Innovative CAR-T Cell Therapy for Solid Tumor; Current Duel between CAR-T Spear and Tumor Shield



Abstract
:1. Introduction
2. Overview of CAR-T Cells
3. Limiting Factors for CAR-T Cell Therapy of Solid Tumors
3.1. CAR-T Cell Trafficking to the Tumor Site
3.2. CAR-T Cell Infiltration into the Tumor Bed
3.3. Immunosuppressive Tumor Microenvironment
3.3.1. Immune Suppressor Cells
3.3.2. Immune Checkpoints
3.3.3. Reactive Oxygen Species (ROS)
3.3.4. Metabolites
3.3.5. Cytokines
3.3.6. pH
3.3.7. Hypoxia
3.4. Shortage of Tumor Antigens
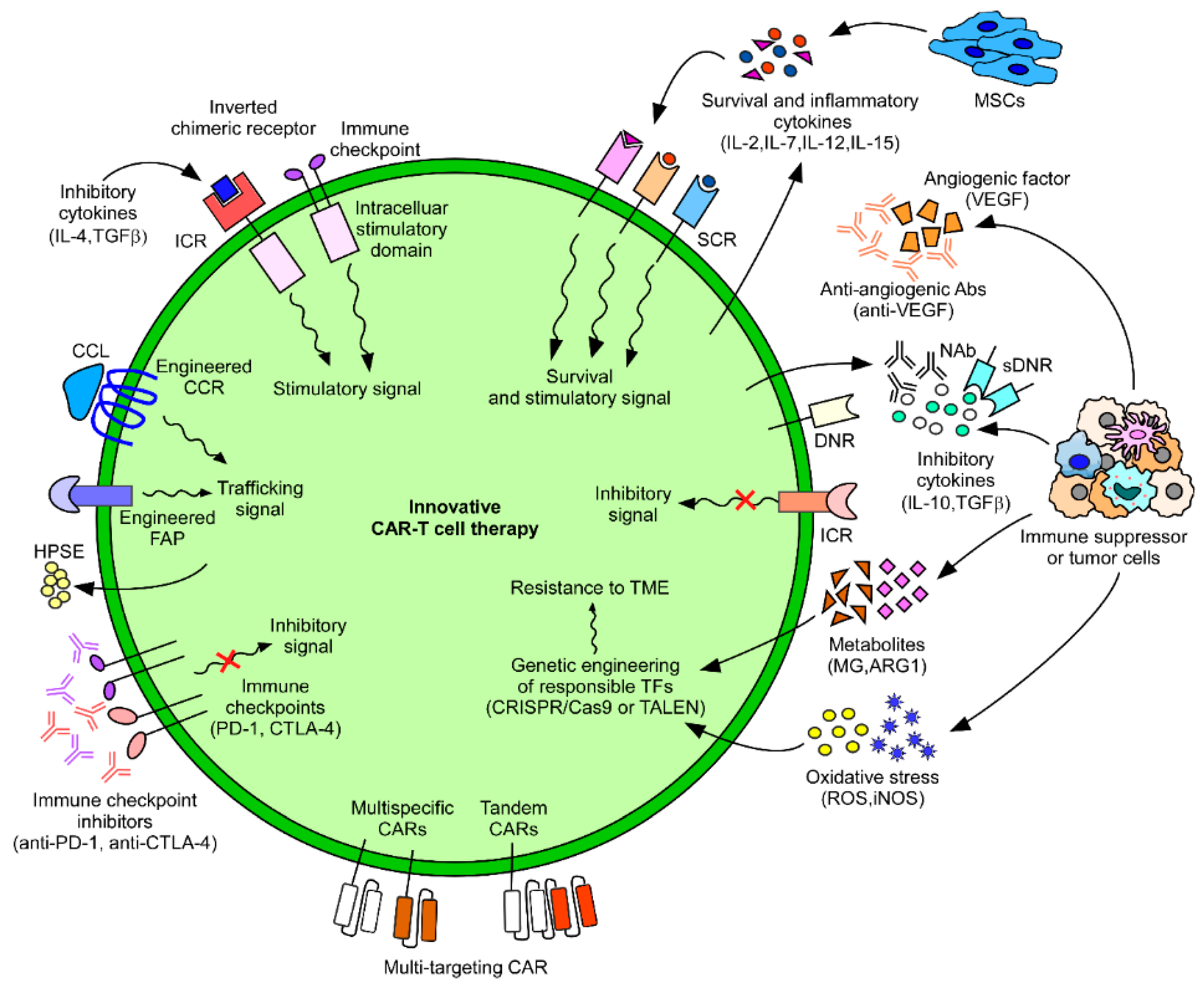
4. Approach to Improve CAR-T Cell Therapy for Solid Tumors
4.1. CAR-T Cell Trafficking and Infiltration
4.2. CAR-T Cell Resistance to the TME
4.3. Multi-Targeting CAR-T Cells
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Baker, S.J.; Reddy, E.P. Targeted inhibition of kinases in cancer therapy. Mt. Sinai J. Med. A J. Transl. Pers. Med. 2010, 77, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Lepore, M.; Mori, L.; De Libero, G. The conventional nature of non-MHC-Restricted T cells. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Miliotou, A.; Papadopoulou, L.C.; Androulla, M.N.; Lefkothea, P.C. CAR T-cell therapy: A new era in cancer immunotherapy. Curr. Pharm. Biotechnol. 2018, 19, 5–18. [Google Scholar] [CrossRef]
- Orlando, E.; Leary, R.; Lacey, S.F.; Fraietta, J.; Bedoya, F.; Ambrose, D.; Wilcox, N.; Maude, S.L.; Frey, N.V.; Levine, B.L.; et al. Gene expression signatures of response to anti-CD19 chimeric antigen receptor (CAR) T-cell therapy in patients with CLL and ALL. J. Clin. Oncol. 2017, 35, 137. [Google Scholar] [CrossRef]
- Liu, B.; Yan, L.; Zhou, M. Target selection of CAR T cell therapy in accordance with the TME for solid tumors. Am. J. Cancer Res. 2019, 9, 228–241. [Google Scholar]
- Ma, S.; Li, X.; Wang, X.; Cheng, L.; Li, Z.; Zhang, C.; Ye, Z.; Qian, Q. Current progress in CAR-T cell therapy for solid tumors. Int. J. Biol. Sci. 2019, 15, 2548–2560. [Google Scholar] [CrossRef] [Green Version]
- Stern, L.A.; Jonsson, V.D.; Priceman, S. CAR T Cell Therapy progress and challenges for solid tumors. Infect. Complicat. Cancer Patients 2020, 180, 297–326. [Google Scholar]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Guedan, S.; Calderon, H.; Posey, A.D.; Maus, M.V. Engineering and design of chimeric antigen receptors. Mol. Ther. Methods Clin. Dev. 2018, 12, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting costimulatory domains for chimeric antigen receptors: Functional and clinical considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef] [Green Version]
- Picanço-Castro, V.; Moço, P.; Mizukami, A.; Vaz, L.D.; Pereira, M.D.S.F.; Silvestre, R.N.; De Azevedo, J.T.C.; Bomfim, A.D.S.; Neto, M.S.D.A.; Malmegrim, K.C.R.; et al. Establishment of a simple and efficient platform for car-t cell generation and expansion: From lentiviral production to in vivo studies. Hematol. Transfus. Cell Ther. 2020, 42, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Izsvak, Z.; Hackett, P.B.; Cooper, L.J.; Ivics, Z. Translating Sleeping Beauty transposition into cellular therapies: Victories and challenges. BioEssays 2010, 32, 756–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep. 2017, 20, 3025–3033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spolski, R.; Leonard, W.J. Interleukin-21: A double-edged sword with therapeutic potential. Nat. Rev. Drug Discov. 2014, 13, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Morgan, R.A.; Beane, J.; Zheng, Z.; Dudley, M.E.; Kassim, S.H.; Nahvi, A.V.; Ngo, L.T.; Sherry, R.M.; Phan, G.Q.; et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin. Cancer Res. 2015, 21, 2278–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadouli, I.; Mueller-Berghaus, J.; Beuneu, C.; Ali, S.; Hofner, B.; Petavy, F.; Tzogani, K.; Miermont, A.; Norga, K.; Kholmanskikh, O.; et al. EMA Review of Axicabtagene Ciloleucel (Yescarta) for the treatment of diffuse large B-Cell Lymphoma. Oncology 2020. [Google Scholar] [CrossRef]
- Ali, S.; Kjeken, R.; Niederlaender, C.; Markey, G.; Saunders, T.S.; Opsata, M.; Moltu, K.; Bremnes, B.; Grønevik, E.; Muusse, M.; et al. The european medicines agency review of Kymriah (Tisagenlecleucel) for the treatment of acute Lymphoblastic Leukemia and diffuse large B-cell Lymphoma. Oncology 2019, 25, e321–e327. [Google Scholar] [CrossRef] [Green Version]
- Boissonnas, A.; Fetler, L. In vivo imaging of cytotoxic T cell infiltration and elimination of a solid tumor. J. Exp. Med. 2007, 204, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Craddock, J.A.; Lu, A.; Bear, A.; Pule, M.; Brenner, M.K.; Rooney, C.M.; Foster, A.E. Enhanced tumor trafficking of GD2 Chimeric antigen receptor T cells by expression of the Chemokine receptor CCR2b. J. Immunother. 2010, 33, 780–788. [Google Scholar] [CrossRef] [Green Version]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8+ T-Cell recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef] [Green Version]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.-C.S.; Kapoor, V.; Predina, J.; Powell, D.J.; Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef] [Green Version]
- Idorn, M.; Straten, P. Chemokine receptors and exercise to tackle the inadequacy of T cell homing to the tumor site. Cells 2018, 7, 108. [Google Scholar] [CrossRef] [Green Version]
- Slaney, C.Y.; Kershaw, M.; Darcy, P.K. Trafficking of T cells into tumors. Cancer Res. 2014, 74, 7168–7174. [Google Scholar] [CrossRef] [Green Version]
- Griffioen, A.W. Anti-angiogenesis: Making the tumor vulnerable to the immune system. Cancer Immunol. Immunother. 2008, 57, 1553–1558. [Google Scholar] [CrossRef] [Green Version]
- Schaaf, M.B.; Garg, A.D.; Agostinis, P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 2018, 9, 115. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Endres, S.; Kobold, S. Enhancing tumor T cell infiltration to enable cancer immunotherapy. Immunotheraphy 2019, 11, 201–213. [Google Scholar] [CrossRef]
- Cho, A.; Howell, V.M.; Colvin, E.K. The extracellular matrix in epithelial ovarian cancer—A piece of a puzzle. Front. Oncol. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Kerbel, R.S. Reappraising antiangiogenic therapy for breast cancer. Breast 2011, 20, S56–S60. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Zinzindohoué, F.; Bruneval, P.; Cugnenc, P.-H.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Pickup, M.W.; Weaver, V. From transformation to metastasis: Deconstructing the extracellular matrix in breast cancer. Cancer Metastasis Rev. 2016, 35, 655–667. [Google Scholar] [CrossRef]
- Kim, S.T.; Jeong, H.; Woo, O.H.; Seo, J.H.; Kim, A.; Lee, E.S.; Shin, S.W.; Kim, Y.H.; Kim, J.S.; Park, K.H. Tumor-infiltrating Lymphocytes, tumor characteristics, and recurrence in patients with early breast cancer. Am. J. Clin. Oncol. 2013, 36, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, J.; Poli, A.; Brons, N.H.; Waha, A.; Eide, G.E.; Enger, P. Øyvind; Zimmer, J.; Chekenya, M. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J. Neuroimmunol. 2013, 264, 71–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wientjes, M.G.; Au, J.L.-S. Pancreatic cancer: Pathobiology, treatment options, and drug delivery. AAPS J. 2010, 12, 223–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piersma, S.J.; Jordanova, E.S.; Van Poelgeest, M.I.; Kwappenberg, K.M.; Van Der Hulst, J.M.; Drijfhout, J.W.; Melief, C.J.; Kenter, G.; Fleuren, G.J.; Offringa, R.; et al. High number of intraepithelial CD8+ tumor-infiltrating lymphocytes is associated with the absence of lymph node metastases in patients with large early-stage cervical cancer. Cancer Res. 2007, 67, 354–361. [Google Scholar] [CrossRef] [Green Version]
- Zhang, E.; Gu, J.; Xu, H. Prospects for chimeric antigen receptor-modified T cell therapy for solid tumors. Mol. Cancer 2018, 17, 7. [Google Scholar] [CrossRef] [Green Version]
- Martinet, L.; Le Guellec, S.; Filleron, T.; Lamant, L.; Meyer, N.; Rochaix, P.; Garrido, I.; Girard, J.-P. High endothelial venules (HEVs) in human melanoma lesions. OncoImmunology 2012, 1, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Digre, A.; Singh, K.; Åbrink, M.; Reijmers, R.M.; Sandler, S.; Vlodavsky, I.; Li, J.-P. Overexpression of heparanase enhances T lymphocyte activities and intensifies the inflammatory response in a model of murine rheumatoid arthritis. Sci. Rep. 2017, 7, 46229. [Google Scholar] [CrossRef] [Green Version]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef]
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010, 70, 6171–6180. [Google Scholar] [CrossRef] [Green Version]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef] [Green Version]
- Burga, R.A.; Thorn, M.; Point, G.R.; Guha, P.; Nguyen, C.T.; Licata, L.A.; DeMatteo, R.P.; Ayala, A.; Espat, N.J.; Junghans, R.P.; et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol. Immunother. 2015, 64, 817–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.-P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and Treg cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T cell Therapy for solid tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Ahmadzadeh, M.; Lu, Y.-C.; Liewehr, D.J.; Dudley, M.E.; Liu, F.; Schrump, D.S.; Steinberg, S.M.; Rosenberg, S.A.; Robbins, P.F. Levels of peripheral CD4+FoxP3+ regulatory T cells are negatively associated with clinical response to adoptive immunotherapy of human cancer. Blood 2012, 119, 5688–5696. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Kang, C.; Jeong, S.-Y.; Song, S.Y.; Choi, E.K. The emerging role of myeloid-derived suppressor cells in radiotherapy. Radiat. Oncol. J. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Tumor-associated macrophages. Curr. Biol. 2020, 30, R246–R248. [Google Scholar] [CrossRef]
- Kosti, P.; Maher, J.; Arnold, J.N. Perspectives on Chimeric antigen receptor T-cell immunotherapy for solid tumors. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized functions in immune regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [Green Version]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, differences, and implications of their inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Parry, R.V.; Chemnitz, J.M. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.A.; June, C.H. Driving gene-engineered T cell immunotherapy of cancer. Cell Res. 2016, 27, 38–58. [Google Scholar] [CrossRef] [Green Version]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T-cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Mirzaei, H.R.; Rodriguez, A.; Shepphird, J.; Brown, C.; Badie, B. Chimeric Antigen Receptors T cell therapy in solid tumor: Challenges and clinical applications. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Butte, M.J.; Pena-Cruz, V.; Kim, M.-J.; Freeman, G.J.; Sharpe, A.H. Interaction of human PD-L1 and B7-1. Mol. Immunol. 2008, 45, 3567–3572. [Google Scholar] [CrossRef] [Green Version]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the Pd-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Sun, Q.; Zhang, X. PD-1 and its ligands are important immune checkpoints in cancer. Oncotarget 2016, 8, 2171–2186. [Google Scholar] [CrossRef] [Green Version]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [Green Version]
- Chambers, C.A.; Kuhns, M.S. CTLA-4-mediated inhibition in regulation of T cell responses: Mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594. [Google Scholar] [CrossRef] [Green Version]
- Egen, J.G.; Kuhns, M.S.; Allison, J.P. CTLA-4: New insights into its biological function and use in tumor immunotherapy. Nat. Immunol. 2002, 3, 611–618. [Google Scholar] [CrossRef]
- McGowan, E.; Lin, Q.; Ma, G.; Yin, H.; Chen, S.; Lin, Y. PD-1 disrupted CAR-T cells in the treatment of solid tumors: Promises and challenges. Biomed. Pharmacother. 2020, 121, 109625. [Google Scholar] [CrossRef] [PubMed]
- Joller, N.; Kuchroo, V.K. Tim-3, Lag-3, and TIGIT. Curr. Top. Microbiol. Immunol. 2017, 410, 127–156. [Google Scholar] [PubMed] [Green Version]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free. Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, F.; Ramnath, N.; Nagrath, D. Reactive oxygen species in the tumor microenvironment: An overview. Cancers 2019, 11, 1191. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Guo, J.; Weng, L.; Tang, W.; Jin, S.; Ma, W. Myeloid-derived suppressor cells—New and exciting players in lung cancer. J. Hematol. Oncol. 2020, 13, 1–17. [Google Scholar] [CrossRef]
- Ohl, K.; Tenbrock, K. Reactive oxygen species as regulators of MDSC-mediated immune suppression. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Corzo, C.A.; Cotter, M.J.; Cheng, P.; Cheng, F.; Kusmartsev, S.; Sotomayor, E.; Padhya, T.; McCaffrey, T.V.; McCaffrey, J.C.; Gabrilovich, D.I. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J. Immunol. 2009, 182, 5693–5701. [Google Scholar] [CrossRef]
- Mazzoni, A.; Bronte, V.; Visintin, A.; Spitzer, J.H.; Apolloni, E.; Serafini, P.; Zanovell, P.; Segal, D.M. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J. Immunol. 2002, 168, 689–695. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 1–13. [Google Scholar] [CrossRef]
- Klaunig, J.E. Oxidative Stress and Cancer. Curr. Pharm. Des. 2019, 24, 4771–4778. [Google Scholar] [CrossRef]
- Baumann, T.; Dunkel, A.; Schmid, C.; Schmitt, S.; Hiltensperger, M.; Lohr, K.; Laketa, V.; Donakonda, S.; Ahting, U.; Lorenz-Depiereux, B.; et al. Regulatory myeloid cells paralyze T cells through cell-cell transfer of the metabolite methylglyoxal. Nat. Immunol. 2020, 21, 555–566. [Google Scholar] [CrossRef] [PubMed]
- De La Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arab, S.; Hadjati, J. Adenosine blockage in tumor microenvironment and improvement of cancer immunotherapy. Immune Netw. 2019, 19, e23. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Xia, J. MiR-153 suppresses IDO1 expression and enhances CAR T cell immunotherapy. J. Hematol. Oncol. 2018, 11, 58. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, P.C. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [Green Version]
- Bronte, V.; Serafini, P.; Mazzoni, A.; Segal, D.M.; Zanovell, P. L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol. 2003, 24, 301–305. [Google Scholar] [CrossRef]
- Cheng, J.; Zhao, L.; Zhang, Y.; Qin, Y.; Guan, Y.; Zhang, T.; Liu, C.; Zhou, J. Understanding the mechanisms of resistance to CAR T-Cell therapy in malignancies. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Latchoumanin, O.; Wu, G.; Zhou, G.; Hebbard, L.; George, J.; Qiao, L. Recent clinical trials utilizing chimeric antigen receptor T cells therapies against solid tumors. Cancer Lett. 2017, 390, 188–200. [Google Scholar] [CrossRef]
- Juillerat, A.; Marechal, A.; Filhol, J.M.; Valogne, Y.; Valton, J.; Duclert, A.; Duchateau, P.; Poirot, L. An oxygen sensitive self-decision making engineered CAR T-cell. Sci. Rep. 2017, 7, 39833. [Google Scholar] [CrossRef]
- Bollong, M.J.; Lee, G.; Coukos, J.S.; Yun, H.; Zambaldo, C.; Chang, J.W.; Chin, E.N.; Ahmad, I.; Chatterjee, A.K.; Lairson, L.L.; et al. A metabolite-derived protein modification integrates glycolysis with KEAP1–NRF2 signalling. Nature 2018, 562, 600–604. [Google Scholar] [CrossRef]
- Nokin, M.J.; Durieux, F. Methylglyoxal, a glycolysis side-product, induces Hsp90 glycation and YAP-mediated tumor growth and metastasis. eLife 2016, 5, e19375. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Gnanaprakasam, J.N.R.; Sherman, J.; Wang, R. A Metabolism toolbox for CAR T therapy. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollard, C.M.; Tripic, T.; Cruz, C.R.; Dotti, G.; Gottschalk, S.; Torrano, V.; Dakhova, O.; Carrum, G.; Ramos, C.A.; Liu, H.; et al. Tumor-specific T-cells engineered to overcome tumor immune evasion induce clinical responses in patients with relapsed hodgkin lymphoma. J. Clin. Oncol. 2018, 36, 1128–1139. [Google Scholar] [CrossRef] [PubMed]
- Chinnasamy, D.; Yu, Z.; Kerkar, S.P.; Zhang, L.; Morgan, R.A.; Restifo, N.P.; Rosenberg, S.A. Local delivery of lnterleukin-12 Using T cells targeting VEGF Receptor-2 Eradicates multiple vascularized tumors in mice. Clin. Cancer Res. 2012, 18, 1672–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloss, C.C.; Lee, J. Dominant-negative TGF-beta receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [Green Version]
- Koneru, M.; Purdon, T.; Spriggs, D.; Koneru, S.; Brentjens, R.J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. OncoImmunology 2015, 4, e994446. [Google Scholar] [CrossRef] [Green Version]
- Wallace, A.; Kapoor, V.; Sun, J.; Mrass, P.; Weninger, W.; Heitjan, D.F.; June, C.; Kaiser, L.R.; Ling, L.E.; Albelda, S.M. Transforming growth factor-beta receptor blockade augments the effectiveness of adoptive T-cell therapy of established solid cancers. Clin. Cancer Res. 2008, 14, 3966–3974. [Google Scholar] [CrossRef] [Green Version]
- Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-derived suppressor cells as a therapeutic target for cancer. Cells 2020, 9, 561. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.; Verhagen, J.; Blaser, K.; Akdis, M.; Akdis, C.A. Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: The role of T regulatory cells. Immunology 2006, 117, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFbeta in cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Yeh, H.-W.; Lee, S.-S.; Chang, C.-Y.; Lang, Y.-D.; Jou, Y.-S. A new switch for TGFβ in cancer. Cancer Res. 2019, 79, 3797–3805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahmani, A.; Delisle, J.-S. TGF-β in T cell biology: Implications for cancer immunotherapy. Cancers 2018, 10, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennis, K.L.; Blatner, N.R.; Gounari, F.; Khazaie, K. Current status of interleukin-10 and regulatory T-cells in cancer. Curr. Opin. Oncol. 2013, 25, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Hong, I.-S. Stimulatory versus suppressive effects of GM-CSF on tumor progression in multiple cancer types. Exp. Mol. Med. 2016, 48, e242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, K.M.; Wojtkowiak, J.W.; Hashim, A.I.; Gillies, R.J. Targeting the metabolic microenvironment of tumors. HIV-1 Mol. Biol. Pathog. 2012, 65, 63–107. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Lin, Y.; Gillies, R.J. Tumor pH and its measurement. J. Nucl. Med. 2010, 51, 1167–1170. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Chen, X.; Cao, J.; Gao, H. Overcoming the biological barriers in the tumor microenvironment for improving drug delivery and efficacy. J. Mater. Chem. B 2020. [Google Scholar] [CrossRef]
- Kato, Y.; Ozawa, S.; Miyamoto, C.; Maehata, Y.; Suzuki, A.; Maeda, T.; Baba, Y. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013, 13, 89. [Google Scholar] [CrossRef] [Green Version]
- Zou, W.; Chen, L. Inhibitory B7-family molecules in the tumour microenvironment. Nat. Rev. Immunol. 2008, 8, 467–477. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T cells for solid tumors: New strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Sfanos, K.S.; Bruno, T.C.; Meeker, A.K.; De Marzo, A.M.; Isaacs, W.B.; Drake, C.G. Human prostate-infiltrating CD8+T lymphocytes are oligoclonal and PD-1+. Prostate 2009, 69, 1694–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadzadeh, M. Johnson, L.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Clin. Immunol. 2009, 131, S38. [Google Scholar] [CrossRef]
- Kusmartsev, S.; Nefedova, Y.; Yoder, D.; I Gabrilovich, D. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J. Immunol. 2004, 172, 989–999. [Google Scholar] [CrossRef] [Green Version]
- Xiang, H.; Ramil, C.P.; Hai, J.; Zhang, C.; Wang, H.; Watkins, A.A.; Afshar, R.; Georgiev, P.; Sze, M.A.; Song, X.S.; et al. Cancer-associated fibroblasts promote immunosuppression by inducing ROS-generating monocytic MDSCs in lung Squamous cell Carcinoma. Cancer Immunol. Res. 2020, 8, 436–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siska, P.J.; Beckermann, K.E.; Mason, F.M.; Andrejeva, G.; Greenplate, A.R.; Sendor, A.B.; Chiang, Y.-C.J.; Corona, A.L.; Gemta, L.F.; Vincent, B.G.; et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jiang, H. An IL-4/21 Inverted Cytokine receptor improving CAR-T cell potency in immunosuppressive solid-tumor microenvironment. Front. Immunol. 2019, 10, 1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schurich, A.; Magalhaes, I.; Mattsson, J. Metabolic regulation of CAR T cell function by the hypoxic microenvironment in solid tumors. Immunotheraphy 2019, 11, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Cao, Y.J. Engineered T Cell Therapy for Cancer in the Clinic. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Richman, S.A.; Nunez-Cruz, S.; Moghimi, B.; Li, L.; Gershenson, Z.T.; Mourelatos, Z.; Barrett, D.M.; Grupp, S.A.; Milone, M.C. High-affinity GD2-Specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol. Res. 2017, 6, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; E Dudley, M.; Laurencot, C.M.; A Rosenberg, S. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Lamers, C.H.J.; Sleijfer, S.; Van Steenbergen, S.; Van Elzakker, P.; Van Krimpen, B.; Groot, C.; Vulto, A.; Bakker, M.D.; Oosterwijk, E.; Debets, R.; et al. Treatment of metastatic renal cell Carcinoma with CAIX CAR-engineered T cells: Clinical evaluation and management of on-target toxicity. Mol. Ther. 2013, 21, 904–912. [Google Scholar] [CrossRef]
- Li, H.; Ding, J.; Lu, M.; Liu, H.; Miao, Y.; Li, L.; Wang, G.; Zheng, J.; Pei, D.; Zhang, Q. CAIX-specific CAR-T cells and Sunitinib show synergistic effects against metastatic renal cancer models. J. Immunother. 2020, 43, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, D.; Simon, M.C. Hypoxia, lipids, and cancer: Surviving the harsh tumor microenvironment. Trends Cell Biol. 2014, 24, 472–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaupel, P.; Mayer, A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Xia, A.-L.; Wang, X.-C.; Lu, Y.-J.; Lu, X.-J.; Sun, B. Chimeric-antigen receptor T (CAR-T) cell therapy for solid tumors: Challenges and opportunities. Oncotarget 2017, 8, 90521–90531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Schalkwyk, M.C.I.; Papa, S.; Jeannon, J.-P.; Urbano, T.G.; Spicer, J.F.; Maher, J. Design of a Phase I clinical trial to evaluate intratumoral delivery of ErbB-targeted chimeric antigen receptor T-cells in locally advanced or recurrent head and neck cancer. Hum. Gene Ther. Clin. Dev. 2013, 24, 134–142. [Google Scholar] [CrossRef]
- Choi, B.D.; Suryadevara, C.M.; Gedeon, P.; Ii, J.E.H.; Sanchez-Perez, L.; Bigner, D.D.; Sampson, J.H.; Herndon, J.E. Intracerebral delivery of a third generation EGFRvIII-specific chimeric antigen receptor is efficacious against human glioma. J. Clin. Neurosci. 2013, 21, 189–190. [Google Scholar] [CrossRef] [Green Version]
- Sridhar, P.; Petrocca, F. Regional delivery of chimeric antigen receptor (CAR) T-cells for cancer therapy. Cancers 2017, 9, 92. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.T.; Moffett, H.F.; Stephan, S.B.; Opel, C.F.; Dumigan, A.; Jiang, X.; Pillarisetty, V.G.; Pillai, S.P.S.; Wittrup, K.D.; Stephan, M.T. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J. Clin. Investig. 2017, 127, 2176–2191. [Google Scholar] [CrossRef] [PubMed]
- Vignali, D.; Kallikourdis, M. Improving homing in T cell therapy. Cytokine Growth Factor Rev. 2017, 36, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Chow, M.T.; Luster, A.D. Chemokines in cancer. Cancer Immunol. Res. 2014, 2, 1125–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacy, P. Editorial: Secretion of Cytokines and Chemokines by innate immune cells. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- McGettrick, H.M.; Butler, L.M.; Buckley, C.D.; Rainger, G.E.; Nash, G.B. Tissue stroma as a regulator of leukocyte recruitment in inflammation. J. Leukoc. Biol. 2012, 91, 385–400. [Google Scholar] [CrossRef]
- Moon, E.K.; Wang, L.-C.S.; Bekdache, K.; Lynn, R.C.; Lo, A.; Thorne, S.H.; Albelda, S.M. Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. OncoImmunology 2018, 7, e1395997. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Wang, G.; Westwood, J.A.; Pachynski, R.K.; Tiffany, H.L.; Marincola, F.M.; Wang, E.; Young, H.A.; Murphy, P.M.; Hwu, P. Redirecting migration of T cells to Chemokine secreted from tumors by genetic modification with CXCR2. Hum. Gene Ther. 2002, 13, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, A.; De Angelis, B.; Rooney, C.M.; Zhang, L.; Mahendravada, A.; Foster, A.E.; Heslop, H.E.; Brenner, M.K.; Dotti, G.; Savoldo, B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 2009, 113, 6392–6402. [Google Scholar] [CrossRef] [Green Version]
- Perera, L.P.; Zhang, M.; Nakagawa, M.; Petrus, M.N.; Maeda, M.; Kadin, M.E.; Waldmann, T.A.; Perera, P.-Y. Chimeric antigen receptor modified T cells that target chemokine receptor CCR4 as a therapeutic modality for T-cell malignancies. Am. J. Hematol. 2017, 92, 892–901. [Google Scholar] [CrossRef]
- Ishitsuka, K.; Yurimoto, S.; Kawamura, K.; Tsuji, Y.; Iwabuchi, M.; Takahashi, T.; Tobinai, K. Safety and efficacy of mogamulizumab in patients with adult T-cell leukemia-lymphoma in Japan: Interim results of postmarketing all-case surveillance. Hematol. Oncol. 2017, 35, 252. [Google Scholar] [CrossRef] [Green Version]
- Kiesgen, S.; Chicaybam, L.; Chintala, N.K.; Adusumilli, P. Chimeric antigen receptor (CAR) T-cell therapy for Thoracic Malignancies. J. Thorac. Oncol. 2017, 13, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Santos, A.M.; Jung, J.; Aziz, N.; Kissil, J.L.; Puré, E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J. Clin. Investig. 2009, 119, 3613–3625. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Rivera, A.; Tao, L.; Zhang, X. Genetically modified T cells targeting neovasculature efficiently destroy tumor blood vessels, shrink established solid tumors and increase nanoparticle delivery. Int. J. Cancer 2013, 133, 2483–2492. [Google Scholar] [CrossRef] [PubMed]
- Gowrishankar, K.; Birtwistle, L.; Micklethwaite, K.P. Manipulating the tumor microenvironment by adoptive cell transfer of CAR T-cells. Mamm. Genome 2018, 29, 739–756. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.; Sukumaran, S.; Bajgain, P.; Watanabe, N.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Fisher, W.E.; Leen, A.M.; Vera, J.F. Improving chimeric antigen receptor-modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Mol. Ther. 2017, 25, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef] [Green Version]
- Chang, Z.L.; Lorenzini, M.H.; Chen, X.; Tran, U.; Bangayan, N.J.; Chen, Y.Y. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat. Methods 2018, 14, 317–324. [Google Scholar] [CrossRef]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1- and CTLA-4-Based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.; Drake, V.; Huang, H.-S.; Chiu, S.; Zheng, L. Reprogramming the tumor microenvironment: Tumor-induced immunosuppressive factors paralyze T cells. OncoImmunology 2015, 4, e1016700. [Google Scholar] [CrossRef]
- Bollard, C.M.; Rössig, C.; Calonge, M.J.; Huls, M.H.; Wagner, H.-J.; Massagué, J.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M. Adapting a transforming growth factor β–related tumor protection strategy to enhance antitumor immunity. Blood 2002, 99, 3179–3187. [Google Scholar] [CrossRef] [Green Version]
- Foster, A.E.; Dotti, G. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-beta receptor. J. Immunother. 2008, 31, 500–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löffek, S. Transforming of the tumor microenvironment: Implications for TGF-β inhibition in the context of immune-checkpoint therapy. J. Oncol. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Siriwon, N.; Zhang, X.; Yang, S.; Jin, T.; He, F.; Kim, Y.J.; Mac, J.; Lu, Z.; Wang, S.; et al. Enhanced cancer immunotherapy by chimeric antigen receptor–modified T cells engineered to secrete checkpoint inhibitors. Clin. Cancer Res. 2017, 23, 6982–6992. [Google Scholar] [CrossRef] [Green Version]
- John, L.B.; Devaud, C.; Duong, C.P.M.; Yong, C.S.; Beavis, P.A.; Haynes, N.M.; Chow, M.T.; Smyth, M.J.; Kershaw, M.; Darcy, P.K. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 2013, 19, 5636–5646. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin. Cancer Res. 2016, 23, 2255–2266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menger, L.; Sledzinska, A.; Bergerhoff, K.; Vargas, F.A.; Smith, J.; Poirot, L.; Pule, M.; Herrero, J.; Peggs, K.S.; Quezada, S.A. TALEN-mediated inactivation of PD-1 in tumor-reactive Lymphocytes promotes Intratumoral T-cell persistence and rejection of established tumors. Cancer Res. 2016, 76, 2087–2093. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.; Schambach, A. Engineering CAR-T cells for improved function against solid tumors. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Lynn, R.C.; Weber, E.W.; Sotillo, E.; Gennert, D.; Xu, P.; Good, Z.; Anbunathan, H.; Lattin, J.; Jones, R.; Tieu, V.; et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 2019, 576, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Krenciute, G.; Prinzing, B.L. Transgenic expression of IL15 improves Antiglioma activity of IL13Ralpha2-CAR T cells but results in antigen loss variants. Cancer Immunol. Res. 2017, 5, 571–581. [Google Scholar] [CrossRef] [Green Version]
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018, 36, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Li, Y.; Liu, W.; Li, X. Engineered IL-7 receptor enhances the therapeutic effect of AXL-CAR-T cells on triple-negative breast cancer. BioMed Res. Int. 2020, 2020, 1–13. [Google Scholar] [CrossRef]
- Hoyos, V.; Savoldo, B.; Quintarelli, C.; Mahendravada, A.; Zhang, M.; Vera, J.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Dotti, G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 2010, 24, 1160–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishio, N.; Dotti, G. Oncolytic virus expressing RANTES and IL-15 enhances function of CAR-modified T cells in solid tumors. OncoImmunology 2015, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alizadeh, D.; Wong, R.A.; Yang, X.; Wang, D.; Pecoraro, J.R.; Kuo, C.-F.; Aguilar, B.; Qi, Y.; Ann, D.K.; Starr, R.; et al. IL15 Enhances CAR-T cell antitumor activity by reducing mTORC1 activity and preserving their stem cell memory phenotype. Cancer Immunol. Res. 2019, 7, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Cieri, N.; Camisa, B.; Cocchiarella, F.; Forcato, M.; Oliveira, G.; Provasi, E.; Bondanza, A.; Bordignon, C.; Peccatori, J.; Ciceri, F.; et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood 2013, 121, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T cells administered in combination with Lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef] [Green Version]
- Suryadevara, C.M.; Desai, R.; Abel, M.L.; Riccione, K.A.; Batich, K.A.; Shen, S.H.; Chongsathidkiet, P.; Gedeon, P.C.; Elsamadicy, A.A.; Snyder, D.J.; et al. Temozolomide lymphodepletion enhances CAR abundance and correlates with antitumor efficacy against established glioblastoma. OncoImmunology 2018, 7, e1434464. [Google Scholar] [CrossRef] [Green Version]
- Bagley, S.J.; O’Rourke, D.M. Clinical investigation of CAR T cells for solid tumors: Lessons learned and future directions. Pharmacol. Ther. 2020, 205, 107419. [Google Scholar] [CrossRef]
- Hombach, A.A.; Geumann, U.; Günther, C.; Hermann, F.G.; Abken, H. IL7-IL12 engineered Mesenchymal stem cells (MSCs) improve a CAR T cell attack against colorectal cancer cells. Cells 2020, 9, 873. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Hao, H.; Yang, G.; Zhang, Y.; Fu, Y. Immunotherapy with CAR-Modified T cells: Toxicities and overcoming strategies. J. Immunol. Res. 2018, 2018. [Google Scholar] [CrossRef]
- Jackson, H.J.; Brentjens, R.J. Overcoming antigen escape with CAR T-cell therapy. Cancer Discov. 2015, 5, 1238–1240. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. New Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Wang, Y.; Wei, J.; Han, W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J. Hematol. Oncol. 2019, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Barrett, D.M.; Kenderian, S.S.; Shestova, O.; Hofmann, T.J.; Perazzelli, J.; Klichinsky, M.; Aikawa, V.; Nazimuddin, F.; Kozlowski, M.; et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Investig. 2016, 126, 3814–3826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, K.-C.; Guo, Y.-L.; Liu, Y.; Dai, H.-R.; Wang, Y.; Lv, H.-Y.; Huang, J.-H.; Yang, Q.-M.; Han, W. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J. Hematol. Oncol. 2017, 10, 4. [Google Scholar] [CrossRef] [Green Version]
- Hombach, A.; Rappl, G.; Abken, H. Blocking CD30 on T Cells by a Dual Specific CAR for CD30 and Colon Cancer Antigens Improves the CAR T cell response against CD30- Tumors. Mol. Ther. 2019, 27, 1825–1835. [Google Scholar] [CrossRef]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.; et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Dichwalkar, T.; Chang, J.Y.; Cossette, B.; Garafola, D.; Zhang, A.Q.; Fichter, M.; Wang, C.; Liang, S.; Silva, M.; et al. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science 2019, 365, 162–168. [Google Scholar] [PubMed]
- Becerra, C.R.; Hoof, P.; Paulson, A.S.; Manji, G.A.; Gardner, O.; Malankar, A.; Shaw, J.; Blass, D.; Ballard, B.; Yi, X.; et al. Ligand-inducible, prostate stem cell antigen (PSCA)-directed GoCAR-T cells in advanced solid tumors: Preliminary results from a dose escalation. J. Clin. Oncol. 2019, 37, 283. [Google Scholar] [CrossRef]
- Labanieh, L.; Majzner, R.G.; Mackall, C.L. Programming CAR-T cells to kill cancer. Nat. Biomed. Eng. 2018, 2, 377–391. [Google Scholar] [CrossRef]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef] [Green Version]
- Raj, D.; Yang, M.-H.; Rodgers, D.; Hampton, E.N.; Begum, J.; Mustafa, A.; Lorizio, D.; Garces, I.; Propper, D.; Kench, J.G.; et al. Switchable CAR-T cells mediate remission in metastatic pancreatic ductal adenocarcinoma. Gut 2018, 68, 1052–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badrinath, N.; Yoo, S.Y. Recent advances in cancer stem cell-targeted immunotherapy. Cancers 2019, 11, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrandina, G.; Petrillo, M.; Bonanno, G.; Scambia, G. Targeting CD133 antigen in cancer. Expert Opin. Ther. Targets 2009, 13, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, M.; Wu, Z.; Tong, C.; Dai, H.; Guo, Y.; Liu, Y.; Huang, J.; Lv, H.; Luo, C.; et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. OncoImmunology 2018, 7, e1440169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.W.; Park, H.W.; Kim, H.; Lee, S.; Choi, S.Y.; Park, Y.; Lee, S.-W. Evaluating antitumor activity of Kiatomab by targeting cancer stem cell-specific KIAA1114 antigen in mice. Immune Netw. 2019, 19, e43. [Google Scholar] [CrossRef] [PubMed]
- Simonetta, F.; Alvarez, M.; Negrin, R.S. Natural killer cells in graft-versus-host-disease after Allogeneic hematopoietic cell transplantation. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Anonymous. Natural killer cells for cancer immunotherapy: A new CAR is catching up. EBioMedicine 2019, 39, 1–2. [Google Scholar] [CrossRef]
- Wang, W.; Wu, C.-P.; Wu, C.-P. CAR-NK for tumor immunotherapy: Clinical transformation and future prospects. Cancer Lett. 2019, 472, 175–180. [Google Scholar] [CrossRef]
- Habib, S.; Tariq, S.M.; Tariq, M. Chimeric Antigen Receptor-Natural Killer Cells: The Future of Cancer Immunotherapy. Ochsner J. 2019, 19, 186–187. [Google Scholar] [CrossRef] [Green Version]
- CAR NK Cells Clinical Trials 2020. Available online: https://clinicaltrials.gov/ct2/results?cond=CAR+NK+cells&term=&cntry=&state=&city=&dist= (accessed on 7 July 2020).
- Ren, J.; Zhang, X.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget 2017, 8, 17002–17011. [Google Scholar] [CrossRef] [Green Version]
- Ruella, M.; Kenderian, S.S. Next-generation chimeric antigen receptor T-cell therapy: Going off the shelf. BioDrugs 2017, 31, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9, eaaj2013. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Wang, G.; Cheng, H.; Wei, C.; Qi, K.; Sang, W.; Zhenyu, L.; Shi, M.; Li, H.; Qiao, J.; et al. Potent anti-leukemia activities of humanized CD19-targeted chimeric antigen receptor T (CAR-T) cells in patients with relapsed/refractory acute lymphoblastic leukemia. Am. J. Hematol. 2018, 93, 851–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.; Hwang, H.; Jo, Y.; Lee, B.; Lee, Y.-H.; Kim, C.H.; Hong, C. Soluble γc receptor attenuates anti-tumor responses of CD8+ T cells in T cell immunotherapy. Int. J. Cancer 2018, 143, 1212–1223. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Disease | Brain Cancer | Breast Cancer | Cervical Cancer | Colorectal Cancer | Esophageal Cancer | Bone Cancer | Gastric Cancer | Liver Cancer | Lung Cancer | Lymphoma | Mesothelioma | Nasopharyngeal Cancer | Ovarian Cancer | Pancreatic Cancer | Prostate Cancer | Reanal Cell Carcinoma | Skin Cancer | Thymoma | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Limiting Factor | |||||||||||||||||||
| Trafficking | ✓ [19] | ✓ [21] | ✓ [21] | ✓ [21] | ✓ [20] | ✓ [18] | |||||||||||||
| Infiltration | ✓ [32] | ✓ [30,31] | ✓ [34] | ✓ [29] | ✓ [27] | ✓ [33] | ✓ [28] | ✓ [36] | |||||||||||
| Immune suppressive TME | Immune suppressor cells | ✓ [41] | ✓ [42] | ✓ [44] | |||||||||||||||
| Immune checkpoints | ✓ [99] | ✓ [99] | ✓ [99] | ✓ [99] | ✓ [100] | ✓ [99] | ✓ [99] | ✓ [99] | ✓ [101] | ✓ [99] | ✓ [102] | ✓ [99] | |||||||
| ROS | ✓ [103] | ✓ [104] | ✓ [105] | ||||||||||||||||
| Metabolites | ✓ [74] | ✓ [100,105] | |||||||||||||||||
| Cytokine | ✓ [106] | ✓ [83] | ✓ [86] | ✓ [85] | ✓ [84] | ✓ [87] | |||||||||||||
| pH | ✓ [98] | ✓ [98] | ✓ [98] | ||||||||||||||||
| Hypoxia | ✓ [107] | ✓ [42] | ✓ [107] | ||||||||||||||||
| Shortage of tumor antigen (ClinicalTrials.gov) [108] | Mesothelin | ✓ | ✓ [53] | ✓ | ✓ | ||||||||||||||
| EGFR | ✓ | ✓ | |||||||||||||||||
| GPC3 | ✓ | ||||||||||||||||||
| MUC1 | ✓ | ✓ | |||||||||||||||||
| HER2 (Human Epidermal Growth Factor Receptor 2) | ✓ | ✓ | |||||||||||||||||
| GD2 | ✓ | ||||||||||||||||||
| CEA | ✓ | ✓ | |||||||||||||||||
| EpCAM | ✓ | ✓ [78] | ✓ | ||||||||||||||||
| PSCA | ✓ | ||||||||||||||||||
| Non-specific antigen On-target, off-tumor toxicity | ✓ [109] | ✓ [110] | ✓ [111,112] | ✓ [52,108] | |||||||||||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jo, Y.; Ali, L.A.; Shim, J.A.; Lee, B.H.; Hong, C. Innovative CAR-T Cell Therapy for Solid Tumor; Current Duel between CAR-T Spear and Tumor Shield. Cancers 2020, 12, 2087. https://doi.org/10.3390/cancers12082087
Jo Y, Ali LA, Shim JA, Lee BH, Hong C. Innovative CAR-T Cell Therapy for Solid Tumor; Current Duel between CAR-T Spear and Tumor Shield. Cancers. 2020; 12(8):2087. https://doi.org/10.3390/cancers12082087
Chicago/Turabian StyleJo, Yuna, Laraib Amir Ali, Ju A Shim, Byung Ha Lee, and Changwan Hong. 2020. "Innovative CAR-T Cell Therapy for Solid Tumor; Current Duel between CAR-T Spear and Tumor Shield" Cancers 12, no. 8: 2087. https://doi.org/10.3390/cancers12082087
APA StyleJo, Y., Ali, L. A., Shim, J. A., Lee, B. H., & Hong, C. (2020). Innovative CAR-T Cell Therapy for Solid Tumor; Current Duel between CAR-T Spear and Tumor Shield. Cancers, 12(8), 2087. https://doi.org/10.3390/cancers12082087