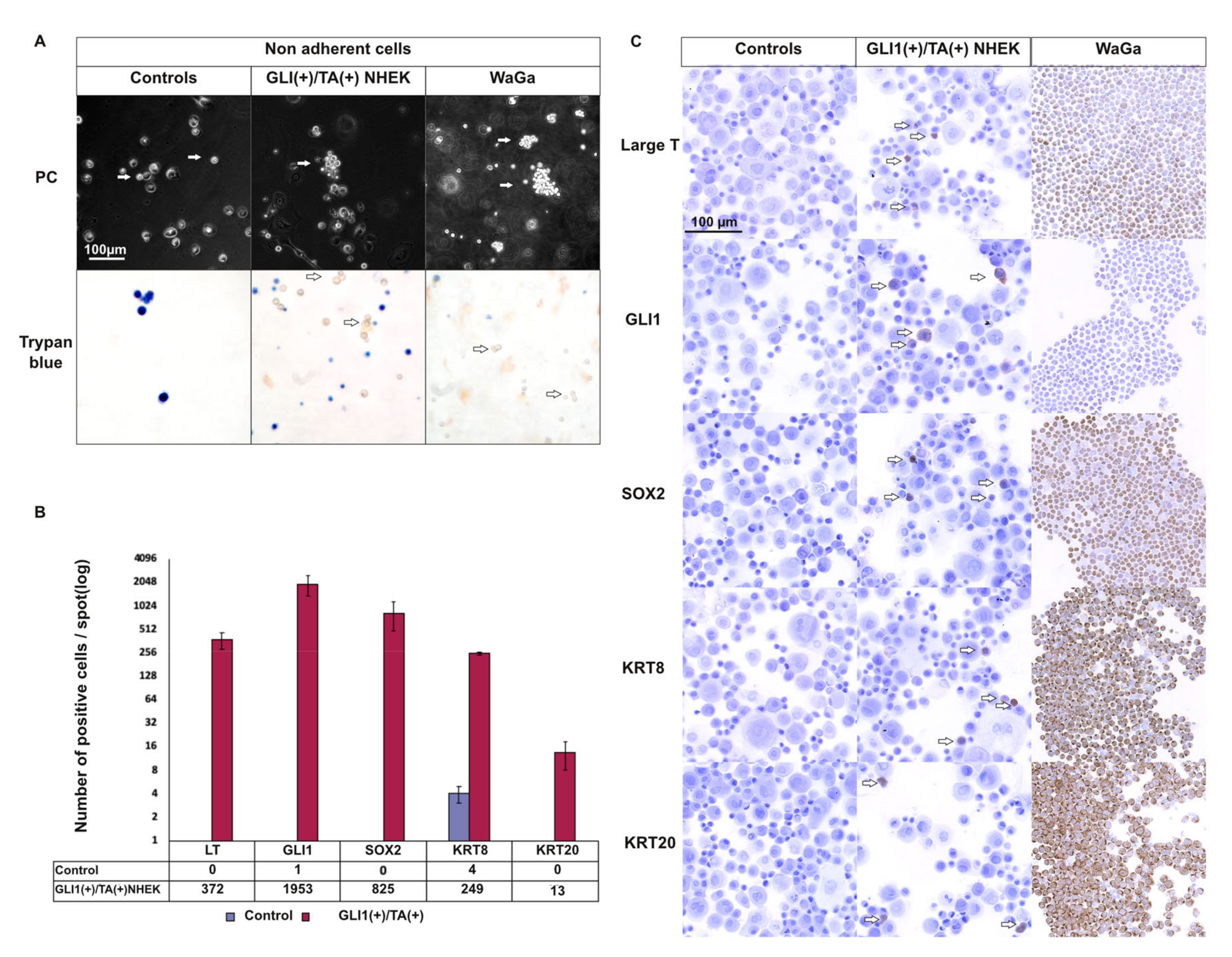
2.6. T Antigens Induce Late MC Markers in GLI1-Expressing NHEK
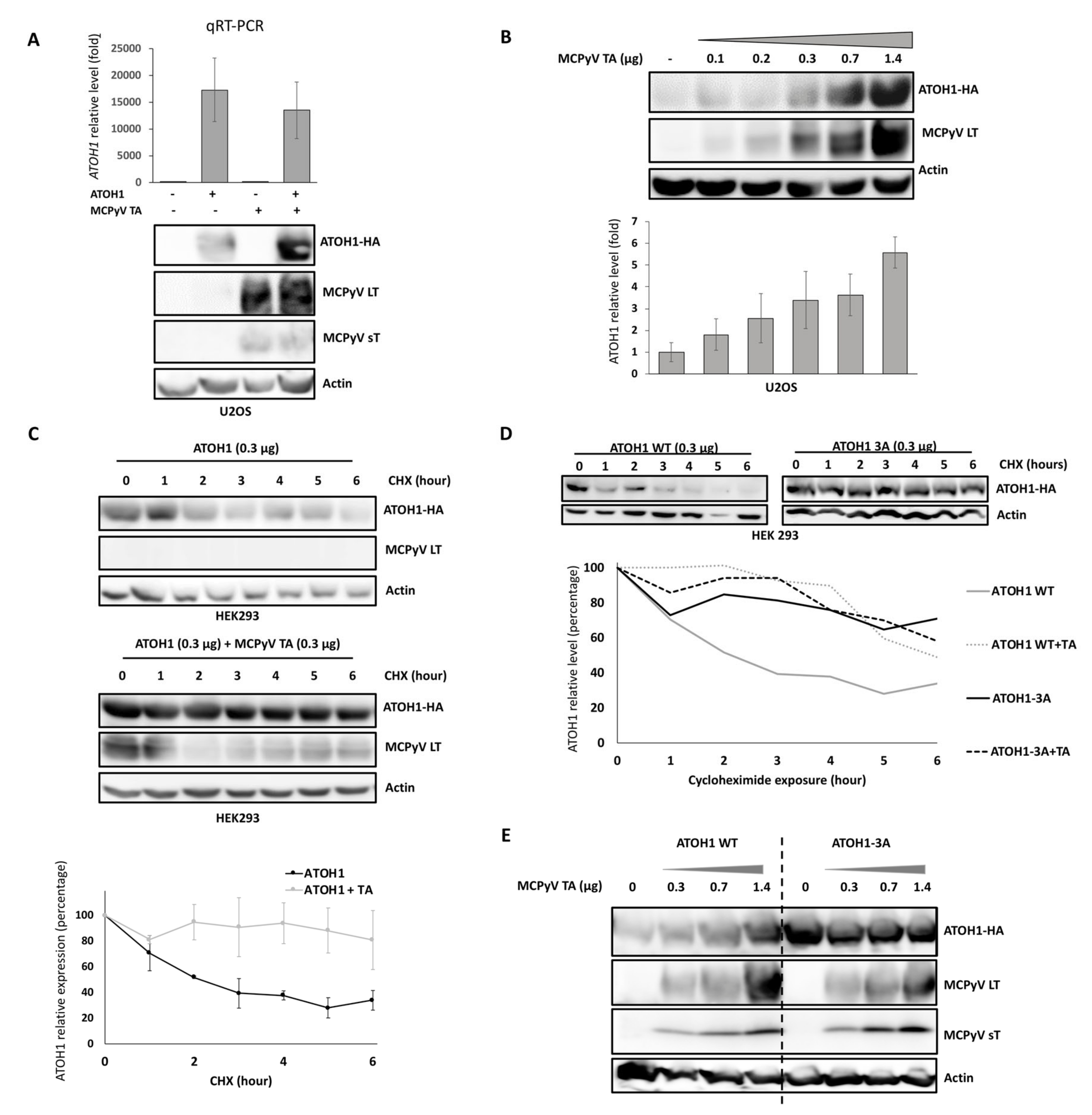
2.7. T Antigens Prevent ATOH1 Degradation
2.8. The MCPyV Unique Region 1 (MUR1) in MCPyV LT Contributes to ATOH1 Stabilization
3. Discussion
4. Material and Methods
4.1. Human Samples
4.2. Immunohistochemistry
4.3. Samples’ Management and Interpretation of Immunohistochemical Staining
4.4. Primary Keratinocytes and Cell Lines
4.5. Lentiviral Vectors’ Generation and Transduction Protocol
4.6. Gene Expression Analyses
4.7. Immunoblot
4.8. Transient Transfection and ATOH1 Half-Life Evaluation
4.9. Flow Cytometry
4.10. Image Analysis and Expression Score Determination
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Institutional Review Board
References
- Lemos, B.D.; Storer, B.E.; Iyer, J.G.; Phillips, J.L.; Bichakjian, C.K.; Fang, L.C.; Johnson, T.M.; Liegeois-Kwon, N.J.; Otley, C.C.; Paulson, K.G.; et al. Pathologic nodal evaluation improves prognostic accuracy in Merkel cell carcinoma: Analysis of 5823 cases as the basis of the first consensus staging system. J. Am. Acad. Dermatol. 2010, 63, 751–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaegen, M.E.; Mangelberger, D.; Harms, P.W.; Eberl, M.; Wilbert, D.M.; Meireles, J.; Bichakjian, C.K.; Saunders, T.L.; Wong, S.Y.; Dlugosz, A.A. Merkel cell polyomavirus small T antigen initiates Merkel cell carcinoma-like tumor development in mice. Cancer Res. 2017, 77, 3151–3157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilling, T.; Moll, I. Which are the cells of origin in merkel cell carcinoma? J. Skin Cancer 2012, 2012, 680410. [Google Scholar] [CrossRef] [Green Version]
- Becker, J.C.; Zur Hausen, A. Cells of origin in skin cancer. J. Investig. Dermatol. 2014, 134, 2491–2493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harold, A.; Amako, Y.; Hachisuka, J.; Bai, Y.; Li, M.Y.; Kubat, L.; Gravemeyer, J.; Franks, J.; Gibbs, J.R.; Park, H.J.; et al. Conversion of Sox2-dependent Merkel cell carcinoma to a differentiated neuron-like phenotype by T antigen inhibition. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, R.; Cha, M.; Ling, J.; Jia, Z.; Coyle, D.; Gu, J.G. Merkel cells transduce and encode tactile stimuli to drive Aβ-afferent impulses. Cell 2014, 157, 664–675. [Google Scholar] [CrossRef] [Green Version]
- Laga, A.C.; Lai, C.-Y.; Zhan, Q.; Huang, S.J.; Velazquez, E.F.; Yang, Q.; Hsu, M.-Y.; Murphy, G.F. Expression of the embryonic stem cell transcription factor SOX2 in human skin: Relevance to melanocyte and merkel cell biology. Am. J. Pathol. 2010, 176, 903–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdigoto, C.N.; Bardot, E.S.; Valdes, V.J.; Santoriello, F.J.; Ezhkova, E. Embryonic maturation of epidermal Merkel cells is controlled by a redundant transcription factor network. Dev. Camb. Engl. 2014, 141, 4690–4696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moll, R.; Moll, I.; Franke, W.W. Identification of Merkel cells in human skin by specific cytokeratin antibodies: Changes of cell density and distribution in fetal and adult plantar epidermis. Differ. Res. Biol. Divers. 1984, 28, 136–154. [Google Scholar] [CrossRef]
- Moll, I.; Paus, R.; Moll, R. Merkel cells in mouse skin: Intermediate filament pattern, localization, and hair cycle-dependent density. J. Investig. Dermatol. 1996, 106, 281–286. [Google Scholar] [CrossRef] [Green Version]
- Van Keymeulen, A.; Mascre, G.; Youseff, K.K.; Harel, I.; Michaux, C.; De Geest, N.; Szpalski, C.; Achouri, Y.; Bloch, W.; Hassan, B.A.; et al. Epidermal progenitors give rise to Merkel cells during embryonic development and adult homeostasis. J. Cell Biol. 2009, 187, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Halata, Z.; Grim, M.; Bauman, K.I. Friedrich Sigmund Merkel and his “Merkel cell”, morphology, development, and physiology: Review and new results. Anat. Rec. A. Discov. Mol. Cell. Evol. Biol. 2003, 271, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Morrison, K.M.; Miesegaes, G.R.; Lumpkin, E.A.; Maricich, S.M. Mammalian Merkel cells are descended from the epidermal lineage. Dev. Biol. 2009, 336, 76–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moll, I.; Lane, A.T.; Franke, W.W.; Moll, R. Intraepidermal formation of Merkel cells in xenografts of human fetal skin. J. Investig. Dermatol. 1990, 94, 359–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrowski, S.M.; Wright, M.C.; Bolock, A.M.; Geng, X.; Maricich, S.M. Ectopic Atoh1 expression drives Merkel cell production in embryonic, postnatal and adult mouse epidermis. Dev. Camb. Engl. 2015, 142, 2533–2544. [Google Scholar] [CrossRef] [Green Version]
- Bardot, E.S.; Valdes, V.J.; Zhang, J.; Perdigoto, C.N.; Nicolis, S.; Hearn, S.A.; Silva, J.M.; Ezhkova, E. Polycomb subunits Ezh1 and Ezh2 regulate the Merkel cell differentiation program in skin stem cells. EMBO J. 2013, 32, 1990–2000. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Thoresen, D.T.; Williams, J.S.; Wang, C.; Perna, J.; Petrova, R.; Brownell, I. Neural Hedgehog signaling maintains stem cell renewal in the sensory touch dome epithelium. Proc. Natl. Acad. Sci. USA 2015, 112, 7195–7200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Thoresen, D.T.; Miao, L.; Williams, J.S.; Wang, C.; Atit, R.P.; Wong, S.Y.; Brownell, I. A Cascade of Wnt, Eda, and Shh Signaling Is Essential for Touch Dome Merkel Cell Development. PLoS Genet. 2016, 12, e1006150. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.B.; Cohen, I.; Kumar, V.; Xu, Z.; Bar, C.; Dauber-Decker, K.L.; Tsai, P.-C.; Marangoni, P.; Klein, O.D.; Hsu, Y.-C.; et al. FGF signalling controls the specification of hair placode-derived SOX9 positive progenitors to Merkel cells. Nat. Commun. 2018, 9, 2333. [Google Scholar] [CrossRef]
- Woo, S.-H.; Stumpfova, M.; Jensen, U.B.; Lumpkin, E.A.; Owens, D.M. Identification of epidermal progenitors for the Merkel cell lineage. Dev. Camb. Engl. 2010, 137, 3965–3971. [Google Scholar] [CrossRef] [Green Version]
- Moll, I.; Troyanovsky, S.M.; Moll, R. Special program of differentiation expressed in keratinocytes of human haarscheiben: An analysis of individual cytokeratin polypeptides. J. Investig. Dermatol. 1993, 100, 69–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, S.C.; Eberl, M.; Vagnozzi, A.N.; Belkadi, A.; Veniaminova, N.A.; Verhaegen, M.E.; Bichakjian, C.K.; Ward, N.L.; Dlugosz, A.A.; Wong, S.Y. Basal cell carcinoma preferentially arises from stem cells within hair follicle and mechanosensory niches. Cell Stem Cell 2015, 16, 400–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kervarrec, T.; Samimi, M.; Guyétant, S.; Sarma, B.; Chéret, J.; Blanchard, E.; Berthon, P.; Schrama, D.; Houben, R.; Touzé, A. Histogenesis of Merkel Cell Carcinoma: A Comprehensive Review. Front. Oncol. 2019, 9, 451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moll, I.; Zieger, W.; Schmelz, M. Proliferative Merkel cells were not detected in human skin. Arch. Dermatol. Res. 1996, 288, 184–187. [Google Scholar] [CrossRef]
- Shuda, M.; Guastafierro, A.; Geng, X.; Shuda, Y.; Ostrowski, S.M.; Lukianov, S.; Jenkins, F.J.; Honda, K.; Maricich, S.M.; Moore, P.S.; et al. Merkel Cell Polyomavirus Small T Antigen Induces Cancer and Embryonic Merkel Cell Proliferation in a Transgenic Mouse Model. PLoS ONE 2015, 10, e0142329. [Google Scholar] [CrossRef] [Green Version]
- Kurzen, H.; Esposito, L.; Langbein, L.; Hartschuh, W. Cytokeratins as markers of follicular differentiation: An immunohistochemical study of trichoblastoma and basal cell carcinoma. Am. J. Dermatopathol. 2001, 23, 501–509. [Google Scholar] [CrossRef]
- Leblebici, C.; Bambul Sığırcı, B.; Kelten Talu, C.; Koca, S.B.; Huq, G.E. CD10, TDAG51, CK20, AR, INSM1, and Nestin Expression in the Differential Diagnosis of Trichoblastoma and Basal Cell Carcinoma. Int. J. Surg. Pathol. 2019, 27, 19–27. [Google Scholar] [CrossRef]
- McNiff, J.M.; Eisen, R.N.; Glusac, E.J. Immunohistochemical comparison of cutaneous lymphadenoma, trichoblastoma, and basal cell carcinoma: Support for classification of lymphadenoma as a variant of trichoblastoma. J. Cutan. Pathol. 1999, 26, 119–124. [Google Scholar] [CrossRef]
- Collina, G.; Eusebi, V.; Capella, C.; Rosai, J. Merkel cell differentiation in trichoblastoma. Virchows Arch. Int. J. Pathol. 1998, 433, 291–296. [Google Scholar] [CrossRef]
- Kervarrec, T.; Aljundi, M.; Appenzeller, S.; Samimi, M.; Maubec, E.; Cribier, B.; Deschamps, L.; Sarma, B.; Sarosi, E.-M.; Berthon, P.; et al. Polyomavirus-positive Merkel cell carcinoma derived from a trichoblastoma suggests an epithelial origin of this Merkel cell carcinoma. J. Investig. Dermatol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Foschini, M.P.; Eusebi, V. Divergent differentiation in endocrine and nonendocrine tumors of the skin. Semin. Diagn. Pathol. 2000, 17, 162–168. [Google Scholar] [PubMed]
- Kervarrec, T.; Tallet, A.; Miquelestorena-Standley, E.; Houben, R.; Schrama, D.; Gambichler, T.; Berthon, P.; Le Corre, Y.; Hainaut-Wierzbicka, E.; Aubin, F.; et al. Morphologic and immunophenotypical features distinguishing Merkel cell polyomavirus-positive and -negative Merkel cell carcinoma. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc. 2019, 32, 1605–1616. [Google Scholar] [CrossRef]
- Kervarrec, T.; Tallet, A.; Miquelestorena-Standley, E.; Houben, R.; Schrama, D.; Gambichler, T.; Berthon, P.; Le Corre, Y.; Hainaut-Wierzbicka, E.; Aubin, F.; et al. Diagnostic accuracy of a panel of immunohistochemical and molecular markers to distinguish Merkel cell carcinoma from other neuroendocrine carcinomas. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc. 2019, 32, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Fan, K.; Gravemeyer, J.; Ritter, C.; Rasheed, K.; Gambichler, T.; Moens, U.; Shuda, M.; Schrama, D.; Becker, J.C. MCPyV Large T antigen induced atonal homolog 1 (ATOH1) is a lineage-dependency oncogene in Merkel cell carcinoma. J. Investig. Dermatol. 2020, 140, 56–65.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forget, A.; Bihannic, L.; Cigna, S.M.; Lefevre, C.; Remke, M.; Barnat, M.; Dodier, S.; Shirvani, H.; Mercier, A.; Mensah, A.; et al. Shh signaling protects Atoh1 from degradation mediated by the E3 ubiquitin ligase Huwe1 in neural precursors. Dev. Cell 2014, 29, 649–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.-F.; Tong, M.; Edge, A.S.B. Destabilization of Atoh1 by E3 Ubiquitin Ligase Huwe1 and Casein Kinase 1 Is Essential for Normal Sensory Hair Cell Development. J. Biol. Chem. 2016, 291, 21096–21109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houben, R.; Angermeyer, S.; Haferkamp, S.; Aue, A.; Goebeler, M.; Schrama, D.; Hesbacher, S. Characterization of functional domains in the Merkel cell polyoma virus Large T antigen. Int. J. Cancer 2015, 136, E290–E300. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; Guastafierro, A.; Shuda, M.; Meinke, G.; Bohm, A.; Moore, P.S.; Chang, Y. The minimum replication origin of merkel cell polyomavirus has a unique large T-antigen loading architecture and requires small T-antigen expression for optimal replication. J. Virol. 2009, 83, 12118–12128. [Google Scholar] [CrossRef] [Green Version]
- Chéret, J.; Bertolini, M.; Ponce, L.; Lehmann, J.; Tsai, T.; Alam, M.; Hatt, H.; Paus, R. Olfactory receptor OR2AT4 regulates human hair growth. Nat. Commun. 2018, 9, 3624. [Google Scholar] [CrossRef]
- Vidal, V.P.I.; Ortonne, N.; Schedl, A. SOX9 expression is a general marker of basal cell carcinoma and adnexal-related neoplasms. J. Cutan. Pathol. 2008, 35, 373–379. [Google Scholar] [CrossRef]
- Mikami, Y.; Fujii, S.; Nagata, K.; Wada, H.; Hasegawa, K.; Abe, M.; Yoshimoto, R.U.; Kawano, S.; Nakamura, S.; Kiyoshima, T. GLI-mediated Keratin 17 expression promotes tumor cell growth through the anti-apoptotic function in oral squamous cell carcinomas. J. Cancer Res. Clin. Oncol. 2017, 143, 1381–1393. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Gu, D.; Wan, J.; Yu, B.; Zhang, X.; Chiorean, E.G.; Wang, Y.; Xie, J. The role of GLI-SOX2 signaling axis for gemcitabine resistance in pancreatic cancer. Oncogene 2019, 38, 1764–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santini, R.; Pietrobono, S.; Pandolfi, S.; Montagnani, V.; D’Amico, M.; Penachioni, J.Y.; Vinci, M.C.; Borgognoni, L.; Stecca, B. SOX2 regulates self-renewal and tumorigenicity of human melanoma-initiating cells. Oncogene 2014, 33, 4697–4708. [Google Scholar] [CrossRef] [Green Version]
- Lesko, M.H.; Driskell, R.R.; Kretzschmar, K.; Goldie, S.J.; Watt, F.M. Sox2 modulates the function of two distinct cell lineages in mouse skin. Dev. Biol. 2013, 382, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Carroll, T.M.; Williams, J.S.; Daily, K.; Rogers, T.; Gelb, T.; Coxon, A.; Wang, S.Q.; Crago, A.M.; Busam, K.J.; Brownell, I. Hedgehog Signaling Inhibitors Fail to Reduce Merkel Cell Carcinoma Viability. J. Investig. Dermatol. 2017, 137, 1187–1190. [Google Scholar] [CrossRef] [Green Version]
- Spurgeon, M.E.; Cheng, J.; Bronson, R.T.; Lambert, P.F.; DeCaprio, J.A. Tumorigenic activity of merkel cell polyomavirus T antigens expressed in the stratified epithelium of mice. Cancer Res. 2015, 75, 1068–1079. [Google Scholar] [CrossRef] [Green Version]
- Starrett, G.J.; Marcelus, C.; Cantalupo, P.G.; Katz, J.P.; Cheng, J.; Akagi, K.; Thakuria, M.; Rabinowits, G.; Wang, L.C.; Symer, D.E.; et al. Merkel Cell Polyomavirus Exhibits Dominant Control of the Tumor Genome and Transcriptome in Virus-Associated Merkel Cell Carcinoma. mBio 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Kwun, H.J.; Shuda, M.; Feng, H.; Camacho, C.J.; Moore, P.S.; Chang, Y. Merkel cell polyomavirus small T antigen controls viral replication and oncoprotein expression by targeting the cellular ubiquitin ligase SCFFbw7. Cell Host Microbe 2013, 14, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Dye, K.N.; Welcker, M.; Clurman, B.E.; Roman, A.; Galloway, D.A. Merkel cell polyomavirus Tumor antigens expressed in Merkel cell carcinoma function independently of the ubiquitin ligases Fbw7 and β-TrCP. PLoS Pathog. 2019, 15, e1007543. [Google Scholar] [CrossRef]
- Cheng, Y.-F. Atoh1 regulation in the cochlea: More than just transcription. J. Zhejiang Univ. Sci. B 2019, 20, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.C.; Stang, A.; Hausen, A.Z.; Fischer, N.; DeCaprio, J.A.; Tothill, R.W.; Lyngaa, R.; Hansen, U.K.; Ritter, C.; Nghiem, P.; et al. Epidemiology, biology and therapy of Merkel cell carcinoma: Conclusions from the EU project IMMOMEC. Cancer Immunol. Immunother. CII 2018, 67, 341–351. [Google Scholar] [CrossRef]
- Kervarrec, T.; Samimi, M.; Gaboriaud, P.; Gheit, T.; Beby-Defaux, A.; Houben, R.; Schrama, D.; Fromont, G.; Tommasino, M.; Le Corre, Y.; et al. Detection of the Merkel cell polyomavirus in the neuroendocrine component of combined Merkel cell carcinoma. Virchows Arch. Int. J. Pathol. 2018. [Google Scholar] [CrossRef]
- Boniface, K.; Bernard, F.-X.; Garcia, M.; Gurney, A.L.; Lecron, J.-C.; Morel, F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J. Immunol. Baltim. Md 1950 2005, 174, 3695–3702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couderc, E.; Morel, F.; Levillain, P.; Buffière-Morgado, A.; Camus, M.; Paquier, C.; Bodet, C.; Jégou, J.-F.; Pohin, M.; Favot, L.; et al. Interleukin-17A-induced production of acute serum amyloid A by keratinocytes contributes to psoriasis pathogenesis. PLoS ONE 2017, 12, e0181486. [Google Scholar] [CrossRef]
- Langan, E.A.; Philpott, M.P.; Kloepper, J.E.; Paus, R. Human hair follicle organ culture: Theory, application and perspectives. Exp. Dermatol. 2015, 24, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Schrama, D.; Sarosi, E.-M.; Adam, C.; Ritter, C.; Kaemmerer, U.; Klopocki, E.; König, E.-M.; Utikal, J.; Becker, J.C.; Houben, R. Characterization of six Merkel cell polyomavirus-positive Merkel cell carcinoma cell lines: Integration pattern suggest that large T antigen truncating events occur before or during integration. Int. J. Cancer 2019. [Google Scholar] [CrossRef] [PubMed]
- Vlčková, K.; Ondrušová, L.; Vachtenheim, J.; Réda, J.; Dundr, P.; Zadinová, M.; Žáková, P.; Poučková, P. Survivin, a novel target of the Hedgehog/GLI signaling pathway in human tumor cells. Cell Death Dis. 2016, 7, e2048. [Google Scholar] [CrossRef]
- Houben, R.; Adam, C.; Baeurle, A.; Hesbacher, S.; Grimm, J.; Angermeyer, S.; Henzel, K.; Hauser, S.; Elling, R.; Bröcker, E.-B.; et al. An intact retinoblastoma protein-binding site in Merkel cell polyomavirus large T antigen is required for promoting growth of Merkel cell carcinoma cells. Int. J. Cancer 2012, 130, 847–856. [Google Scholar] [CrossRef]
- Angermeyer, S.; Hesbacher, S.; Becker, J.C.; Schrama, D.; Houben, R. Merkel cell polyomavirus-positive Merkel cell carcinoma cells do not require expression of the viral small T antigen. J. Investig. Dermatol. 2013, 133, 2059–2064. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MC Progenitor Markers | TB (n = 8 Cases) | MCC (n = 103 Cases) |
|---|---|---|
| GLI1 | ||
| Negative | 1 (13%) | 60 (67%) |
| Positive (nuclear) | 7 (87%) | 29 (33%) |
| No data available | 0 | 14 |
| KRT17 | ||
| Negative | 0 | 94 (100%) |
| Positive (cytoplasmic) | 8 (100%) | 0 |
| No data available | 0 | 9 |
| SOX9 | ||
| Negative | 0 | 7 (8%) |
| Dot-like (cytoplasmic) | 0 | 59 (64%) |
| Patchy (nuclear) | 0 | 26 (28%) |
| Diffuse (nuclear) | 8 (100%) | 0 |
| No data available | 0 | 11 |
| MC markers | TB | MCC |
| SOX2 | ||
| Negative | 1 (17%) | 2 (2%) |
| Positive (nuclear) | 5 (83%) | 94 (98%) |
| No data available | 2 | 7 |
| KRT20 | ||
| Negative | 0 | 8 |
| Diffuse (cytoplasmic) | 8 (100%) | 2 |
| Mixed (cytoplasmic) | 0 | 66 |
| Dot-like pattern (cytoplasmic) | 0 | 19 |
| No data available | 0 | 8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kervarrec, T.; Samimi, M.; Hesbacher, S.; Berthon, P.; Wobser, M.; Sallot, A.; Sarma, B.; Schweinitzer, S.; Gandon, T.; Destrieux, C.; et al. Merkel Cell Polyomavirus T Antigens Induce Merkel Cell-Like Differentiation in GLI1-Expressing Epithelial Cells. Cancers 2020, 12, 1989. https://doi.org/10.3390/cancers12071989
Kervarrec T, Samimi M, Hesbacher S, Berthon P, Wobser M, Sallot A, Sarma B, Schweinitzer S, Gandon T, Destrieux C, et al. Merkel Cell Polyomavirus T Antigens Induce Merkel Cell-Like Differentiation in GLI1-Expressing Epithelial Cells. Cancers. 2020; 12(7):1989. https://doi.org/10.3390/cancers12071989
Chicago/Turabian StyleKervarrec, Thibault, Mahtab Samimi, Sonja Hesbacher, Patricia Berthon, Marion Wobser, Aurélie Sallot, Bhavishya Sarma, Sophie Schweinitzer, Théo Gandon, Christophe Destrieux, and et al. 2020. "Merkel Cell Polyomavirus T Antigens Induce Merkel Cell-Like Differentiation in GLI1-Expressing Epithelial Cells" Cancers 12, no. 7: 1989. https://doi.org/10.3390/cancers12071989
APA StyleKervarrec, T., Samimi, M., Hesbacher, S., Berthon, P., Wobser, M., Sallot, A., Sarma, B., Schweinitzer, S., Gandon, T., Destrieux, C., Pasqualin, C., Guyétant, S., Touzé, A., Houben, R., & Schrama, D. (2020). Merkel Cell Polyomavirus T Antigens Induce Merkel Cell-Like Differentiation in GLI1-Expressing Epithelial Cells. Cancers, 12(7), 1989. https://doi.org/10.3390/cancers12071989