Downregulation of the DNA Repair Gene DDB2 by Arecoline Is through p53’s DNA-Binding Domain and Is Correlated with Poor Outcome of Head and Neck Cancer Patients with Betel Quid Consumption

 , and
, and 
Abstract
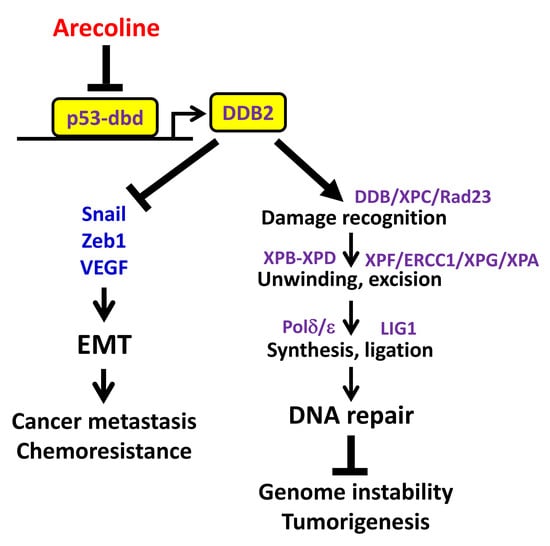
:
1. Introduction
2. Results
2.1. Arecoline Downregulates the Expression of DDB2
2.2. Reconstituted DDB2 Expression Restores Arecoline-Inhibited NER Activity
2.3. Arecoline Inhibits the Recruitment of p53 and RNA Polymerase II to the Promoters of DDB2 and p21cip1 in HEp-2 Cells
2.4. Arecoline Inhibits p53-Induced DDB2 Promoter Activity
2.5. Arecoline Inhibits p53-Regulated Promoters through p53’s DNA-Binding Domain
2.6. The DDB2 Promoter and NER Activities Are Decreased in Long-Term Arecoline-Treated Cells
2.7. DDB2 mRNA Is Downregulated in Oral Submucous Fibroblasts (OSFs) and HNC in BQ-Epidemic Areas and Is Correlated with Lymph Node Invasion and Patient Outcome
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Arecoline Treatment
4.2. Analysis of DNA Repair Activity Using Host Cell Reactivation (HCR) Assay
4.3. Promoter-Luciferase Reporters and DDB2- and p53-Expressing Plasmids
4.4. Dual-Luciferase Assay
4.5. Chromatin Immunoprecipitation (ChIP) Assay
4.6. RNA Extraction, Reverse Transcription and Real-Time Quantitative PCR (RT-qPCR)
4.7. Western Blot
4.8. Simulation of BQ-Chewing Habit by Long-Term Repetitive Arecoline Treatment
4.9. Analysis of DDB2 mRNA Expression in BQ-Associated HNC Specimens
4.10. Acquisition of HNC Dataset from the Cancer Genome Atlas (TCGA)
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- IARC. Betel-quid and Areca-nut Chewing and Some Areca-nut-derived Nitrosamines-Summary of Data Reported and Evaluation. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 2004; p. 85. [Google Scholar]
- Gupta, P.C.; Warnakulasuriya, S. Global epidemiology of areca nut usage. Addict. Biol. 2002, 7, 77–83. [Google Scholar] [CrossRef]
- Winstock, A.R.; Trivedy, C.R.; Warnakulasuriya, K.A.; Peters, T.J. A dependency syndrome related to areca nut use: Some medical and psychological aspects among areca nut users in the Gujarat community in the UK. Addict. Biol. 2000, 5, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.C.; Huang, Y.L.; Lee, C.H.; Chen, M.J.; Lin, L.M.; Tsai, C.C. Betel quid chewing, cigarette smoking and alcohol consumption related to oral cancer in Taiwan. J. Oral Pathol. Med. 1995, 24, 450–453. [Google Scholar] [CrossRef]
- Lee, K.W.; Kuo, W.R.; Tsai, S.M.; Wu, D.C.; Wang, W.M.; Fang, F.M.; Chiang, F.Y.; Ho, K.Y.; Wang, L.F.; Tai, C.F.; et al. Different impact from betel quid, alcohol and cigarette: Risk factors for pharyngeal and laryngeal cancer. Int. J. Cancer 2005, 117, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.T.; Lee, Y.C.; Chen, C.J.; Yang, P.W.; Lee, C.J.; Wu, D.C.; Hsu, H.K.; Ho, C.K.; Kao, E.L.; Lee, J.M. Risk of betel chewing for oesophageal cancer in Taiwan. Br. J. Cancer 2001, 85, 658–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.H.; Boucher, B.J.; Chiu, Y.H.; Liao, C.S.; Chen, T.H. Impact of chewing betel-nut (Areca catechu) on liver cirrhosis and hepatocellular carcinoma: A population-based study from an area with a high prevalence of hepatitis B and C infections. Public Health Nutr. 2009, 12, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Wen, C.P.; Tsai, M.K.; Chung, W.S.; Hsu, H.L.; Chang, Y.C.; Chan, H.T.; Chiang, P.H.; Cheng, T.Y.; Tsai, S.P. Cancer risks from betel quid chewing beyond oral cancer: A multiple-site carcinogen when acting with smoking. Cancer Causes Control. 2010, 21, 1427–1435. [Google Scholar] [CrossRef]
- Sharan, R.N.; Mehrotra, R.; Choudhury, Y.; Asotra, K. Association of betel nut with carcinogenesis: Revisit with a clinical perspective. PLoS ONE 2012, 7, e42759. [Google Scholar] [CrossRef] [Green Version]
- Shirname, L.P.; Menon, M.M.; Bhide, S.V. Mutagenicity of betel quid and its ingredients using mammalian test systems. Carcinogenesis 1984, 5, 501–503. [Google Scholar] [CrossRef]
- Stich, H.F.; Stich, W.; Lam, P.P. Potentiation of genotoxicity by concurrent application of compounds found in betel quid: Arecoline, eugenol, quercetin, chlorogenic acid and Mn2+. Mutat. Res. 1981, 90, 355–363. [Google Scholar] [CrossRef]
- Panigrahi, G.B.; Rao, A.R. Chromosome-breaking ability of arecoline, a major betel-nut alkaloid, in mouse bone-marrow cells in vivo. Mutat. Res. 1982, 103, 197–204. [Google Scholar] [CrossRef]
- Sharan, R.N.; Wary, K.K. Study of unscheduled DNA synthesis following exposure of human cells to arecoline and extracts of betel nut in vitro. Mutat. Res. 1992, 278, 271–276. [Google Scholar] [CrossRef]
- Stich, H.F.; Stich, W. Chromosome-damaging activity of saliva of betel nut and tobacco chewers. Cancer Lett. 1982, 15, 193–202. [Google Scholar] [CrossRef]
- Stich, H.F.; Stich, W.; Parida, B.B. Elevated frequency of micronucleated cells in the buccal mucosa of individuals at high risk for oral cancer: Betel quid chewers. Cancer Lett. 1982, 17, 125–134. [Google Scholar] [CrossRef]
- Sundqvist, K.; Liu, Y.; Nair, J.; Bartsch, H.; Arvidson, K.; Grafstrom, R.C. Cytotoxic and genotoxic effects of areca nut-related compounds in cultured human buccal epithelial cells. Cancer Res. 1989, 49, 5294–5298. [Google Scholar]
- Shih, Y.T.; Chen, P.S.; Wu, C.H.; Tseng, Y.T.; Wu, Y.C.; Lo, Y.C. Arecoline, a major alkaloid of the areca nut, causes neurotoxicity through enhancement of oxidative stress and suppression of the antioxidant protective system. Free Radic. Biol. Med. 2010, 49, 1471–1479. [Google Scholar] [CrossRef]
- Thangjam, G.S.; Kondaiah, P. Regulation of oxidative-stress responsive genes by arecoline in human keratinocytes. J. Periodontal Res. 2009, 44, 673–682. [Google Scholar] [CrossRef]
- Chang, M.C.; Ho, Y.S.; Lee, P.H.; Chan, C.P.; Lee, J.J.; Hahn, L.J.; Wang, Y.J.; Jeng, J.H. Areca nut extract and arecoline induced the cell cycle arrest but not apoptosis of cultured oral KB epithelial cells: Association of glutathione, reactive oxygen species and mitochondrial membrane potential. Carcinogenesis 2001, 22, 1527–1535. [Google Scholar] [CrossRef]
- Tsai, Y.S.; Lee, K.W.; Huang, J.L.; Liu, Y.S.; Juo, S.H.; Kuo, W.R.; Chang, J.G.; Lin, C.S.; Jong, Y.J. Arecoline, a major alkaloid of areca nut, inhibits p53, represses DNA repair, and triggers DNA damage response in human epithelial cells. Toxicology 2008, 249, 230–237. [Google Scholar] [CrossRef]
- Tsai, Y.S.; Lin, C.S.; Chiang, S.L.; Lee, C.H.; Lee, K.W.; Ko, Y.C. Areca nut induces miR-23a and inhibits repair of DNA double-strand breaks by targeting FANCG. Toxicol. Sci. 2011, 123, 480–490. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.C.; Tsai, Y.S.; Huang, J.L.; Lee, K.W.; Kuo, C.C.; Wang, C.S.; Huang, A.M.; Chang, J.Y.; Jong, Y.J.; Lin, C.S. Arecoline arrests cells at prometaphase by deregulating mitotic spindle assembly and spindle assembly checkpoint: Implication for carcinogenesis. Oral Oncol. 2010, 46, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Nikonova, A.S.; Astsaturov, I.; Serebriiskii, I.G.; Dunbrack, R.L., Jr.; Golemis, E.A. Aurora A kinase (AURKA) in normal and pathological cell division. Cell. Mol. Life Sci. 2013, 70, 661–687. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.L.; Lu, H.H.; Lu, Y.N.; Hung, P.S.; Lin, Y.J.; Lin, C.C.; Yang, C.C.; Wong, T.Y.; Lu, S.Y.; Lin, C.S. Enhancement of the genotoxicity of benzo[a]pyrene by arecoline through suppression of DNA repair in HEp-2 cells. Toxicol. In Vitro 2016, 33, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.W.; Yeh, H.; Schaeffer, L.; Roy, R.; Moncollin, V.; Egly, J.M.; Wang, Z.; Freidberg, E.C.; Evans, M.K.; Taffe, B.G.; et al. p53 modulation of TFIIH-associated nucleotide excision repair activity. Nat. Genet. 1995, 10, 188–195. [Google Scholar] [CrossRef]
- Liu, M.T.; Chang, Y.T.; Chen, S.C.; Chuang, Y.C.; Chen, Y.R.; Lin, C.S.; Chen, J.Y. Epstein-Barr virus latent membrane protein 1 represses p53-mediated DNA repair and transcriptional activity. Oncogene 2005, 24, 2635–2646. [Google Scholar] [CrossRef] [Green Version]
- Adimoolam, S.; Ford, J.M. p53 and DNA damage-inducible expression of the xeroderma pigmentosum group C gene. Proc. Natl. Acad. Sci. USA 2002, 99, 12985–12990. [Google Scholar] [CrossRef] [Green Version]
- Hwang, B.J.; Ford, J.M.; Hanawalt, P.C.; Chu, G. Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc. Natl. Acad. Sci. USA 1999, 96, 424–428. [Google Scholar] [CrossRef] [Green Version]
- Tan, T.; Chu, G. p53 binds and activates the xeroderma pigmentosum DDB2 gene in humans but not mice. Mol. Cell. Biol. 2002, 22, 3247–3254. [Google Scholar] [CrossRef] [Green Version]
- Hastak, K.; Adimoolam, S.; Trinklein, N.D.; Myers, R.M.; Ford, J.M. Identification of a Functional In Vivo p53 Response Element in the Coding Sequence of the Xeroderma Pigmentosum Group C Gene. Genes Cancer 2012, 3, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhai, L.; Xu, J.; Joo, H.Y.; Jackson, S.; Erdjument-Bromage, H.; Tempst, P.; Xiong, Y.; Zhang, Y. Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol. Cell 2006, 22, 383–394. [Google Scholar] [CrossRef]
- Lan, L.; Nakajima, S.; Kapetanaki, M.G.; Hsieh, C.L.; Fagerburg, M.; Thickman, K.; Rodriguez-Collazo, P.; Leuba, S.H.; Levine, A.S.; Rapic-Otrin, V. Monoubiquitinated histone H2A destabilizes photolesion-containing nucleosomes with concomitant release of UV-damaged DNA-binding protein E3 ligase. J. Biol. Chem. 2012, 287, 12036–12049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapetanaki, M.G.; Guerrero-Santoro, J.; Bisi, D.C.; Hsieh, C.L.; Rapic-Otrin, V.; Levine, A.S. The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc. Natl. Acad. Sci. USA 2006, 103, 2588–2593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugasawa, K. Xeroderma pigmentosum genes: Functions inside and outside DNA repair. Carcinogenesis 2008, 29, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Yoon, T.; Chakrabortty, A.; Franks, R.; Valli, T.; Kiyokawa, H.; Raychaudhuri, P. Tumor-prone phenotype of the DDB2-deficient mice. Oncogene 2005, 24, 469–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, T.; Cado, D.; Kamide, R.; Linn, S. DDB2 gene disruption leads to skin tumors and resistance to apoptosis after exposure to ultraviolet light but not a chemical carcinogen. Proc. Natl. Acad. Sci. USA 2004, 101, 2052–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pant, I.; Kumar, N.; Khan, I.; Rao, S.G.; Kondaiah, P. Role of Areca Nut Induced TGF-beta and Epithelial-Mesenchymal Interaction in the Pathogenesis of Oral Submucous Fibrosis. PLoS ONE 2015, 10, e0129252. [Google Scholar] [CrossRef]
- Lin, C.S.; Kuo, H.H.; Chen, J.Y.; Yang, C.S.; Wang, W.B. Epstein-barr virus nuclear antigen 2 retards cell growth, induces p21(WAF1) expression, and modulates p53 activity post-translationally. J. Mol. Biol. 2000, 303, 7–23. [Google Scholar] [CrossRef]
- Khan, I.; Agarwal, P.; Thangjam, G.S.; Radhesh, R.; Rao, S.G.; Kondaiah, P. Role of TGF-beta and BMP7 in the pathogenesis of oral submucous fibrosis. Growth Factors 2011, 29, 119–127. [Google Scholar] [CrossRef]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Gorgoulis, V.G.; Vassiliou, L.V.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A., Jr.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef]
- Bergink, S.; Jentsch, S. Principles of ubiquitin and SUMO modifications in DNA repair. Nature 2009, 458, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Sugasawa, K.; Okuda, Y.; Saijo, M.; Nishi, R.; Matsuda, N.; Chu, G.; Mori, T.; Iwai, S.; Tanaka, K.; Hanaoka, F. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell 2005, 121, 387–400. [Google Scholar] [CrossRef] [Green Version]
- Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Argiris, A.; Karamouzis, M.V.; Raben, D.; Ferris, R.L. Head and neck cancer. Lancet 2008, 371, 1695–1709. [Google Scholar] [CrossRef]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tan, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Networks, T.C.G.A. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar]
- Smeets, S.J.; van der Plas, M.; Schaaij-Visser, T.B.; van Veen, E.A.; van Meerloo, J.; Braakhuis, B.J.; Steenbergen, R.D.; Brakenhoff, R.H. Immortalization of oral keratinocytes by functional inactivation of the p53 and pRb pathways. Int. J. Cancer 2011, 128, 1596–1605. [Google Scholar] [CrossRef]
- Zhou, G.; Liu, Z.; Myers, J.N. TP53 Mutations in Head and Neck Squamous Cell Carcinoma and Their Impact on Disease Progression and Treatment Response. J. Cell. Biochem. 2016, 117, 2682–2692. [Google Scholar] [CrossRef] [Green Version]
- Gu, B.; Zhu, W.G. Surf the post-translational modification network of p53 regulation. Int. J. Biol. Sci. 2012, 8, 672–684. [Google Scholar] [CrossRef] [Green Version]
- Sykes, S.M.; Mellert, H.S.; Holbert, M.A.; Li, K.; Marmorstein, R.; Lane, W.S.; McMahon, S.B. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol. Cell 2006, 24, 841–851. [Google Scholar] [CrossRef] [Green Version]
- Arbely, E.; Natan, E.; Brandt, T.; Allen, M.D.; Veprintsev, D.B.; Robinson, C.V.; Chin, J.W.; Joerger, A.C.; Fersht, A.R. Acetylation of lysine 120 of p53 endows DNA-binding specificity at effective physiological salt concentration. Proc. Natl. Acad. Sci. USA 2011, 108, 8251–8256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Somasundaram, K.; Peng, Y.; Tian, H.; Zhang, H.; Bi, D.; Weber, B.L.; El-Deiry, W.S. BRCA1 physically associates with p53 and stimulates its transcriptional activity. Oncogene 1998, 16, 1713–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takimoto, R.; MacLachlan, T.K.; Dicker, D.T.; Niitsu, Y.; Mori, T.; el-Deiry, W.S. BRCA1 transcriptionally regulates damaged DNA binding protein (DDB2) in the DNA repair response following UV-irradiation. Cancer Biol. 2002, 1, 177–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLachlan, T.K.; Takimoto, R.; El-Deiry, W.S. BRCA1 directs a selective p53-dependent transcriptional response towards growth arrest and DNA repair targets. Mol. Cell. Biol. 2002, 22, 4280–4292. [Google Scholar] [CrossRef] [Green Version]
- Chiang, S.L.; Jiang, S.S.; Wang, Y.J.; Chiang, H.C.; Chen, P.H.; Tu, H.P.; Ho, K.Y.; Tsai, Y.S.; Chang, I.S.; Ko, Y.C. Characterization of arecoline-induced effects on cytotoxicity in normal human gingival fibroblasts by global gene expression profiling. Toxicol. Sci. 2007, 100, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhury, Y.; Sharan, R.N. Altered p53 response and enhanced transgenerational transmission of carcinogenic risk upon exposure of mice to betel nut. Environ. Toxicol. Pharmacol. 2009, 27, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, Y.; Sharan, R.N. Altered BRCA1 and BRCA2 responses and mutation of BRCA1 gene in mice exposed chronically and transgenerationally to aqueous extract of betel nut (AEBN). Environ. Toxicol. Pharmacol. 2011, 31, 57–69. [Google Scholar] [CrossRef]
- Miyashita, H.; Mori, S.; Tanda, N.; Nakayama, K.; Kanzaki, A.; Sato, A.; Morikawa, H.; Motegi, K.; Takebayashi, Y.; Fukumoto, M. Loss of heterozygosity of nucleotide excision repair factors in sporadic oral squamous cell carcinoma using microdissected tissue. Oncol. Rep. 2001, 8, 1133–1138. [Google Scholar] [CrossRef]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254 e6. [Google Scholar] [CrossRef] [Green Version]
- Chou, S.T.; Peng, H.Y.; Mo, K.C.; Hsu, Y.M.; Wu, G.H.; Hsiao, J.R.; Lin, S.F.; Wang, H.D.; Shiah, S.G. MicroRNA-486–3p functions as a tumor suppressor in oral cancer by targeting DDR1. J. Exp. Clin. Cancer Res. Cr 2019, 38, 281. [Google Scholar] [CrossRef]
- Shiah, S.G.; Hsiao, J.R.; Chang, H.J.; Hsu, Y.M.; Wu, G.H.; Peng, H.Y.; Chou, S.T.; Kuo, C.C.; Chang, J.Y. MiR-30a and miR-379 modulate retinoic acid pathway by targeting DNA methyltransferase 3B in oral cancer. J. Biomed. Sci. 2020, 27, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Liu, J.; Jing, J.; Wang, Z.; Li, Y.; Gou, K.; Feng, X.; Yuan, Y.; Xing, C. Expression of DDB2 Protein in the Initiation, Progression, and Prognosis of Colorectal Cancer. Dig. Dis. Sci. 2018, 63, 2959–2968. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, J.F.; Torrieri, R.; Serafim, R.B.; Di Cristofaro, L.F.; Escanfella, F.D.; Ribeiro, R.; Zanette, D.L.; Paco-Larson, M.L.; da Silva, W.A., Jr.; Tirapelli, D.P.; et al. Expression signatures of DNA repair genes correlate with survival prognosis of astrocytoma patients. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2017, 39, 1010428317694552. [Google Scholar] [CrossRef] [Green Version]
- Bommi, P.V.; Ravindran, S.; Raychaudhuri, P.; Bagchi, S. DDB2 regulates Epithelial-to-Mesenchymal Transition (EMT) in Oral/Head and Neck Squamous Cell Carcinoma. Oncotarget 2018, 9, 34708–34718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, N.; Bommi, P.V.; Bhat, U.G.; Bhattacharjee, S.; Elangovan, I.; Li, J.; Patra, K.C.; Kopanja, D.; Blunier, A.; Benya, R.; et al. DDB2 suppresses epithelial-to-mesenchymal transition in colon cancer. Cancer Res. 2013, 73, 3771–3782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Staalduinen, J.; Baker, D.; Ten Dijke, P.; van Dam, H. Epithelial-mesenchymal-transition-inducing transcription factors: New targets for tackling chemoresistance in cancer? Oncogene 2018, 37, 6195–6211. [Google Scholar] [CrossRef] [PubMed]
- Ennen, M.; Klotz, R.; Touche, N.; Pinel, S.; Barbieux, C.; Besancenot, V.; Brunner, E.; Thiebaut, D.; Jung, A.C.; Ledrappier, S.; et al. DDB2: A novel regulator of NF-kappaB and breast tumor invasion. Cancer Res. 2013, 73, 5040–5052. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.C.; Tsai, C.H.; Lai, Y.L.; Yu, C.C.; Chi, W.Y.; Li, J.J.; Chang, W.W. Arecoline-induced myofibroblast transdifferentiation from human buccal mucosal fibroblasts is mediated by ZEB1. J. Cell. Mol. Med. 2014, 18, 698–708. [Google Scholar] [CrossRef]
- Wang, T.Y.; Peng, C.Y.; Lee, S.S.; Chou, M.Y.; Yu, C.C.; Chang, Y.C. Acquisition cancer stemness, mesenchymal transdifferentiation, and chemoresistance properties by chronic exposure of oral epithelial cells to arecoline. Oncotarget 2016, 7, 84072–84081. [Google Scholar] [CrossRef]
- Zheng, L.; Jian, X.; Guo, F.; Li, N.; Jiang, C.; Yin, P.; Min, A.J.; Huang, L. miR-203 inhibits arecoline-induced epithelial-mesenchymal transition by regulating secreted frizzled-related protein 4 and transmembrane-4 L six family member 1 in oral submucous fibrosis. Oncol. Rep. 2015, 33, 2753–2760. [Google Scholar] [CrossRef]
- Lin, C.S.; Wang, Y.C.; Huang, J.L.; Hung, C.C.; Chen, J.Y. Autophagy and reactive oxygen species modulate cytotoxicity induced by suppression of ATM kinase activity in head and neck cancer cells. Oral Oncol. 2012, 48, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.; Ohshima, H.; Friesen, M.; Croisy, A.; Bhide, S.V.; Bartsch, H. Tobacco-specific and betel nut-specific N-nitroso compounds: Occurrence in saliva and urine of betel quid chewers and formation in vitro by nitrosation of betel quid. Carcinogenesis 1985, 6, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Chiou, W.Y.; Lee, K.W.; Chen, T.F.; Lin, Y.J.; Huang, J.L. Xeroderma pigmentosum, complementation group D expression in H1299 lung cancer cells following benzo[a]pyrene exposure as well as in head and neck cancer patients. J. Toxicol. Environ. Health A 2016, 79, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Kuo, K.K.; Lee, K.T.; Chen, K.K.; Yang, Y.H.; Lin, Y.C.; Tsai, M.H.; Wuputra, K.; Lee, Y.L.; Ku, C.C.; Miyoshi, H.; et al. Positive Feedback Loop of OCT4 and c-JUN Expedites Cancer Stemness in Liver Cancer. Stem Cells 2016, 34, 2613–2624. [Google Scholar] [CrossRef] [Green Version]
- Donner, A.J.; Szostek, S.; Hoover, J.M.; Espinosa, J.M. CDK8 is a stimulus-specific positive coregulator of p53 target genes. Mol. Cell 2007, 27, 121–133. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.W.; Tsai, Y.S.; Chiang, F.Y.; Huang, J.L.; Ho, K.Y.; Yang, Y.H.; Kuo, W.R.; Chen, M.K.; Lin, C.S. Lower ataxia telangiectasia mutated (ATM) mRNA expression is correlated with poor outcome of laryngeal and pharyngeal cancer patients. Ann. Oncol. 2011, 22, 1088–1093. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | DDB2 mRNA a | pb | ||||
|---|---|---|---|---|---|---|
| <0.24 | >0.24 | |||||
| Gender | Male | 38 | (43.7%) | 49 | (56.3%) | 0.391 c |
| Female | 1 | (20.0%) | 4 | (80.0%) | ||
| Age | <60 | 21 | (38.9%) | 33 | (61.1%) | 0.418 |
| >60 | 18 | (47.4%) | 20 | (52.6%) | ||
| T | 1–2 | 19 | (35.8%) | 34 | (64.2%) | 0.139 |
| 3–4 | 20 | (51.3%) | 19 | (48.7%) | ||
| N | 0 | 18 | (31.6%) | 39 | (68.4%) | 0.007 |
| 1–3 | 21 | (60.0%) | 14 | (40.0%) | ||
| Stage | I–II | 12 | (30.0%) | 28 | (70.0%) | 0.035 |
| III–IV | 27 | (51.9%) | 25 | (48.1%) | ||
| Death | No | 14 | (28.0%) | 36 | (72.0%) | 0.002 |
| Yes | 25 | (59.5%) | 17 | (40.5%) | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.-C.; Huang, J.-L.; Lee, K.-W.; Lu, H.-H.; Lin, Y.-J.; Chen, L.-F.; Wang, C.-S.; Cheng, Y.-C.; Zeng, Z.-T.; Chu, P.-Y.; et al. Downregulation of the DNA Repair Gene DDB2 by Arecoline Is through p53’s DNA-Binding Domain and Is Correlated with Poor Outcome of Head and Neck Cancer Patients with Betel Quid Consumption. Cancers 2020, 12, 2053. https://doi.org/10.3390/cancers12082053
Wang Y-C, Huang J-L, Lee K-W, Lu H-H, Lin Y-J, Chen L-F, Wang C-S, Cheng Y-C, Zeng Z-T, Chu P-Y, et al. Downregulation of the DNA Repair Gene DDB2 by Arecoline Is through p53’s DNA-Binding Domain and Is Correlated with Poor Outcome of Head and Neck Cancer Patients with Betel Quid Consumption. Cancers. 2020; 12(8):2053. https://doi.org/10.3390/cancers12082053
Chicago/Turabian StyleWang, Yu-Chu, Jau-Ling Huang, Ka-Wo Lee, Hsing-Han Lu, Yuan-Jen Lin, Long-Fong Chen, Chung-Sheng Wang, Yun-Chiao Cheng, Zih-Ting Zeng, Pei-Yi Chu, and et al. 2020. "Downregulation of the DNA Repair Gene DDB2 by Arecoline Is through p53’s DNA-Binding Domain and Is Correlated with Poor Outcome of Head and Neck Cancer Patients with Betel Quid Consumption" Cancers 12, no. 8: 2053. https://doi.org/10.3390/cancers12082053
APA StyleWang, Y.-C., Huang, J.-L., Lee, K.-W., Lu, H.-H., Lin, Y.-J., Chen, L.-F., Wang, C.-S., Cheng, Y.-C., Zeng, Z.-T., Chu, P.-Y., & Lin, C.-S. (2020). Downregulation of the DNA Repair Gene DDB2 by Arecoline Is through p53’s DNA-Binding Domain and Is Correlated with Poor Outcome of Head and Neck Cancer Patients with Betel Quid Consumption. Cancers, 12(8), 2053. https://doi.org/10.3390/cancers12082053