Cancer Cell Acid Adaptation Gene Expression Response Is Correlated to Tumor-Specific Tissue Expression Profiles and Patient Survival

 , and
, and
Abstract
:1. Introduction
2. Results
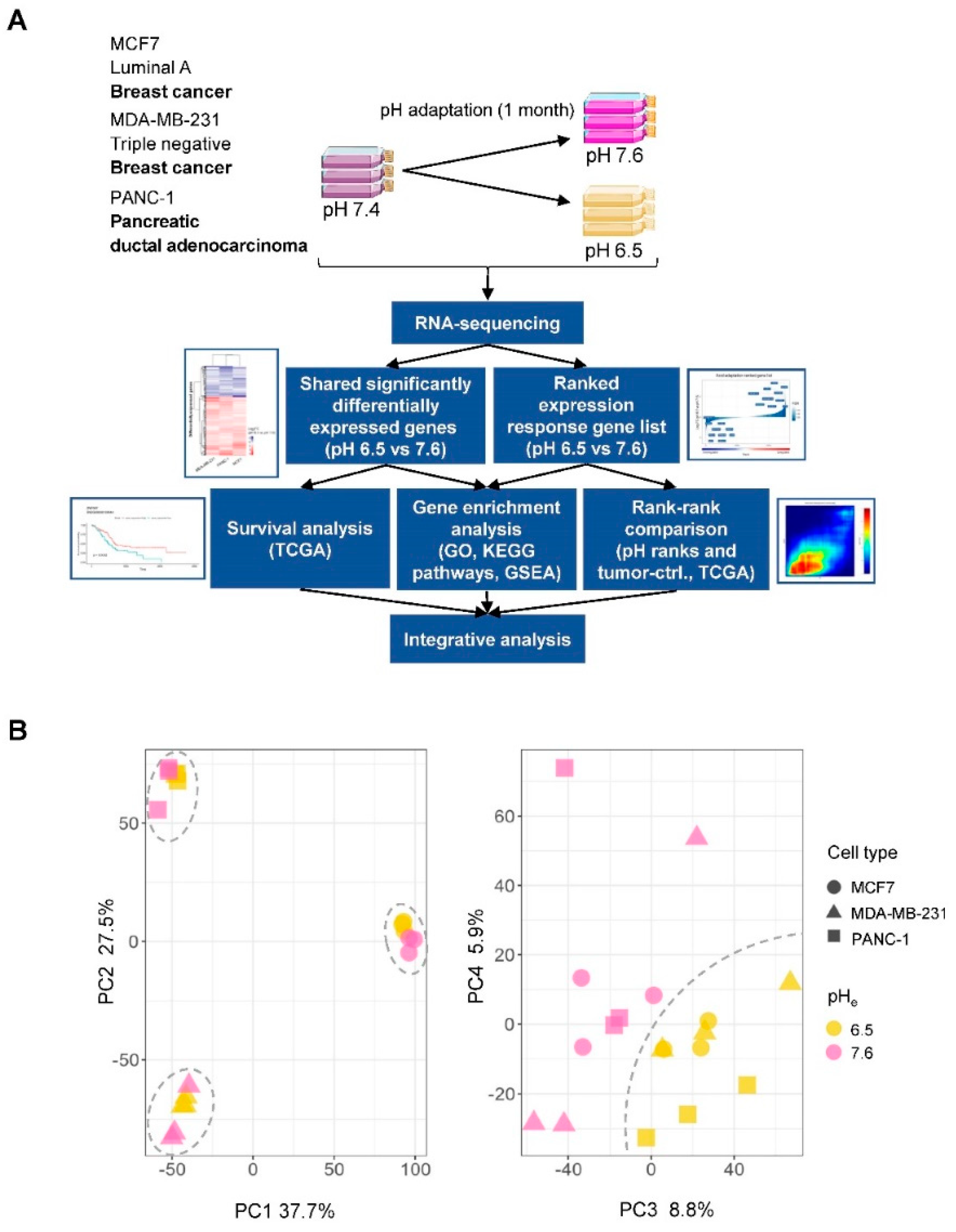
2.1. Gene Expression Changes Induced by Chronic Adaptation of Cancer Cells to Acidosis
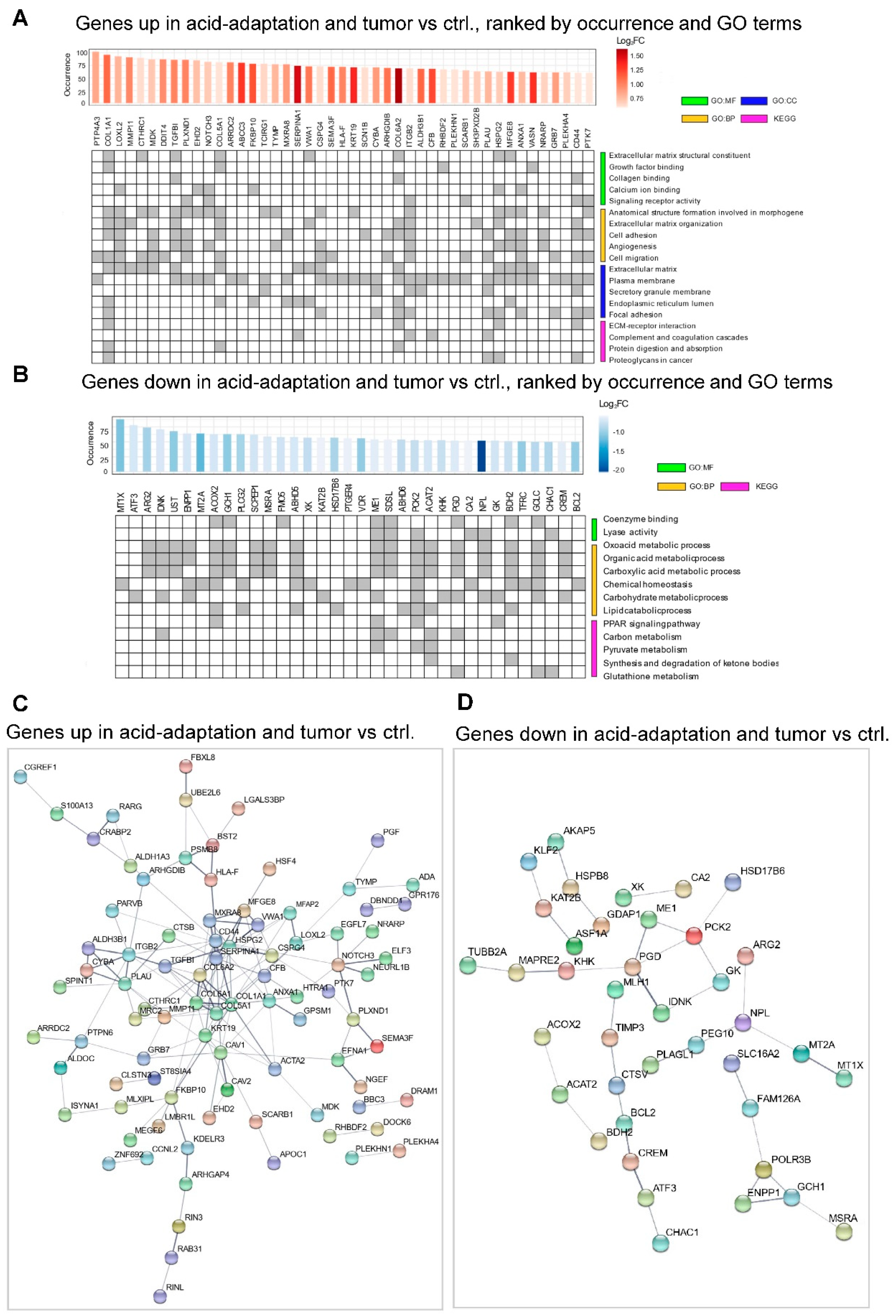
2.2. Identification of a Shared Acid Adaptation Gene Response Profile
2.3. In Vivo Expression of Genes Reacting to Chronic Acidosis Is Predictive of Cancer Patient Survival
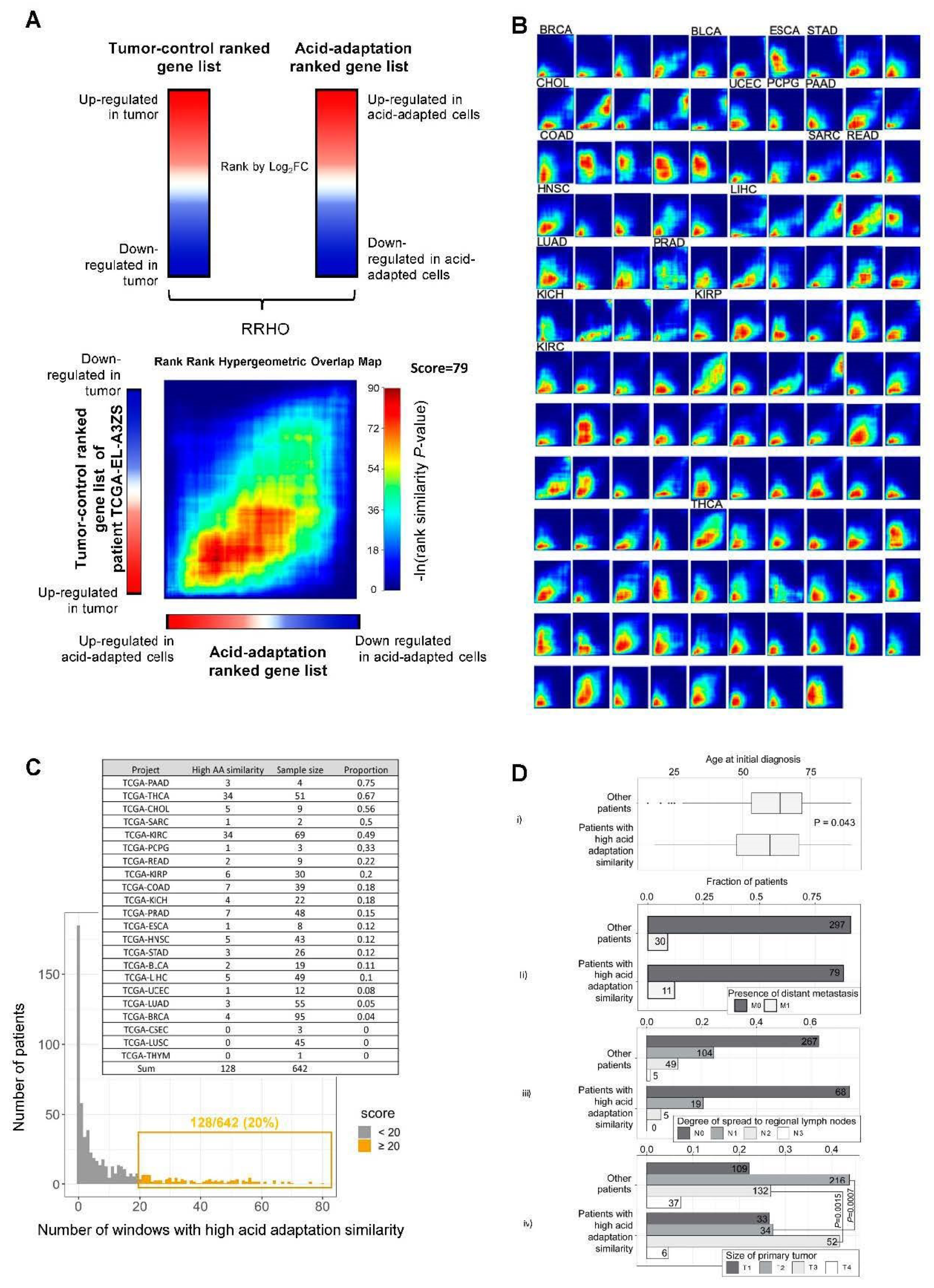
2.4. RRHO Analysis Identifies Substantial Overlap between Genes Upregulated in Acid Adapted Cells and Patient Tumor Tissue
2.5. Integration of Survival Data and Rank–Rank Analysis
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Acid Adaptation
4.2. RNA Isolation and RNA Sequencing
4.3. Analyses of RNA-Seq Data
4.4. GO Enrichment Analysis and Gene Set Enrichment Analysis (GSEA)
4.5. RRHO Analysis
4.6. Patient Survival Analysis
4.7. Data Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation 1. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gillies, R.J. A microenvironmental model of carcinogenesis. Nat. Rev. Cancer 2008, 8, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Merlo, L.M.; Pepper, J.W.; Reid, B.J.; Maley, C.C. Cancer as an evolutionary and ecological process. Nat. Rev. Cancer 2006, 6, 924–935. [Google Scholar] [CrossRef] [PubMed]
- Hashim, A.I.; Zhang, X.; Wojtkowiak, J.W.; Martinez, G.V.; Gillies, R.J. Imaging pH and metastasis. NMR Biomed. 2011, 24, 582–591. [Google Scholar] [CrossRef]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: A review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef]
- Elingaard-Larsen, L.O.; Rolver, M.G.; Sorensen, E.E.; Pedersen, S.F. How reciprocal interactions between the tumor microenvironment and ion transport proteins drive cancer progression. In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Boedtkjer, E.; Pedersen, S.F. The Acidic Tumor Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy 12. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Zhou, H.Y.; Deng, S.C.; Deng, S.J.; He, C.; Li, X.; Chen, J.Y.; Jin, Y.; Hu, Z.L.; Wang, F.; et al. ASIC1 and ASIC3 contribute to acidity-induced EMT of pancreatic cancer through activating Ca2+/RhoA pathway. Cell Death Dis. 2017, 8, e2806. [Google Scholar] [CrossRef]
- Peppicelli, S.; Bianchini, F.; Torre, E.; Calorini, L. Contribution of acidic melanoma cells undergoing epithelial-to-mesenchymal transition to aggressiveness of non-acidic melanoma cells. Clin. Exp. Metastasis 2014, 31, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Moellering, R.E.; Black, K.C.; Krishnamurty, C.; Baggett, B.K.; Stafford, P.; Rain, M.; Gatenby, R.A.; Gillies, R.J. Acid treatment of melanoma cells selects for invasive phenotypes. Clin. Exp. Metastasis 2008, 25, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbet, C.; Bastien, E.; de Jesus, J.P.S.; Dierge, E.; Martherus, R.; Vander Linden, C.; Doix, B.; Degavre, C.; Guilbaud, C.; Petit, L.; et al. TGFβ2-induced formation of lipid droplets supports acidosis-driven EMT and the metastatic spreading of cancer cells. Nat. Commun. 2020, 11, 454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Zaguilán, R.; Seftor, E.A.; Seftor, R.E.; Chu, Y.W.; Gillies, R.J.; Hendrix, M.J. Acidic pH enhances the invasive behavior of human melanoma cells. Clin. Exp. Metastasis 1996, 14, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Rohani, N.; Hao, L.; Alexis, M.S.; Joughin, B.A.; Krismer, K.; Moufarrej, M.N.; Soltis, A.R.; Lauffenburger, D.A.; Yaffe, M.B.; Burge, C.B.; et al. Acidification of Tumor at Stromal Boundaries Drives Transcriptome Alterations Associated with Aggressive Phenotypes. Cancer Res. 2019, 79, 1952–1966. [Google Scholar] [CrossRef] [Green Version]
- LaMonte, G.; Tang, X.; Chen, J.L.; Wu, J.; Ding, C.K.; Keenan, M.M.; Sangokoya, C.; Kung, H.N.; Ilkayeva, O.; Boros, L.G.; et al. Acidosis induces reprogramming of cellular metabolism to mitigate oxidative stress. Cancer Metab. 2013, 1, 23. [Google Scholar] [CrossRef] [Green Version]
- Corbet, C.; Draoui, N.; Polet, F.; Pinto, A.; Drozak, X.; Riant, O.; Feron, O. The SIRT1/HIF2alpha axis drives reductive glutamine metabolism under chronic acidosis and alters tumor response to therapy. Cancer Res. 2014, 74, 5507–5519. [Google Scholar] [CrossRef] [Green Version]
- Corbet, C.; Feron, O. Tumour acidosis: From the passenger to the driver’s seat. Nat. Rev. Cancer 2017, 17, 577–593. [Google Scholar] [CrossRef]
- Pellegrini, P.; Serviss, J.T.; Lundbäck, T.; Bancaro, N.; Mazurkiewicz, M.; Kolosenko, I.; Yu, D.; Haraldsson, M.; D’Arcy, P.; Linder, S.; et al. A drug screening assay on cancer cells chronically adapted to acidosis. Cancer Cell Int. 2018, 18, 147. [Google Scholar] [CrossRef]
- Chen, J.L.; Merl, D.; Peterson, C.W.; Wu, J.; Liu, P.Y.; Yin, H.; Muoio, D.M.; Ayer, D.E.; West, M.; Chi, J.T. Lactic acidosis triggers starvation response with paradoxical induction of TXNIP through MondoA. PLoS Genet 2010, 6, e1001093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michl, J.; Park, K.C.; Swietach, P. Evidence-based guidelines for controlling pH in mammalian live-cell culture systems. Commun. Biol. 2019, 2, 144. [Google Scholar] [CrossRef] [PubMed]
- Wilde, B.R.; Ye, Z.; Lim, T.Y.; Ayer, D.E. Cellular acidosis triggers human MondoA transcriptional activity by driving mitochondrial ATP production. Elife 2019, 8, e40199. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.J.; Geng, W.S.; Wang, Z.Q.; Chen, L.; Zeng, X.S. The role of thioredoxin system in cancer: Strategy for cancer therapy. Cancer Chemother. Pharmacol. 2019, 84, 453–470. [Google Scholar] [CrossRef]
- Cadenas, C.; Franckenstein, D.; Schmidt, M.; Gehrmann, M.; Hermes, M.; Geppert, B.; Schormann, W.; Maccoux, L.J.; Schug, M.; Schumann, A.; et al. Role of thioredoxin reductase 1 and thioredoxin interacting protein in prognosis of breast cancer. Breast Cancer Res. 2010, 12, R44. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; O’Shea, J.M.; Kaadige, M.R.; Cunha, S.; Wilde, B.R.; Cohen, A.L.; Welm, A.L.; Ayer, D.E. Metabolic reprogramming in triple-negative breast cancer through Myc suppression of TXNIP. Proc. Natl. Acad. Sci. USA 2015, 112, 5425–5430. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.H.; Song, J.W.; Li, W.; Liu, X.; Cao, L.; Wan, L.M.; Tan, Y.X.; Ji, S.P.; Liang, Y.M.; Gong, F. The acid-sensing ion channel, ASIC2, promotes invasion and metastasis of colorectal cancer under acidosis by activating the calcineurin/NFAT1 axis. J. Exp. Clin. Cancer Res. 2017, 36, 130. [Google Scholar] [CrossRef]
- Waldmann, R.; Champigny, G.; Bassilana, F.; Heurteaux, C.; Lazdunski, M. A proton-gated cation channel involved in acid-sensing. Nature 1997, 386, 173–177. [Google Scholar] [CrossRef]
- Collier, D.M.; Snyder, P.M. Extracellular protons regulate human ENaC by modulating Na+ self-inhibition. J. Biol. Chem. 2009, 284, 792–798. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Ling, Z.H.; Wang, H.; Wang, X.Y.; Gui, J. Upregulation of SCNN1A Promotes Cell Proliferation, Migration, and Predicts Poor Prognosis in Ovarian Cancer Through Regulating Epithelial-Mesenchymal Transformation. Cancer Biother. Radiopharm. 2019, 34, 642–649. [Google Scholar] [CrossRef]
- Kanwar, N.; Carmine-Simmen, K.; Nair, R.; Wang, C.; Moghadas-Jafari, S.; Blaser, H.; Tran-Thanh, D.; Wang, D.; Wang, P.; Wang, J.; et al. Amplification of a calcium channel subunit CACNG4 increases breast cancer metastasis. EBioMedicine 2020, 52, 102646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, M.; Millican-Slater, R.; Forrest, L.C.; Brackenbury, W.J. The sodium channel β1 subunit mediates outgrowth of neurite-like processes on breast cancer cells and promotes tumour growth and metastasis. Int. J. Cancer 2014, 135, 2338–2351. [Google Scholar] [CrossRef] [PubMed]
- Lui, A.J.; Geanes, E.S.; Ogony, J.; Behbod, F.; Marquess, J.; Valdez, K.; Jewell, W.; Tawfik, O.; Lewis-Wambi, J. IFITM1 suppression blocks proliferation and invasion of aromatase inhibitor-resistant breast cancer in vivo by JAK/STAT-mediated induction of p21. Cancer Lett. 2017, 399, 29–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sari, I.N.; Yang, Y.G.; Phi, L.T.; Kim, H.; Baek, M.J.; Jeong, D.; Kwon, H.Y. Interferon-induced transmembrane protein 1 (IFITM1) is required for the progression of colorectal cancer. Oncotarget 2016, 7, 86039–86050. [Google Scholar] [CrossRef] [PubMed]
- Moy, I.; Todorovic, V.; Dubash, A.D.; Coon, J.S.; Parker, J.B.; Buranapramest, M.; Huang, C.C.; Zhao, H.; Green, K.J.; Bulun, S.E. Estrogen-dependent sushi domain containing 3 regulates cytoskeleton organization and migration in breast cancer cells. Oncogene 2015, 34, 323–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inamori, K.I.; Beedle, A.M.; de Bernabe, D.B.; Wright, M.E.; Campbell, K.P. LARGE2-dependent glycosylation confers laminin-binding ability on proteoglycans. Glycobiology 2016, 26, 1284–1296. [Google Scholar] [CrossRef] [Green Version]
- Hinke, S.A.; Navedo, M.F.; Ulman, A.; Whiting, J.L.; Nygren, P.J.; Tian, G.; Jimenez-Caliani, A.J.; Langeberg, L.K.; Cirulli, V.; Tengholm, A.; et al. Anchored phosphatases modulate glucose homeostasis. EMBO J. 2012, 31, 3991–4004. [Google Scholar] [CrossRef]
- Zhong, Z.; Ye, Z.; He, G.; Zhang, W.; Wang, J.; Huang, S. Low expression of A-kinase anchor protein 5 predicts poor prognosis in non-mucin producing stomach adenocarcinoma based on TCGA data. Ann. Transl. Med. 2020, 8, 115. [Google Scholar] [CrossRef]
- Feng, X.; Yan, N.; Sun, W.; Zheng, S.; Jiang, S.; Wang, J.; Guo, C.; Hao, L.; Tian, Y.; Liu, S.; et al. miR-4521-FAM129A axial regulation on ccRCC progression through TIMP-1/MMP2/MMP9 and MDM2/p53/Bcl2/Bax pathways. Cell Death Discov. 2019, 5, 89. [Google Scholar] [CrossRef] [Green Version]
- Nozima, B.H.; Mendes, T.B.; Pereira, G.; Araldi, R.P.; Iwamura, E.S.M.; Smaili, S.S.; Carvalheira, G.M.G.; Cerutti, J.M. FAM129A regulates autophagy in thyroid carcinomas in an oncogene-dependent manner. Endocr. Relat. Cancer 2019, 26, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Lelouvier, B.; Puertollano, R. Mucolipin-3 regulates luminal calcium, acidification, and membrane fusion in the endosomal pathway. J. Biol. Chem. 2011, 286, 9826–9832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, Z.E.; Patel, C.H.; Brooks, R.C.; Yu, Y.; Ibrahim-Hashim, A.; Riddle, M.; Porcu, A.; Jiang, T.; Ecker, B.L.; Tameire, F.; et al. Acid Suspends the Circadian Clock in Hypoxia through Inhibition of mTOR. Cell 2018, 174, 72–87.e32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, Z.E.; Brooks, R.C.; Dang, C.V. mTOR Senses Intracellular pH through Lysosome Dispersion from RHEB. Bioessays 2019, 41, e1800265. [Google Scholar] [CrossRef]
- Zhou, Z.H.; Wang, Q.L.; Mao, L.H.; Li, X.Q.; Liu, P.; Song, J.W.; Liu, X.; Xu, F.; Lei, J.; He, S. Chromatin accessibility changes are associated with enhanced growth and liver metastasis capacity of acid-adapted colorectal cancer cells. Cell Cycle 2019, 18, 511–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngeow, J.; Yu, W.; Yehia, L.; Niazi, F.; Chen, J.; Tang, X.; Heald, B.; Lei, J.; Romigh, T.; Tucker-Kellogg, L.; et al. Exome Sequencing Reveals Germline SMAD9 Mutation That Reduces Phosphatase and Tensin Homolog Expression and Is Associated With Hamartomatous Polyposis and Gastrointestinal Ganglioneuromas. Gastroenterology 2015, 149, 886–889.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Burton, E.M.; Koganti, S.; Zhi, J.; Doyle, F.; Tenenbaum, S.A.; Horn, B.; Bhaduri-McIntosh, S. KRAB-ZFP Repressors Enforce Quiescence of Oncogenic Human Herpesviruses. J. Virol. 2018, 92, e00298-18. [Google Scholar] [CrossRef] [Green Version]
- The Universal Protein Resource (UniProt). Available online: https://www.uniprot.org/ (accessed on 3 June 2020).
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Jiang, J.; Huang, L.; Feng, W.; Zhang, Z.; Jin, L.; Xing, X. Sperm associated antigen 4 (SPAG4) as a new cancer marker interacts with Nesprin3 to regulate cell migration in lung carcinoma. Oncol. Rep. 2018, 40, 783–792. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.K.; Wang, W.J.; Cai, H.Y.; Du, B.B.; Mai, P.; Zhang, L.J.; Ma, W.; Hu, Y.G.; Feng, S.F.; Miao, G.Y. MFAP2 promotes epithelial-mesenchymal transition in gastric cancer cells by activating TGF-beta/SMAD2/3 signaling pathway. Onco. Targets Ther. 2018, 11, 4001–4017. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Zhang, K.N.; Chai, R.C.; Wang, K.Y.; Huang, R.Y.; Li, G.Z.; Wang, Y.Z.; Chen, J.; Jiang, T. ADAMTSL4, a Secreted Glycoprotein, Is a Novel Immune-Related Biomarker for Primary Glioblastoma Multiforme. Dis. Markers 2019, 2019, 1802620. [Google Scholar] [CrossRef] [Green Version]
- Kaikkonen, E.; Rantapero, T.; Zhang, Q.; Taimen, P.; Laitinen, V.; Kallajoki, M.; Jambulingam, D.; Ettala, O.; Knaapila, J.; Bostrom, P.J.; et al. ANO7 is associated with aggressive prostate cancer. Int. J. Cancer 2018, 143, 2479–2487. [Google Scholar] [CrossRef] [PubMed]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L.; Hulikova, A. The chemistry, physiology and pathology of pH in cancer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damaghi, M.; Gillies, R. Phenotypic changes of acid adapted cancer cells push them toward aggressiveness in their evolution in the tumor microenvironment. Cell Cycle 2016, 16, 1739–1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, S.F.; Novak, I.; Alves, F.; Schwab, A.; Pardo, L.A. Alternating pH landscapes shape epithelial cancer initiation and progression: Focus on pancreatic cancer. Bioessays 2017, 39, 1600253. [Google Scholar] [CrossRef] [PubMed]
- Wojtkowiak, J.W.; Verduzco, D.; Schramm, K.J.; Gillies, R.J. Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Mol. Pharm. 2011, 8, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Pilon-Thomas, S.; Kodumudi, K.N.; El-Kenawi, A.E.; Russell, S.; Weber, A.M.; Luddy, K.; Damaghi, M.; Wojtkowiak, J.W.; Mule, J.J.; Ibrahim-Hashim, A.; et al. Neutralization of Tumor Acidity Improves Antitumor Responses to Immunotherapy. Cancer Res. 2016, 76, 1381–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damaghi, M.; Tafreshi, N.K.; Lloyd, M.C.; Sprung, R.; Estrella, V.; Wojtkowiak, J.W.; Morse, D.L.; Koomen, J.M.; Bui, M.M.; Gatenby, R.A.; et al. Chronic acidosis in the tumour microenvironment selects for overexpression of LAMP2 in the plasma membrane. Nat. Commun. 2015, 6, 8752. [Google Scholar] [CrossRef] [Green Version]
- Bon, E.; Driffort, V.; Gradek, F.; Martinez-Caceres, C.; Anchelin, M.; Pelegrin, P.; Cayuela, M.L.; Marionneau-Lambot, S.; Oullier, T.; Guibon, R.; et al. SCN4B acts as a metastasis-suppressor gene preventing hyperactivation of cell migration in breast cancer. Nat. Commun. 2016, 7, 13648. [Google Scholar] [CrossRef] [Green Version]
- Haworth, A.S.; Brackenbury, W.J. Emerging roles for multifunctional ion channel auxiliary subunits in cancer. Cell Calcium 2019, 80, 125–140. [Google Scholar] [CrossRef]
- Corbet, C.; Pinto, A.; Martherus, R.; de Jesus, J.P.S.; Polet, F.; Feron, O. Acidosis Drives the Reprogramming of Fatty Acid Metabolism in Cancer Cells through Changes in Mitochondrial and Histone Acetylation. Cell Metab. 2018, 24, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Malone, C.F.; Emerson, C.; Ingraham, R.; Barbosa, W.; Guerra, S.; Yoon, H.; Liu, L.L.; Michor, F.; Haigis, M.; Macleod, K.F.; et al. mTOR and HDAC Inhibitors Converge on the TXNIP/Thioredoxin Pathway to Cause Catastrophic Oxidative Stress and Regression of RAS-Driven Tumors. Cancer Discov. 2017, 7, 1450–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flinck, M.; Kramer, S.H.; Pedersen, S.F. Roles of pH in control of cell proliferation. Acta Physiol. (Oxf.) 2018, 223, e13068. [Google Scholar] [CrossRef] [PubMed]
- Peppicelli, S.; Andreucci, E.; Ruzzolini, J.; Laurenzana, A.; Margheri, F.; Fibbi, G.; Del Rosso, M.; Bianchini, F.; Calorini, L. The acidic microenvironment as a possible niche of dormant tumor cells. Cell Mol. Life Sci. 2017, 74, 2761–2771. [Google Scholar] [CrossRef] [PubMed]
- Robey, I.F.; Baggett, B.K.; Kirkpatrick, N.D.; Roe, D.J.; Dosescu, J.; Sloane, B.F.; Hashim, A.I.; Morse, D.L.; Raghunand, N.; Gatenby, R.A.; et al. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. 2009, 69, 2260–2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim-Hashim, A.; Cornnell, H.H.; Abrahams, D.; Lloyd, M.; Bui, M.; Gillies, R.J.; Gatenby, R.A. Systemic buffers inhibit carcinogenesis in TRAMP mice. J. Urol. 2012, 188, 624–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas (TCGA). Available online: http://cancergenome.nih.gov/ (accessed on 3 June 2020).
- University of California Santa Cruz (UCSC) Xena Functional Genomic Browser. Available online: https://xenabrowser.net/ (accessed on 3 June 2020).
- R Package (Version 1.28.0) for Rank-Rank Hypergeometric Overlap (RRHO) Test. Available online: https://bioconductor.org/packages/release/bioc/html/RRHO.html (accessed on 3 June 2020).
- Plaisier, S.B.; Taschereau, R.; Wong, J.A.; Graeber, T.G. Rank-rank hypergeometric overlap: Identification of statistically significant overlap between gene-expression signatures. Nucleic Acids Res. 2010, 38, e169. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types of Cancer | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene Name | Log2FC | FDR | Correlation with Poor OS | PAAD | BRCA | LUAD | GBM | COAD | OV | THCA | STAD | Molecular Function/Biological Process (UniProt) |
| 4 OVERLAPS | ||||||||||||
| SMAD9 | 1.1723 | 0.0091 | Low expression | DNA-binding, transcription regulation | ||||||||
| 3 OVERLAPS | ||||||||||||
| LGR4 | 0.7849 | 0.0127 | High expression | Differentiation, immunity | ||||||||
| RARG | 0.6840 | 0.0117 | High expression | DNA-binding, transcription regulation | ||||||||
| PNISR | 0.5890 | 0.0258 | Low expression | RNA-binding | ||||||||
| PCOLCE2 | 0.5538 | 0.0408 | High expression | Collagen biosynthesis | ||||||||
| RALGDS | 0.5128 | 0.0249 | Low expression | GTPase regulator, signal transduction | ||||||||
| 2 OVERLAPS | ||||||||||||
| SCNN1A | 2.0490 | 0.0100 | High expression | Ion transport | ||||||||
| PLA2R1 | 1.6361 | 0.0249 | High expression | Endocytosis | ||||||||
| PDK4 | 1.6088 | 0.0351 | High expression | Ser/Thr protein kinase, metabolism | ||||||||
| SNED1 | 1.5574 | 0.0302 | High expression | Cell-matrix adhesion | ||||||||
| ADAMTSL4 | 1.5083 | 0.0176 | High expression | Peptidase, apoptosis | ||||||||
| RAMP1 | 1.4724 | 0.0169 | High expression | Transport, angiogenesis | ||||||||
| MFAP2 | 1.3770 | 0.0154 | High expression | Embryonic morphogenesis | ||||||||
| HSPG2 | 1.2833 | 0.0073 | High expression | Angiogenesis, morphogenesis, | ||||||||
| GNG7 | 1.2239 | 0.0142 | Low expression | GTPase activity | ||||||||
| HOXA11 | 1.1569 | 0.0346 | High expression | DNA-binding, transcription regulation | ||||||||
| ALDH3B1 | 1.1061 | 0.0081 | High expression | Oxidoreductase, metabolism | ||||||||
| CORO2A | 1.0708 | 0.0321 | High expression | Actin-binding, signal transduction | ||||||||
| LRP1 | 1.0403 | 0.0380 | High expression | Endocytosis | ||||||||
| TMEM8B | 0.9897 | 0.0045 | Low expression | Cell adhesion, growth regulation | ||||||||
| ZNF710-AS1 | 0.9413 | 0.0330 | Low expression | lncRNA | ||||||||
| PYGL | 0.8856 | 0.0251 | High expression | Transferase, metabolism | ||||||||
| TMED7-TICAM2 | 0.8634 | 0.0325 | High expression | Golgi organization, protein transport | ||||||||
| NPEPL1 | 0.8764 | 0.0117 | Low expression | Aminopeptidase | ||||||||
| SMARCD3 | 0.8743 | 0.0058 | Low expression | Chromatin regulator, neurogenesis | ||||||||
| RHOBTB1 | 0.8684 | 0.0234 | Low expression | GTPase activity, actin organization | ||||||||
| UNC13D | 0.8587 | 0.0368 | High expression | Exocytosis | ||||||||
| DBP | 0.8034 | 0.0249 | Low expression | DNA-binding, transcription regulation | ||||||||
| PDCD4 | 0.7330 | 0.0077 | Low expression | RNA-binding, apoptosis | ||||||||
| MTMR7 | 0.7273 | 0.0329 | Low expression | Phosphatase | ||||||||
| OTX1 | 0.6797 | 0.0091 | High expression | DNA-binding, morphogenesis | ||||||||
| TXNRD3 | 0.6584 | 0.0125 | High expression | Differentiation, electron transport | ||||||||
| PPOX | 0.6399 | 0.0279 | Low expression | Oxidoreductase, heme biosynthesis | ||||||||
| SPAG4 | 0.6392 | 0.0404 | High expression | Differentiation, spermatogenesis | ||||||||
| ANO7 | 0.6260 | 0.0329 | Low expression | Lipid transport | ||||||||
| FAHD2B | 0.6164 | 0.0069 | High expression | Hydrolase | ||||||||
| ORAI3 | 0.6063 | 0.0224 | Low expression | Calcium channel | ||||||||
| NOP53 | 0.5938 | 0.0156 | High expression | Host-virus interaction, ribosome biogenesis | ||||||||
| C17orf49 | 0.5910 | 0.0081 | Low expression | Chromatin regulator, DNA-binding | ||||||||
| VAMP2 | 0.5901 | 0.0073 | Low expression | Exocytosis, protein transport | ||||||||
| ZNF487 | 0.5872 | 0.0377 | Low expression | DNA-binding, transcription regulation | ||||||||
| TDRD3 | 0.5832 | 0.0058 | Low expression | Chromatin regulator, RNA-binding | ||||||||
| FRG1HP | 0.5302 | 0.0376 | Low expression | Pseudogene | ||||||||
| PLD2 | 0.5226 | 0.0306 | High expression | Hydrolase, lipid metabolism, motility | ||||||||
| MLLT6 | 0.5118 | 0.0038 | Low expression | Histone-binding, metal ion-binding | ||||||||
| Types of Cancer | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene Name | Log2FC | FDR | Correlation with Poor OS | PAAD | BRCA | LUAD | GBM | COAD | OV | THCA | STAD | Molecular Function/Biological Process (UniProt) |
| 5 OVERLAPS | ||||||||||||
| ZNF557 | −0.5999 | 0.0069 | Low expression | DNA-binding, transcription factor, metal on binding | ||||||||
| 3 OVERLAPS | ||||||||||||
| MDGA1 | −1.2003 | 0.0153 | High expression | Differentiation, neurogenesis | ||||||||
| VEGFC | −0.8963 | 0.0345 | High expression | Growth factor, angiogenesis, differentiation | ||||||||
| MCM10 | −0.8486 | 0.0249 | High expression | DNA-binding, DNA replication, DNA damage | ||||||||
| CTBP1-DT | −0.5490 | 0.0098 | Low expression | Oxidoreductase, differentiation, transcription | ||||||||
| CHMP4A | −0.5182 | 0.0247 | Low expression | Protein transport | ||||||||
| 2 OVERLAPS | ||||||||||||
| CGN | −1.5661 | 0.0112 | High expression | Tight junction regulation | ||||||||
| AKAP5 | −1.4671 | 0.0408 | Low expression | Calmodulin-binding | ||||||||
| HIST1H4H | −1.3143 | 0.0276 | High expression | DNA-binding | ||||||||
| STYK1 | −1.0844 | 0.0290 | High expression | Protein tyrosine kinase | ||||||||
| CDC25A | −1.0023 | 0.0182 | Low expression | Protein phosphatase, cell cycle, cell division | ||||||||
| RRM2 | −0.9925 | 0.0325 | High expression | Oxidoreductase, DNA replication | ||||||||
| VDR | −0.9901 | 0.0131 | High expression | Vitamin D3 receptor, DNA-binding, transcription | ||||||||
| CDC6 | −0.8805 | 0.0110 | High expression | Cell cycle, cell division, DNA replication | ||||||||
| GINS4 | −0.8574 | 0.0376 | High expression | DNA replication | ||||||||
| AC019069.1 | −0.8118 | 0.0467 | High expression | lncRNA | ||||||||
| TCF19 | −0.7835 | 0.0296 | High expression | DNA-binding, transcription factor | ||||||||
| GINS2 | −0.7784 | 0.0182 | Low expression | DNA replication | ||||||||
| SOWAHC | −0.7653 | 0.0201 | High expression | Ankyrin repeat domain-containing protein | ||||||||
| BRCA2 | −0.7332 | 0.0375 | High expression | DNA-binding, DNA recombination, DNA damage | ||||||||
| AC092279.1 | −0.6853 | 0.0203 | Low expression | lncRNA | ||||||||
| CDCA5 | −0.6652 | 0.0425 | High expression | Cell cycle, cell division | ||||||||
| TUBB2A | −0.6559 | 0.0360 | High expression | GTP-binding, microtubule cytoskeleton organization | ||||||||
| OAS3 | −0.6518 | 0.0117 | High expression | RNA-binding, antiviral defense, immunity | ||||||||
| SFN | −0.6382 | 0.0158 | High expression | DNA damage response | ||||||||
| MTHFD1 | −0.6333 | 0.0107 | High expression | Embryonic morphogenesis | ||||||||
| ZNF562 | −0.6041 | 0.0069 | Low expression | DNA-binding, transcription factor, metal ion binding | ||||||||
| MASTL | −0.6008 | 0.0482 | High expression | Ser/Thr protein kinase, cell division | ||||||||
| CDCA4 | −0.5907 | 0.0236 | High expression | Cell division | ||||||||
| MMS22L | −0.5725 | 0.0418 | Low expression | DNA repair | ||||||||
| RMI1 | −0.5661 | 0.0443 | Low expression | DNA replication | ||||||||
| C1orf112 | −0.5581 | 0.0415 | High expression | Uncharacterized protein C1orf112 | ||||||||
| MIR4435-2HG | −0.5537 | 0.0105 | High expression | lncRNA | ||||||||
| MCM4 | −0.5328 | 0.0482 | High expression | DNA-binding, cell cycle, DNA replication | ||||||||
| DSCC1 | −0.5268 | 0.0492 | High expression | DNA-binding, cell cycle, DNA replication | ||||||||
| USP1 | −0.5192 | 0.0330 | Low expression | Protease, DNA repair | ||||||||
| MCM3 | −0.5129 | 0.0471 | Low expression | Transferase, mRNA transport, immunity | ||||||||
| EIF6 | −0.5008 | 0.0100 | High expression | Ribosome biogenesis, protein synthesis | ||||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, J.; Czaplinska, D.; Ialchina, R.; Schnipper, J.; Liu, B.; Sandelin, A.; Pedersen, S.F. Cancer Cell Acid Adaptation Gene Expression Response Is Correlated to Tumor-Specific Tissue Expression Profiles and Patient Survival. Cancers 2020, 12, 2183. https://doi.org/10.3390/cancers12082183
Yao J, Czaplinska D, Ialchina R, Schnipper J, Liu B, Sandelin A, Pedersen SF. Cancer Cell Acid Adaptation Gene Expression Response Is Correlated to Tumor-Specific Tissue Expression Profiles and Patient Survival. Cancers. 2020; 12(8):2183. https://doi.org/10.3390/cancers12082183
Chicago/Turabian StyleYao, Jiayi, Dominika Czaplinska, Renata Ialchina, Julie Schnipper, Bin Liu, Albin Sandelin, and Stine Falsig Pedersen. 2020. "Cancer Cell Acid Adaptation Gene Expression Response Is Correlated to Tumor-Specific Tissue Expression Profiles and Patient Survival" Cancers 12, no. 8: 2183. https://doi.org/10.3390/cancers12082183
APA StyleYao, J., Czaplinska, D., Ialchina, R., Schnipper, J., Liu, B., Sandelin, A., & Pedersen, S. F. (2020). Cancer Cell Acid Adaptation Gene Expression Response Is Correlated to Tumor-Specific Tissue Expression Profiles and Patient Survival. Cancers, 12(8), 2183. https://doi.org/10.3390/cancers12082183