PARP Inhibitor Resistance Mechanisms and Implications for Post-Progression Combination Therapies


{kind=link}
{kind=link}
Abstract
:1. Introduction
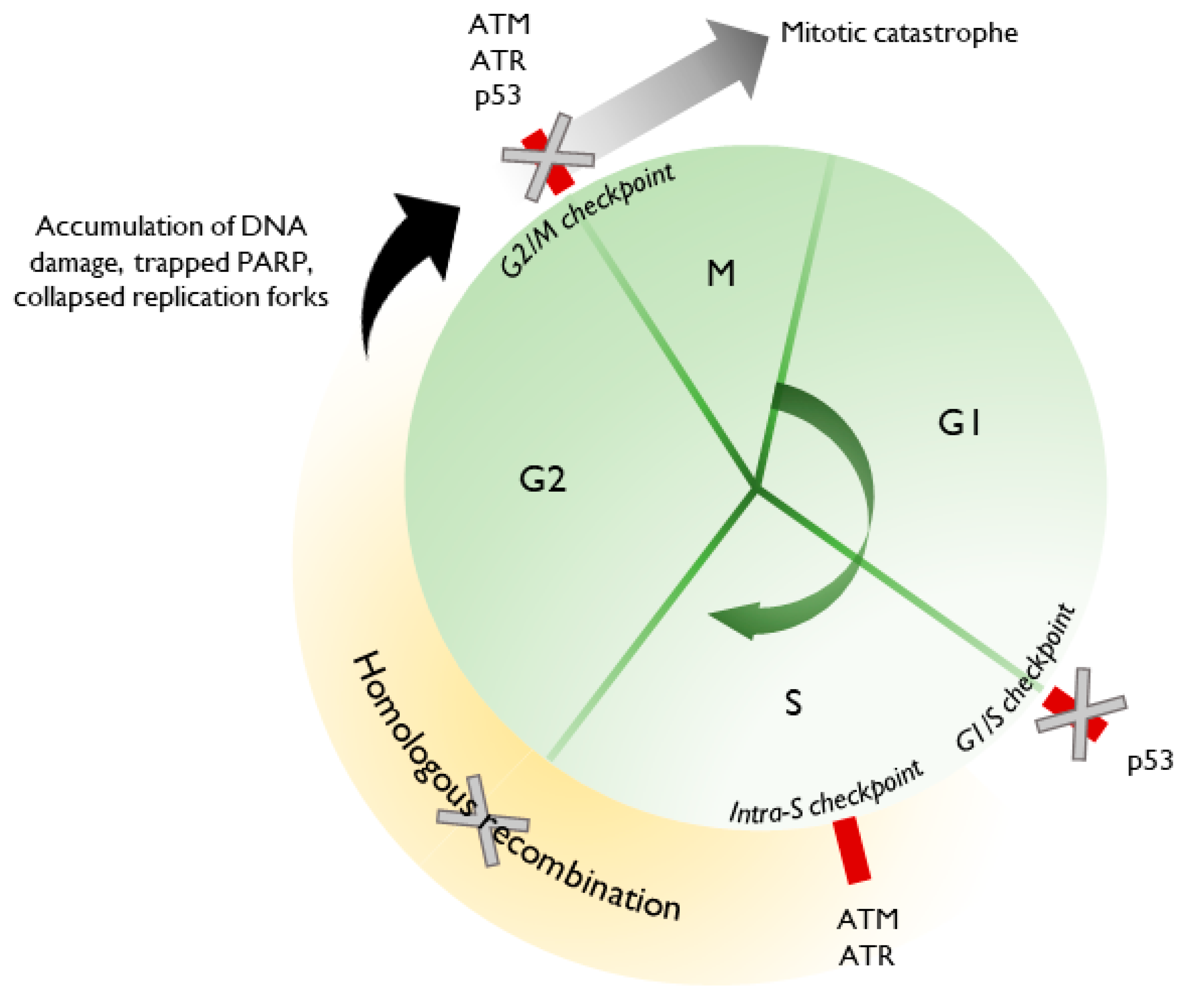
2. DNA Damage Repair and the Role of PARP
3. Replication Fork Dynamics and the Role of PARP
4. Effects of PARP Inhibitors
5. Biomarkers of PARP Inhibition
6. PARP Inhibitors in Ovarian Cancer
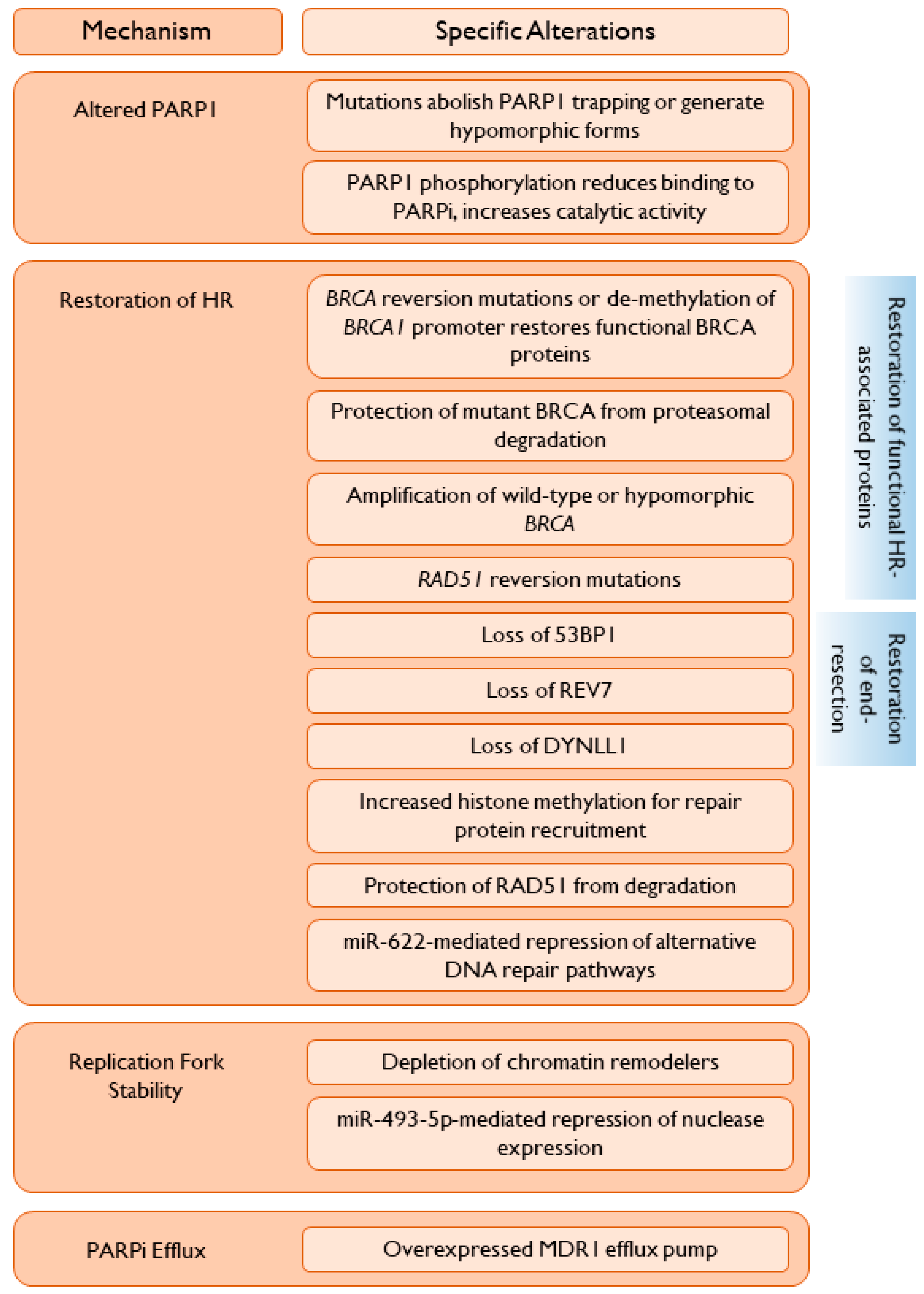
7. Mechanisms of PARP Inhibitor Resistance
7.1. Alterations in PARP1
7.1.1. PARP1 Mutations
7.1.2. Post-Translational Modifications of PARP1
7.2. Restoration of Homologous Recombination
7.2.1. Restoration of Functional HR-Associated Proteins
Reversion Mutations
BRCA1 Promoter Alterations
Generation of Hypomorphic BRCA Proteins
Decreased Proteasomal Degradation
Amplification of Wild-Type BRCA
7.2.2. Restoration of End-Resection
7.2.3. Promotion of Repair Protein Recruitment
7.2.4. Repression of Alternative DNA Repair Pathways
7.3. Alteration of Replication Fork Dynamics
7.4. PARPi Efflux
8. Implications for Post-Progression PARP Inhibitor Combination Therapies
8.1. PARPi and Anti-Angiogenic Agents
8.2. PARPi and Hsp90 Inhibition
8.3. PARPi and PI3K Pathway Inhibition
8.4. PARPi and MEK Inhibition
8.5. PARPi and Inhibition of ATR, Chk1, and Wee1
8.6. PARPi and BET/BRD4 Inhibition
8.7. PARPi and CDK12 Inhibition
8.8. PARPi and Immune Checkpoint Inhibition
9. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| altNHEJ | Alternative non-homologous end-joining |
| BER | Base excision repair |
| cfDNA | Cell-free DNA |
| cNHEJ | Classical non-homologous end-joining |
| DSB | Double strand break |
| EOC | Epithelial ovarian cancer |
| HGSOC | High grade serous ovarian cancer |
| HR | Homologous recombination |
| HRD | Homologous recombination deficient |
| HRP | Homologous recombination proficient |
| ICI | Immune checkpoint inhibition |
| NER | Nucleotide excision repair |
| NHEJ | Non-homologous end-joining |
| PARP | Poly(ADP-ribose) polymerase |
| PARPi | PARP inhibitor |
| PDX | Patient-derived xenograft |
| SSA | Single strand annealing |
| SSB | Single strand break |
| ssDNA | Single stranded DNA |
| VEGF/R | Vascular endothelial growth factor/receptor |
References
- Jachimowicz, R.D.; Goergens, J.; Reinhardt, H.C. DNA double-strand break repair pathway choice - from basic biology to clinical exploitation. Cell Cycle 2019, 18, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Feng, W.; Lim, P.X.; Kass, E.M.; Jasin, M. Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu. Rev. Cancer Biol 2018, 2, 313–349. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Álvarez-Quilón, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Ghezraoui, H.; Oliveira, C.; Becker, J.R.; Bilham, K.; Moralli, D.; Anzilotti, C.; Fischer, R.; Deobagkar-Lele, M.; Sanchiz-Calvo, M.; Fueyo-Marcos, E.; et al. 53BP1 cooperation with the REV7–shieldin complex underpins DNA structure-specific NHEJ. Nature 2018, 560, 122–127. [Google Scholar] [CrossRef]
- Findlay, S.; Heath, J.; Luo, V.M.; Malina, A.; Morin, T.; Coulombe, Y.; Djerir, B.; Li, Z.; Samiei, A.; Simo-Cheyou, E.; et al. SHLD 2/FAM 35A co-operates with REV 7 to coordinate DNA double-strand break repair pathway choice. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Gupta, R.; Somyajit, K.; Narita, T.; Maskey, E.; Stanlie, A.; Kremer, M.; Typas, D.; Lammers, M.; Mailand, N.; Nussenzweig, A.; et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 2018, 173, 972–988.e23. [Google Scholar] [CrossRef] [Green Version]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.R.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single Strand Annealing and its role in genome maintenance Chromosomal break repair by the Single Strand Annealing (SSA) pathway HHS Public Access. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dantzer, F.; Schreiber, V.; Niedergang, C.; Trucco, C.; Flatter, E.; Rubia, G.D.L.; Oliver, J.; Rolli, V.; Ménissier-de Murcia, J.; De Murcia, G. Involvement of poly(ADP-ribose) polymerase in base excision repair. Biochimie 1999, 81, 69–75. [Google Scholar] [CrossRef]
- Vodenicharov, M.D. Base excision repair is efficient in cells lacking poly(ADP-ribose) polymerase 1. Nucleic Acids Res. 2000, 28, 3887–3896. [Google Scholar] [CrossRef] [PubMed]
- Pines, A.; Vrouwe, M.G.; Marteijn, J.A.; Typas, D.; Luijsterburg, M.S.; Cansoy, M.; Hensbergen, P.; Deelder, A.; de Groot, A.; Matsumoto, S.; et al. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J. Cell Biol. 2012, 199, 235–249. [Google Scholar] [CrossRef] [Green Version]
- Messner, S.; Altmeyer, M.; Zhao, H.; Pozivil, A.; Roschitzki, B.; Gehrig, P.; Rutishauser, D.; Huang, D.; Caflisch, A.; Hottiger, M.O. PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic Acids Res. 2010, 38, 6350–6362. [Google Scholar] [CrossRef] [Green Version]
- Poirier, G.G.; de Murcia, G.; Jongstra-Bilen, J.; Niedergang, C.; Mandel, P. Poly (ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc. Natl. Acad. Sci. USA 1982, 79, 3423–3427. [Google Scholar] [CrossRef] [Green Version]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Chaudhuri, A.R.; Callen, E.; Pang, Y.; Biswas, K.; Klarmann, K.D.; Martin, B.K.; Burkett, S.; Cleveland, L.; Stauffer, S.; et al. Synthetic viability by BRCA2 and PARP1/ARTD1 deficiencies. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, A.; Drew, Y.; Matheson, E.; Salehan, M.; Gentles, L.; Pachter, J.A.; Curtin, N.J. Evaluating the potential of kinase inhibitors to suppress DNA repair and sensitise ovarian cancer cells to PARP inhibitors. Biochem. Pharmacol. 2019, 125–132. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Cancer, O.; Group, S.; Sharma, R.; Stewart, C.; Fereday, S.; et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurman, R.J. Origin and molecular pathogenesis of ovarian high-grade serous carcinoma. Ann. Oncol. 2013, 24. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the {cBioPortal.}. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (Amst). 2018, 71, 172–176. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, D.R.; Dougherty, B.A.; Lai, Z.; Fielding, A.; Grinsted, L.; Spencer, S.; O’Connor, M.J.; Ho, T.W.; Robertson, J.D.; Lanchbury, J.S.; et al. Candidate biomarkers of PARP inhibitor sensitivity in ovarian cancer beyond the BRCA genes. Br. J. Cancer 2018, 119, 1401–1409. [Google Scholar] [CrossRef] [Green Version]
- Stover, E.H.; Konstantinopoulos, P.A.; Matulonis, U.A.; Swisher, E.M. Biomarkers of response and resistance to DNA repair targeted therapies. Clin. Cancer Res. 2016, 22, 5651–5660. [Google Scholar] [CrossRef] [Green Version]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Spentzos, D.; Karlan, B.Y.; Taniguchi, T.; Fountzilas, E.; Francoeur, N.; Levine, D.A.; Cannistra, S.A. Gene Expression Profile of BRCAness That Correlates With Responsiveness to Chemotherapy and With Outcome in Patients With Epithelial Ovarian Cancer. J. Clin. Oncol. 2010, 28, 3555–3561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matulonis, U.A.; Penson, R.T.; Domchek, S.M.; Kaufman, B.; Shapira-Frommer, R.; Audeh, M.W.; Kaye, S.; Molife, L.R.; Gelmon, K.A.; Robertson, J.D.; et al. Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: A multistudy analysis of response rates and safety. Ann. Oncol. 2016, 27, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Hjortkjær, M.; Waldstrøm, M.; Jakobsen, A.; Kanstrup, H.; Søgaard-Andersen, E.; Steffensen, K.D. The prognostic value of BRCA1 and PARP expression in epithelial ovarian carcinoma: Immunohistochemical detection. Int. J. Gynecol. Pathol. 2017, 36, 180–189. [Google Scholar] [CrossRef]
- Makvandi, M.; Xu, K.; Lieberman, B.P.; Anderson, R.C.; Effron, S.S.; Winters, H.D.; Zeng, C.; McDonald, E.S.; Pryma, D.A.; Greenberg, R.A.; et al. A radiotracer strategy to quantify PARP-1 expression in vivo provides a biomarker that can enable patient selection for PARP inhibitor therapy. Cancer Res. 2016, 76, 4516–4524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA approval summary: Olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef] [Green Version]
- Balasubramaniam, S.; Beaver, J.A.; Horton, S.; Fernandes, L.L.; Tang, S.; Horne, H.N.; Liu, J.; Liu, C.; Schrieber, S.J.; Yu, J.; et al. FDA approval summary: Rucaparib for the treatment of patients with deleterious BRCA mutation–associated advanced ovarian cancer. Clin. Cancer Res. 2017, 23, 7165–7170. [Google Scholar] [CrossRef] [Green Version]
- FDA Approves Niraparib for HRD-Positive Advanced Ovarian Cancer | FDA. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-niraparib-hrd-positive-advanced-ovarian-cancer (accessed on 11 February 2020).
- Ison, G.; Howie, L.J.; Amiri-Kordestani, L.; Zhang, L.; Tang, S.; Sridhara, R.; Pierre, V.; Charlab, R.; Ramamoorthy, A.; Song, P.; et al. FDA approval summary: Niraparib for the maintenance treatment of patients with recurrent ovarian cancer in response to platinum-based chemotherapy. Clin. Cancer Res. 2018, 24, 4066–4071. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food & Drug Administration FDA Approved Olaparib (LYNPARZA) for the Maintenance Treatment of Adult Patients with Deleterious or Suspected Deleterious Germline or Somatic BRCA-Mutated (gBRCAm or sBRCAm) Advanced Epithelial Ovarian, Fallopian Tube or Primary Peritoneal Cancer. Available online: https://www.fda.gov/drugs/fda-approved-olaparib-lynparza-astrazeneca-pharmaceuticals-lp-maintenance-treatment-adult-patients (accessed on 17 December 2019).
- FDA Approves Niraparib for First-Line Maintenance of Advanced Ovarian Cancer | FDA. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-niraparib-first-line-maintenance-advanced-ovarian-cancer (accessed on 5 May 2020).
- FDA Approves Olaparib Plus Bevacizumab as Maintenance Treatment for Ovarian, Fallopian Tube, or Primary Peritoneal Cancers | FDA. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-olaparib-plus-bevacizumab-maintenance-treatment-ovarian-fallopian-tube-or-primary (accessed on 11 May 2020).
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019. [Google Scholar] [CrossRef] [Green Version]
- Wakefield, M.J.; Nesic, K.; Kondrashova, O.; Scott, C.L. Diverse mechanisms of PARP inhibitor resistance in ovarian cancer. Biochim. Biophys. Acta Rev. Cancer 2019, 1872. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Yamaguchi, H.; Wei, Y.; Hsu, J.L.; Wang, H.L.; Hsu, Y.H.; Lin, W.C.; Yu, W.H.; Leonard, P.G.; Lee, G.R.; et al. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat. Med. 2016, 22, 194–201. [Google Scholar] [CrossRef]
- Dong, Q.; Du, Y.; Li, H.; Liu, C.; Wei, Y.; Chen, M.K.; Zhao, X.; Chu, Y.Y.; Qiu, Y.; Qin, L.; et al. EGFR and c-Met cooperate to enhance resistance to PARP inhibitors in hepatocellular carcinoma. Cancer Res. 2019, 79, 819–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigelt, B.; Comino-Méndez, I.; De Bruijn, I.; Tian, L.; Meisel, J.L.; García-Murillas, I.; Fribbens, C.; Cutts, R.; Martelotto, L.G.; Ng, C.K.Y.; et al. Diverse BRCA1 and BRCA2 reversion mutations in circulating cell-free DNA of therapy-resistant breast or ovarian cancer. Clin. Cancer Res. 2017, 23, 6708–6720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, K.K.; Harrell, M.I.; Oza, A.M.; Oaknin, A.; Ray-Coquard, I.; Tinker, A.V.; Helman, E.; Radke, M.R.; Say, C.; Vo, L.T.; et al. BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2019, 9, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Norquist, B.; Wurz, K.A.; Pennil, C.C.; Garcia, R.; Gross, J.; Sakai, W.; Karlan, B.Y.; Taniguchi, T.; Swisher, E.M. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J. Clin. Oncol. 2011, 29, 3008–3015. [Google Scholar] [CrossRef] [Green Version]
- Mayor, P.; Gay, L.M.; Gornstein, E.; Morley, S.; Frampton, G.M.; Heilmann, A.; Sun, J.; Chung, J.; Daniel, S.; Ramkissoon, S.; et al. BRCA1/2 reversion mutations revealed in breast and gynecologic cancers sequenced during routine clinical care using tissue or liquid biopsy. J. Clin. Oncol. 2017, 35, 5551. [Google Scholar] [CrossRef]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef]
- Goodall, J.; Mateo, J.; Yuan, W.; Mossop, H.; Porta, N.; Miranda, S.; Perez-Lopez, R.; Dolling, D.; Robinson, D.R.; Sandhu, S.; et al. Circulating cell-free DNA to guide prostate cancer treatment with PARP inhibition. Cancer Discov. 2017, 7, 1006–1017. [Google Scholar] [CrossRef] [Green Version]
- Gornstein, E.L.; Sandefur, S.; Chung, J.H.; Gay, L.M.; Holmes, O.; Erlich, R.L.; Soman, S.; Martin, L.K.; Rose, A.V.; Stephens, P.J.; et al. BRCA2 Reversion Mutation Associated With Acquired Resistance to Olaparib in Estrogen Receptor-positive Breast Cancer Detected by Genomic Profiling of Tissue and Liquid Biopsy. Clin. Breast Cancer 2018, 18, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swisher, E.M.; Sakai, W.; Karlan, B.Y.; Wurz, K.; Urban, N.; Taniguchi, T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008, 68, 2581–2586. [Google Scholar] [CrossRef] [Green Version]
- Quigley, D.; Alumkal, J.J.; Wyatt, A.W.; Kothari, V.; Foye, A.; Lloyd, P.; Aggarwal, R.; Kim, W.; Lu, E.; Schwartzman, J.; et al. Analysis of Circulating Cell-Free DNA Identifies Multiclonal Heterogeneity of BRCA2 Reversion Mutations Associated with Resistance to PARP Inhibitors. Cancer Discov. 2017, 7, 999–1005. [Google Scholar] [CrossRef] [Green Version]
- Ter Brugge, P.; Kristel, P.; Van Der Burg, E.; Boon, U.; De Maaker, M.; Lips, E.; Mulder, L.; De Ruiter, J.; Moutinho, C.; Gevensleben, H.; et al. Mechanisms of therapy resistance in patient-derived xenograft models of brca1-deficient breast cancer. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Simmons, A.D.; Nguyen, M.; Pintus, E. Polyclonal BRCA2 mutations following carboplatin treatment confer resistance to the PARP inhibitor rucaparib in a patient with mCRPC: A case report. BMC Cancer 2020, 20. [Google Scholar] [CrossRef]
- Christie, E.L.; Fereday, S.; Doig, K.; Pattnaik, S.; Dawson, S.J.; Bowtell, D.D.L. Reversion of BRCA1/2 germline mutations detected in circulating tumor DNA from patients with high-grade serous ovarian cancer. J. Clin. Oncol. 2017, 35, 1274–1280. [Google Scholar] [CrossRef]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Burness, M.L.; Martin-Trevino, R.; Guy, J.; Bai, S.; Harouaka, R.; Brooks, M.D.; Shang, L.; Fox, A.; Luther, T.K.; et al. RAD51 mediates resistance of cancer stem cells to PARP inhibition in triple-negative breast cancer. Clin. Cancer Res. 2017, 23, 514–522. [Google Scholar] [CrossRef] [Green Version]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Bernhardy, A.J.; Cruz, C.; Krais, J.J.; Nacson, J.; Nicolas, E.; Peri, S.; Van Der Gulden, H.; Van Der Heijden, I.; O’Brien, S.W.; et al. The BRCA1-Δ11q alternative splice isoform bypasses germline mutations and promotes therapeutic resistance to PARP inhibition and cisplatin. Cancer Res. 2016, 76, 2778–2790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Krais, J.J.; Bernhardy, A.J.; Nicolas, E.; Cai, K.Q.; Harrell, M.I.; Kim, H.H.; George, E.; Swisher, E.M.; Simpkins, F.; et al. RING domain-deficient BRCA1 promotes PARP inhibitor and platinum resistance. J. Clin. Invest. 2016, 126, 3145–3157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Bernhardy, A.J.; Nacson, J.; Krais, J.J.; Tan, Y.F.; Nicolas, E.; Radke, M.R.; Handorf, E.; Llop-Guevara, A.; Balmaña, J.; et al. BRCA1 intronic Alu elements drive gene rearrangements and PARP inhibitor resistance. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, R.S.; Glover, J.N.M. Structural consequences of a cancer-causing BRCA1-BRCT missense mutation. J. Biol. Chem. 2003, 278, 2630–2635. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.S.; Green, R.; Marsillac, S.M.; Coquelle, N.; Williams, R.S.; Yeung, T.; Foo, D.; Hau, D.D.; Hui, B.; Monteiro, A.N.A.; et al. Comprehensive analysis of missense variations in the BRCT domain of BRCA1 by structural and functional assays. Cancer Res. 2010, 70, 4880–4890. [Google Scholar] [CrossRef] [Green Version]
- Johnson, N.; Johnson, S.F.; Yao, W.; Li, Y.C.; Choi, Y.E.; Bernhardy, A.J.; Wang, Y.; Capelletti, M.; Sarosiek, K.A.; Moreau, L.A.; et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc. Natl. Acad. Sci. USA 2013, 110, 17041–17046. [Google Scholar] [CrossRef] [Green Version]
- Lheureux, S.; Bruce, J.P.; Burnier, J.V.; Karakasis, K.; Shaw, P.A.; Clarke, B.A.; Yang, S.Y.C.; Quevedo, R.; Li, T.; Dowar, M.; et al. Somatic BRCA1/2 recovery as a resistance mechanism after exceptional response to poly (ADP-ribose) polymerase inhibition. J. Clin. Oncol. 2017, 35, 1240–1249. [Google Scholar] [CrossRef]
- Park, P.H.; Yamamoto, T.M.; Li, H.; Alcivar, A.L.; Xia, B.; Wang, Y.; Bernhardy, A.J.; Turner, K.M.; Kossenkov, A.V.; Watson, Z.L.; et al. Amplification of the Mutation-Carrying BRCA2 Allele Promotes RAD51 Loading and PARP Inhibitor Resistance in the Absence of Reversion Mutations. Mol. Cancer Ther. 2020, 19, 602–613. [Google Scholar] [CrossRef] [Green Version]
- Bunting, S.F.; Callén, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Belotserkovskaya, R.; Raga Gil, E.; Lawrence, N.; Butler, R.; Clifford, G.; Wilson, M.D.; Jackson, S.P. PALB2 chromatin recruitment restores homologous recombination in BRCA1-deficient cells depleted of 53BP1. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.H.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.M.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Cohen, O.; Kochupurakkal, B.; Kim, D.; Dunn, C.E.; Buendia Buendia, J.; Wander, S.; Helvie, K.; Lloyd, M.R.; Marini, L.; et al. Reversion and non-reversion mechanisms of resistance to PARP inhibitor or platinum chemotherapy in BRCA1/2-mutant metastatic breast cancer. Ann. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; Van Der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Publ. Gr. 2010, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Xu, X.; Bunting, S.F.; Liu, J.; Wang, R.H.; Cao, L.L.; Wu, J.J.; Peng, T.N.; Chen, J.; Nussenzweig, A.; et al. A Selective Requirement for 53BP1 in the Biological Response to Genomic Instability Induced by Brca1 Deficiency. Mol. Cell 2009, 35, 534–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; Van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53BP1 causes PARP inhibitor resistance in BRCA1-mutated mouse mammary tumors. Cancer Discov. 2013, 3, 68–81. [Google Scholar] [CrossRef] [Green Version]
- Nacson, J.; Krais, J.J.; Bernhardy, A.J.; Clausen, E.; Feng, W.; Wang, Y.; Nicolas, E.; Cai, K.Q.; Tricarico, R.; Hua, X.; et al. BRCA1 Mutation-Specific Responses to 53BP1 Loss-Induced Homologous Recombination and PARP Inhibitor Resistance. Cell Rep. 2018, 24, 3513–3527.e7. [Google Scholar] [CrossRef] [Green Version]
- Duarte, A.A.; Gogola, E.; Sachs, N.; Barazas, M.; Annunziato, S.; De Ruiter, J.R.; Velds, A.; Blatter, S.; Houthuijzen, J.M.; Van De Ven, M.; et al. BrcA-deficient mouse mammary tumor organoids to study cancer-drug resistance. Articles 2018, 134. [Google Scholar] [CrossRef]
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 2015, 521, 541–544. [Google Scholar] [CrossRef] [Green Version]
- He, Y.J.; Meghani, K.; Caron, M.C.; Yang, C.; Ronato, D.A.; Bian, J.; Sharma, A.; Moore, J.; Niraj, J.; Detappe, A.; et al. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature 2018, 563, 522–526. [Google Scholar] [CrossRef]
- Watson, Z.L.; Yamamoto, T.M.; McMellen, A.; Kim, H.; Hughes, C.J.; Wheeler, L.J.; Post, M.D.; Behbakht, K.; Bitler, B.G. Histone methyltransferases EHMT1 and EHMT2 (GLP/G9A) maintain PARP inhibitor resistance in high-grade serous ovarian carcinoma. Clin. Epigenetics 2019, 11. [Google Scholar] [CrossRef]
- Yang, Q.; Zhu, Q.; Lu, X.; Du, Y.; Cao, L.; Shen, C.; Hou, T.; Li, M.; Li, Z.; Liu, C.; et al. G9a coordinates with the RPA complex to promote DNA damage repair and cell survival. Proc. Natl. Acad. Sci. USA 2017, 114, E6054–E6063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginjala, V.; Rodriguez-Colon, L.; Ganguly, B.; Gangidi, P.; Gallina, P.; Al-Hraishawi, H.; Kulkarni, A.; Tang, J.; Gheeya, J.; Simhadri, S.; et al. Protein-lysine methyltransferases G9a and GLP1 promote responses to DNA damage. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Dungrawala, H.; Rose, K.L.; Bhat, K.P.; Mohni, K.N.; Glick, G.G.; Couch, F.B.; Cortez, D. The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol. Cell 2015, 59, 998–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzio, A.; Puccini, J.; Kwon, Y.; Sung, P.; Bar-Sagi, D.; Correspondence, M.P. The F-Box Domain-Dependent Activity of EMI1 Regulates PARPi Sensitivity in Triple-Negative Breast Cancers. Mol. Cell 2019, 73, 224–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamovich, A.I.; Toland, A.E.; Parvin, J.D. F-Box Protein-Mediated Resistance to PARP Inhibitor Therapy. Mol. Cell 2019, 73, 195–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.E.; Meghani, K.; Brault, M.E.; Leclerc, L.; He, Y.J.; Day, T.A.; Elias, K.M.; Drapkin, R.; Weinstock, D.M.; Dao, F.; et al. Platinum and PARP Inhibitor Resistance Due to Overexpression of MicroRNA-622 in BRCA1-Mutant Ovarian Cancer. Cell Rep. 2016, 14, 429–439. [Google Scholar] [CrossRef] [Green Version]
- Fukumoto, T.; Zhu, H.; Nacarelli, T.; Karakashev, S.; Fatkhutdinov, N.; Wu, S.; Liu, P.; Kossenkov, A.V.; Showe, L.C.; Jean, S.; et al. N6-methylation of adenosine of FZD10 mRNA contributes to PARP inhibitor resistance. Cancer Res. 2019, 79, 2812–2820. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.M.; McMellen, A.; Watson, Z.L.; Aguilera, J.; Ferguson, R.; Nurmemmedov, E.; Thakar, T.; Moldovan, G.L.; Kim, H.; Cittelly, D.M.; et al. Activation of Wnt signaling promotes olaparib resistant ovarian cancer. Mol. Carcinog. 2019, 58, 1770–1782. [Google Scholar] [CrossRef]
- Yamamoto, K.N.; Hirota, K.; Takeda, S.; Haeno, H. Evolution of pre-existing versus acquired resistance to platinum drugs and PARP inhibitors in BRCA-associated cancers. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Jun, S.; Jung, Y.S.; Suh, H.N.; Wang, W.; Kim, M.J.; Oh, Y.S.; Lien, E.M.; Shen, X.; Matsumoto, Y.; McCrea, P.D.; et al. LIG4 mediates Wnt signalling-induced radioresistance. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, R.; Tang, J.; Ma, J.G.; Chen, S.P.; Xia, L.P.; Zhou, W.J.; Li, D.D.; Feng, G.K.; Zeng, Y.X.; Zhu, X.F. PKB/Akt promotes DSB repair in cancer cells through upregulating Mre11 expression following ionizing radiation. Oncogene 2011, 30, 944–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhuri, A.R.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 2017, 68, 414–430.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clements, K.E.; Thakar, T.; Nicolae, C.M.; Liang, X.; Wang, H.-G.; Moldovan, G.-L. Loss of E2F7 confers resistance to poly-ADP-ribose polymerase (PARP) inhibitors in BRCA2-deficient cells. Nucleic Acids Res. 2018, 46, 8898–8907. [Google Scholar] [CrossRef]
- Westendorp, B.; Mokry, M.; Groot Koerkamp, M.J.A.; Holstege, F.C.P.; Cuppen, E.; De Bruin, A. E2F7 represses a network of oscillating cell cycle genes to control S-phase progression. Nucleic Acids Res. 2012, 40, 3511–3523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rondinelli, B.; Gogola, E.; Yücel, H.; Duarte, A.A.; Van De Ven, M.; Van Der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378. [Google Scholar] [CrossRef]
- Murai, J.; Feng, Y.; Yu, G.K.; Ru, Y.; Tang, S.W.; Shen, Y.; Pommier, Y. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget 2016, 7, 76534–76550. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Tang, S.W.; Leo, E.; Baechler, S.A.; Redon, C.E.; Zhang, H.; Al Abo, M.; Rajapakse, V.N.; Nakamura, E.; Jenkins, L.M.M.; et al. SLFN11 Blocks Stressed Replication Forks Independently of ATR. Mol. Cell 2018, 69, 371–384.e6. [Google Scholar] [CrossRef] [Green Version]
- Meghani, K.; Fuchs, W.; Detappe, A.; Drané, P.; Gogola, E.; Rottenberg, S.; Jonkers, J.; Matulonis, U.; Swisher, E.M.; Konstantinopoulos, P.A.; et al. Multifaceted Impact of MicroRNA 493-5p on Genome-Stabilizing Pathways Induces Platinum and PARP Inhibitor Resistance in BRCA2-Mutated Carcinomas. Cell Rep. 2018, 23, 100–111. [Google Scholar] [CrossRef] [Green Version]
- Guillemette, S.; Serra, R.W.; Peng, M.; Hayes, J.A.; Konstantinopoulos, P.A.; Green, M.R.; Green, M.R.; Cantor, S.B. Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4. Genes Dev. 2015, 29, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Christie, E.L.; Pattnaik, S.; Beach, J.; Copeland, A.; Rashoo, N.; Fereday, S.; Hendley, J.; Alsop, K.; Brady, S.L.; Lamb, G.; et al. Multiple ABCB1 transcriptional fusions in drug resistant high-grade serous ovarian and breast cancer. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombard, A.P.; Liu, C.; Armstrong, C.M.; D’Abronzo, L.S.; Lou, W.; Chen, H.; Dall’Era, M.; Ghosh, P.M.; Evans, C.P.; Gao, A.C. Overexpressed ABCB1 Induces Olaparib-Taxane Cross-Resistance in Advanced Prostate Cancer. Transl. Oncol. 2019, 12, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, A.; Sawers, L.; Gannon, A.L.; Chakravarty, P.; Scott, A.L.; Bray, S.E.; Ferguson, M.J.; Smith, G. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br. J. Cancer 2016, 115, 431–441. [Google Scholar] [CrossRef] [Green Version]
- Sundar, R.; Valeri, N.; Harrington, K.J.; Yap, T.A. Combining molecularly targeted agents: Is more always better? Clin. Cancer Res. 2017, 23, 1123–1125. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.; Yang, K.; Taylor-Harding, B.; Ruprecht Wiedemeyer, W.; Buckanovich, R.J. VEGFR3 inhibition chemosensitizes ovarian cancer stemlike cells through down-regulation of BRCA1 and BRCA2. Neoplasia 2014, 16, 343–353.e2. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Roman, N.; Chong, M.Y.; Chahal, S.K.; Caragher, S.P.; Jackson, M.R.; Stevenson, K.H.; Dongre, S.A.; Chalmers, A.J. Radiation Responses of 2D and 3D Glioblastoma Cells: A Novel, 3D-specific Radioprotective Role of VEGF/Akt Signaling through Functional Activation of NHEJ. Mol. Cancer Ther. 2020, 19, 575–589. [Google Scholar] [CrossRef]
- Bindra, R.S.; Schaffer, P.J.; Meng, A.; Woo, J.; Maseide, K.; Roth, M.E.; Lizardi, P.; Hedley, D.W.; Bristow, R.G.; Glazer, P.M. Down-Regulation of Rad51 and Decreased Homologous Recombination in Hypoxic Cancer Cells. Mol. Cell. Biol. 2004, 24, 8504–8518. [Google Scholar] [CrossRef] [Green Version]
- Chan, N.; Koritzinsky, M.; Zhao, H.; Bindra, R.; Glazer, P.M.; Powell, S.; Belmaaza, A.; Wouters, B.; Bristow, R.G. Chronic hypoxia decreases synthesis of homologous recombination proteins to offset chemoresistance and radioresistance. Cancer Res. 2008, 68, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Kumareswaran, R.; Ludkovski, O.; Meng, A.; Sykes, J.; Pintilie, M.; Bristow, R.G. Chronic hypoxia compromises repair of DNA double-strand breaks to drive genetic instability. J. Cell Sci. 2012, 125, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism: Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Oaknin, A.; Garg, S.; Bruce, J.; Dhani, N.C.; Madariaga, A.; Bonilla, L.; Lee, Y.C.; Colombo, I.; Bhat, G.; et al. Evolve: A post PARP inhibitor clinical translational phase II trial of cediranib-olaparib in ovarian cancer—A Princess Margaret Consortium – GCIG Phase II Trial. J. Clin. Oncol. 2019, 37, 5521. [Google Scholar] [CrossRef]
- Kaplan, A.R.; Gueble, S.E.; Liu, Y.; Oeck, S.; Kim, H.; Yun, Z.; Glazer, P.M. Cediranib suppresses homology-directed DNA repair through down-regulation of BRCA1/2 and RAD51. Sci. Transl. Med. 2019, 11, eaav4508. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.J.; Buss, M.K.; Nattam, S.R.; Hurteau, J.; et al. Overall survival and updated progression-free survival outcomes in a randomized phase II study of combination cediranib and olaparib versus olaparib in relapsed platinum-sensitive ovarian cancer. Ann. Oncol. 2019, 30, 551–557. [Google Scholar] [CrossRef]
- Liu, J.F.; Brady, M.F.; Matulonis, U.A.; Miller, A.; Kohn, E.C.; Swisher, E.M.; Tew, W.P.; Cloven, N.G.; Muller, C.; Bender, D.; et al. A phase III study comparing single-agent olaparib or the combination of cediranib and olaparib to standard platinum-based chemotherapy in recurrent platinum-sensitive ovarian cancer. J. Clin. Oncol. 2020, 38, 6003. [Google Scholar] [CrossRef]
- Datta, M.; Coussens, L.M.; Nishikawa, H.; Hodi, F.S.; Jain, R.K. Reprogramming the Tumor Microenvironment to Improve Immunotherapy: Emerging Strategies and Combination Therapies. Am. Soc. Clin. Oncol. Educ. B. 2019, 39, 165–174. [Google Scholar] [CrossRef]
- Jain, R.K. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef] [Green Version]
- Stecklein, S.R.; Kumaraswamy, E.; Behbod, F.; Wang, W.; Chaguturu, V.; Harlan-Williams, L.M.; Jensen, R.A. BRCA1 and HSP90 cooperate in homologous and non-homologous DNA double-strand-break repair and G2/M checkpoint activation. Proc. Natl. Acad. Sci. USA 2012, 109, 13650–13655. [Google Scholar] [CrossRef] [Green Version]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Publ. Gr. 2017, 18. [Google Scholar] [CrossRef]
- Gabbasov, R.; Benrubi, I.D.; O’Brien, S.W.; Krais, J.J.; Johnson, N.; Litwin, S.; Connolly, D.C. Targeted blockade of HSP90 impairs DNA-damage response proteins and increases the sensitivity of ovarian carcinoma cells to PARP inhibition. Cancer Biol. Ther. 2019, 20, 1035–1045. [Google Scholar] [CrossRef]
- Juvekar, A.; Hu, H.; Yadegarynia, S.; Lyssiotis, C.A.; Ullas, S.; Lien, E.C.; Bellinger, G.; Son, J.; Hok, R.C.; Seth, P.; et al. Phosphoinositide 3-kinase inhibitors induce DNA damage through nucleoside depletion. Proc. Natl. Acad. Sci. USA 2016, 113, E4338–E4347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, Y.H.; García-García, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzmán, M.; Grueso, J.; et al. PI3K Inhibition Impairs BRCA1/2 Expression and Sensitizes BRCA-Proficient Triple-Negative Breast Cancer to PARP Inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juvekar, A.; Burga, L.N.; Hu, H.; Lunsford, E.P.; Ibrahim, Y.H.; Balmañà, J.; Rajendran, A.; Papa, A.; Spencer, K.; Lyssiotis, C.A.; et al. Combining a PI3K Inhibitor with a PARP Inhibitor Provides an Effective Therapy for BRCA1-Related Breast Cancer. Cancer Discov. 2012, 2, 1048–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehman, F.L.; Lord, C.J.; Ashworth, A. The Promise of Combining Inhibition of PI3K and PARP as Cancer Therapy. Cancer Discov. 2012, 2, 982–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matulonis, U.A.; Wulf, G.M.; Barry, W.T.; Birrer, M.; Westin, S.N.; Farooq, S.; Bell-McGuinn, K.M.; Obermayer, E.; Whalen, C.; Spagnoletti, T.; et al. Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high-grade serous ovarian and breast cancer. Ann. Oncol. 2017, 28, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Barry, W.T.; Birrer, M.; Westin, S.N.; Cadoo, K.A.; Shapiro, G.I.; Mayer, E.L.; O’Cearbhaill, R.E.; Coleman, R.L.; Kochupurakkal, B.; et al. Olaparib and α-specific PI3K inhibitor alpelisib for patients with epithelial ovarian cancer: A dose-escalation and dose-expansion phase 1b trial. Lancet Oncol. 2019, 20, 570–580. [Google Scholar] [CrossRef]
- Yap, T.A.; Kristeleit, R.; Michalarea, V.; Pettitt, S.J.; Lim, J.S.J.; Carreira, S.; Roda, D.; Miller, R.; Riisnaes, R.; Miranda, S.; et al. Phase I trial of the poly(ADP-ribose) polymerase (PARP) inhibitor olaparib and AKT inhibitor capivasertib in patients with BRCA1/2 and non-BRCA1/2 mutant cancers. Cancer Discov. 2020, CD-20-0163. [Google Scholar] [CrossRef]
- Labrie, M.; Ju, Z.; Litton, J.K.; Kim, T.-B.; Lee, S.; Chen, K.; Soliman, P.T.; Frumovitz, M.; Meyer, L.A.; Moulder, S.; et al. Abstract 2070: Exploration of markers of synergistic lethality of PARP and PI3K-Akt-mTOR inhibitors in women’s cancers. Cancer Res. 2019, 79 (Suppl. 2070), 13. [Google Scholar] [CrossRef]
- Sun, C.; Fang, Y.; Yin, J.; Chen, J.; Ju, Z.; Zhang, D.; Chen, X.; Vellano, C.P.; Jeong, K.J.; Ng, P.K.S.; et al. Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Vena, F.; Jia, R.; Esfandiari, A.; Garcia-Gomez, J.J.; Rodriguez-Justo, M.; Ma, J.; Syed, S.; Crowley, L.; Elenbaas, B.; Goodstal, S.; et al. MEK inhibition leads to BRCA2 downregulation and sensitization to DNA damaging agents in pancreas and ovarian cancer models. Oncotarget 2018, 9, 11592–11603. [Google Scholar] [CrossRef] [Green Version]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.M.; Nguyen, H.D.; Ho, C.K.; Kwan, T.T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Ning, J.F.; Stanciu, M.; Humphrey, M.R.; Gorham, J.; Wakimoto, H.; Nishihara, R.; Lees, J.; Zou, L.; Martuza, R.L.; Wakimoto, H.; et al. Myc targeted CDK18 promotes ATR and homologous recombination to mediate PARP inhibitor resistance in glioblastoma. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Burgess, B.T.; Anderson, A.M.; McCorkle, J.R.; Wu, J.; Ueland, F.R.; Kolesar, J.M. Olaparib combined with an ATR or Chk1 inhibitor as a treatment strategy for acquired olaparib-resistant BRCA1 mutant ovarian cells. Diagnostics 2020, 10, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wengner, A.M.; Siemeister, G.; Lucking, U.; Lefranc, J.; Wortmann, L.; Lienau, P.; Bader, B.; Bomer, U.; Moosmayer, D.; Eberspacher, U.; et al. The novel ATR inhibitor BAY 1895344 is efficacious as monotherapy and combined with DNA damage—inducing or repair—compromising therapies in preclinical cancer models. Mol. Cancer Ther. 2020, 19, 26–38. [Google Scholar] [CrossRef] [Green Version]
- Schoonen, P.M.; Kok, Y.P.; Wierenga, E.; Bakker, B.; Foijer, F.; Spierings, D.C.J.; van Vugt, M.A.T.M. Premature mitotic entry induced by ATR inhibition potentiates olaparib inhibition-mediated genomic instability, inflammatory signaling, and cytotoxicity in BRCA2-deficient cancer cells. Mol. Oncol. 2019, 13, 2422–2440. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; George, E.; Ragland, R.L.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.A.; Herlyn, M.; Brown, E.J.; Simpkins, F. Targeting the ATR/CHK1 axis with PARP inhibition results in tumor regression in BRCA-mutant ovarian cancer models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef] [Green Version]
- Brill, E.; Yokoyama, T.; Nair, J.; Yu, M.; Ahn, Y.R.; Lee, J.M. Prexasertib, a cell cycle checkpoint kinases 1 and 2 inhibitor, increases in vitro toxicity of PARP inhibition by preventing Rad51 foci formation in BRCA wild type high-grade serous ovarian cancer. Oncotarget 2017, 8, 111026–111040. [Google Scholar] [CrossRef]
- Parmar, K.; Kochupurakkal, B.S.; Lazaro, J.B.; Wang, Z.C.; Palakurthi, S.; Kirschmeier, P.T.; Yang, C.; Sambel, L.A.; Farkkila, A.; Reznichenko, E.; et al. The CHK1 inhibitor prexasertib exhibits monotherapy activity in high-grade serous ovarian cancer models and sensitizes to PARP inhibition. Clin. Cancer Res. 2019, 25, 6127–6140. [Google Scholar] [CrossRef]
- Do, K.T.; Hill, S.J.; Kochupurakkal, B.; Supko, J.G.; Gannon, C.; Anderson, A.; Muzikansky, A.; Wolanski, A.; Hedglin, J.; Parmar, K.; et al. Abstract CT232: Phase I combination study of the CHK1 inhibitor prexasertib (LY2606368) and olaparib in patients with high-grade serous ovarian cancer and other advanced solid tumors. Cancer Res. 2019, 79 (Suppl. CT232), 13. [Google Scholar] [CrossRef]
- Karnak, D.; Engelke, C.G.; Parsels, L.A.; Kausar, T.; Wei, D.; Robertson, J.R.; Marsh, K.B.; Davis, M.A.; Zhao, L.; Maybaum, J.; et al. Combined inhibition of Wee1 and PARP1/2 for radiosensitization in pancreatic cancer. Clin. Cancer Res. 2014, 20, 5085–5096. [Google Scholar] [CrossRef] [Green Version]
- Parsels, L.A.; Karnak, D.; Parsels, J.D.; Zhang, Q.; Velez-Padilla, J.; Reichert, Z.R.; Wahl, D.R.; Maybaum, J.; O’Connor, M.J.; Lawrence, T.S.; et al. PARP1 Trapping and DNA Replication Stress Enhance Radiosensitization with Combined WEE1 and PARP Inhibitors. Mol. Cancer Res. 2018, 16, 222–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; McGrail, D.J.; Sun, C.; Labrie, M.; Chen, X.; Zhang, D.; Ju, Z.; Vellano, C.P.; Lu, Y.; Li, Y.; et al. Sequential Therapy with PARP and WEE1 Inhibitors Minimizes Toxicity while Maintaining Efficacy. Cancer Cell 2019, 35, 851–867.e7. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.; Falchook, G.S.; Wang, J.S.; Fu, S.; Oza, A.; Karen, S.; Imedio, E.R.; Kumar, S.; Ottesen, L.; Mugundu, G.M.; et al. Abstract CT025: Phase Ib study of adavosertib in combination with olaparib in patients with refractory solid tumors: Dose escalation. Cancer Res. 2019, 79, CT025. [Google Scholar] [CrossRef]
- Sun, C.; Yin, J.; Fang, Y.; Chen, J.; Jeong, K.J.; Chen, X.; Vellano, C.P.; Ju, Z.; Zhao, W.; Zhang, D.; et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell 2018, 33, 401–416.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, A.J.; Stubbs, M.; Liu, P.; Ruggeri, B.; Khabele, D. The BET inhibitor INCB054329 reduces homologous recombination efficiency and augments PARP inhibitor activity in ovarian cancer. Gynecol. Oncol. 2018, 149, 575–584. [Google Scholar] [CrossRef]
- Krajewska, M.; Dries, R.; Grassetti, A.V.; Dust, S.; Gao, Y.; Huang, H.; Sharma, B.; Day, D.S.; Kwiatkowski, N.; Pomaville, M.; et al. CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat. Commun. 2019, 10, 1757. [Google Scholar] [CrossRef] [Green Version]
- Dubbury, S.J.; Boutz, P.L.; Sharp, P.A. CDK12 regulates DNA repair genes by suppressing intronic polyadenylation. Nature 2018, 564, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Chirackal Manavalan, A.P.; Pilarova, K.; Kluge, M.; Bartholomeeusen, K.; Rajecky, M.; Oppelt, J.; Khirsariya, P.; Paruch, K.; Krejci, L.; Friedel, C.C.; et al. CDK12 controls G1/S progression by regulating RNAPII processivity at core DNA replication genes. EMBO Rep. 2019, 20, e47592. [Google Scholar] [CrossRef]
- Ekumi, K.M.; Paculova, H.; Lenasi, T.; Pospichalova, V.; Osken, C.A.B.; Rybarikova, J.; Bryja, V.; Geyer, M.; Blazek, D.; Barboric, M. Ovarian carcinoma CDK12 mutations misregulate expression of DNA repair genes via deficient formation and function of the Cdk12/CycK complex. Nucleic Acids Res. 2015, 43, 2575–2589. [Google Scholar] [CrossRef]
- Joshi, P.M.; Sutor, S.L.; Huntoon, C.J.; Karnitz, L.M. Ovarian cancer-associated mutations disable catalytic activity of CDK12, a kinase that promotes homologous recombination repair and resistance to cisplatin and poly(ADP-ribose) polymerase inhibitors. J. Biol. Chem. 2014, 289, 9247–9253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quereda, V.; Bayle, S.; Vena, F.; Frydman, S.M.; Monastyrskyi, A.; Roush, W.R.; Duckett, D.R. Therapeutic Targeting of CDK12/CDK13 in Triple-Negative Breast Cancer. Cancer Cell 2019, 36, 545–558.e7. [Google Scholar] [CrossRef] [PubMed]
- Bajrami, I.; Frankum, J.R.; Konde, A.; Miller, R.E.; Rehman, F.L.; Brough, R.; Campbell, J.; Sims, D.; Rafiq, R.; Hooper, S.; et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 2014, 74, 287–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8 + T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Chabanon, R.M.; Lord, C.J.; Postel-Vinay, S. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer The Journal of Clinical Investigation. J Clin Invest 2019, 129. [Google Scholar] [CrossRef]
- Huang, J.; Wang, L.; Cong, Z.; Amoozgar, Z.; Kiner, E.; Xing, D.; Orsulic, S.; Matulonis, U.; Goldberg, M.S. The PARP1 inhibitor BMN 673 exhibits immunoregulatory effects in a Brca1-/- murine model of ovarian cancer. Biochem. Biophys. Res. Commun. 2015, 463, 551–556. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, K.; Xiao, Y.; Feng, B.; Mikule, K.; Ma, X.Y.; Feng, N.; Vellano, C.P.; Federico, L.; Marszalek, J.R.; et al. Niraparib activates interferon signaling and potentiates anti-PD-1 antibody efficacy in tumor models. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Kim, H.J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef] [Green Version]
- Kis-Toth, K.; Szanto, A.; Thai, T.-H.; Tsokos, G.C. Cytosolic DNA-Activated Human Dendritic Cells Are Potent Activators of the Adaptive Immune Response. J. Immunol. 2011, 187, 1222–1234. [Google Scholar] [CrossRef] [Green Version]
- Reisländer, T.; Lombardi, E.P.; Groelly, F.J.; Miar, A.; Porru, M.; Di Vito, S.; Wright, B.; Lockstone, H.; Biroccio, A.; Harris, A.; et al. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Li, L.; Chen, L.; Yuan, W.; Dong, L.; Zhang, Y.; Wu, H.; Wang, C. PARP-1 Mediates LPS-Induced HMGB1 Release by Macrophages through Regulation of HMGB1 Acetylation. J. Immunol. 2014, 193, 6114–6123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ditsworth, D.; Zong, W.X.; Thompson, C.B. Activation of poly(ADP)-ribose polymerase (PARP-1) induces release of the pro-inflammatory mediator HMGB1 from the nucleus. J. Biol. Chem. 2007, 282, 17845–17854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, M.; Porat, A.; He, M.; Suurmond, J.; Santiago-Schwarz, F.; Andersson, U.; Coleman, T.R.; Volpe, B.T.; Tracey, K.J.; Al-Abed, Y.; et al. C1q and HMGB1 reciprocally regulate human macrophage polarization. Blood 2016, 128, 2218–2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Liu, L.; Xie, M.; Sun, X.; Yu, Y.; Kang, R.; Yang, L.; Zhu, S.; Cao, L.; Tang, D. Poly-ADP-ribosylation of HMGB1 regulates TNFSF10/TRAIL resistance through autophagy. Autophagy 2015, 11, 214–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.W.; Koh, B.D.; Zhang, J.S.; Flatten, K.S.; Schneider, P.A.; Billadeau, D.D.; Hess, A.D.; Smith, B.D.; Karp, J.E.; Kaufmann, S.H. Poly(ADP-ribose) polymerase inhibitors sensitize cancer cells to death receptor-mediated apoptosis by enhancing death receptor expression. J. Biol. Chem. 2014, 289, 20543–20558. [Google Scholar] [CrossRef] [Green Version]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [Green Version]
- Li, C.W.; Lim, S.O.; Xia, W.; Lee, H.H.; Chan, L.C.; Kuo, C.W.; Khoo, K.H.; Chang, S.S.; Cha, J.H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination with Pembrolizumab in Patients with Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef] [Green Version]
- Lampert, E.J.; Zimmer, A.S.; Padget, M.R.; Cimino-Mathews, A.; Nair, J.R.; Liu, Y.; Swisher, E.M.; Hodge, J.W.; Nixon, A.B.; Nichols, E.; et al. Combination of PARP inhibitor olaparib, and PD-L1 inhibitor durvalumab, in recurrent ovarian cancer: A proof-of-concept phase 2 study. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Asante, D.B.; Calapre, L.; Ziman, M.; Meniawy, T.M.; Gray, E.S. Liquid biopsy in ovarian cancer using circulating tumor DNA and cells: Ready for prime time? Cancer Lett. 2020, 468, 59–71. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, E.K.; Matulonis, U.A. PARP Inhibitor Resistance Mechanisms and Implications for Post-Progression Combination Therapies. Cancers 2020, 12, 2054. https://doi.org/10.3390/cancers12082054
Lee EK, Matulonis UA. PARP Inhibitor Resistance Mechanisms and Implications for Post-Progression Combination Therapies. Cancers. 2020; 12(8):2054. https://doi.org/10.3390/cancers12082054
Chicago/Turabian StyleLee, Elizabeth K., and Ursula A. Matulonis. 2020. "PARP Inhibitor Resistance Mechanisms and Implications for Post-Progression Combination Therapies" Cancers 12, no. 8: 2054. https://doi.org/10.3390/cancers12082054
APA StyleLee, E. K., & Matulonis, U. A. (2020). PARP Inhibitor Resistance Mechanisms and Implications for Post-Progression Combination Therapies. Cancers, 12(8), 2054. https://doi.org/10.3390/cancers12082054