Altered Organelle Calcium Transport in Ovarian Physiology and Cancer


 ,
,  ,
, 
 and
and
Abstract
:1. Introduction
2. Understanding Ovarian Cancer
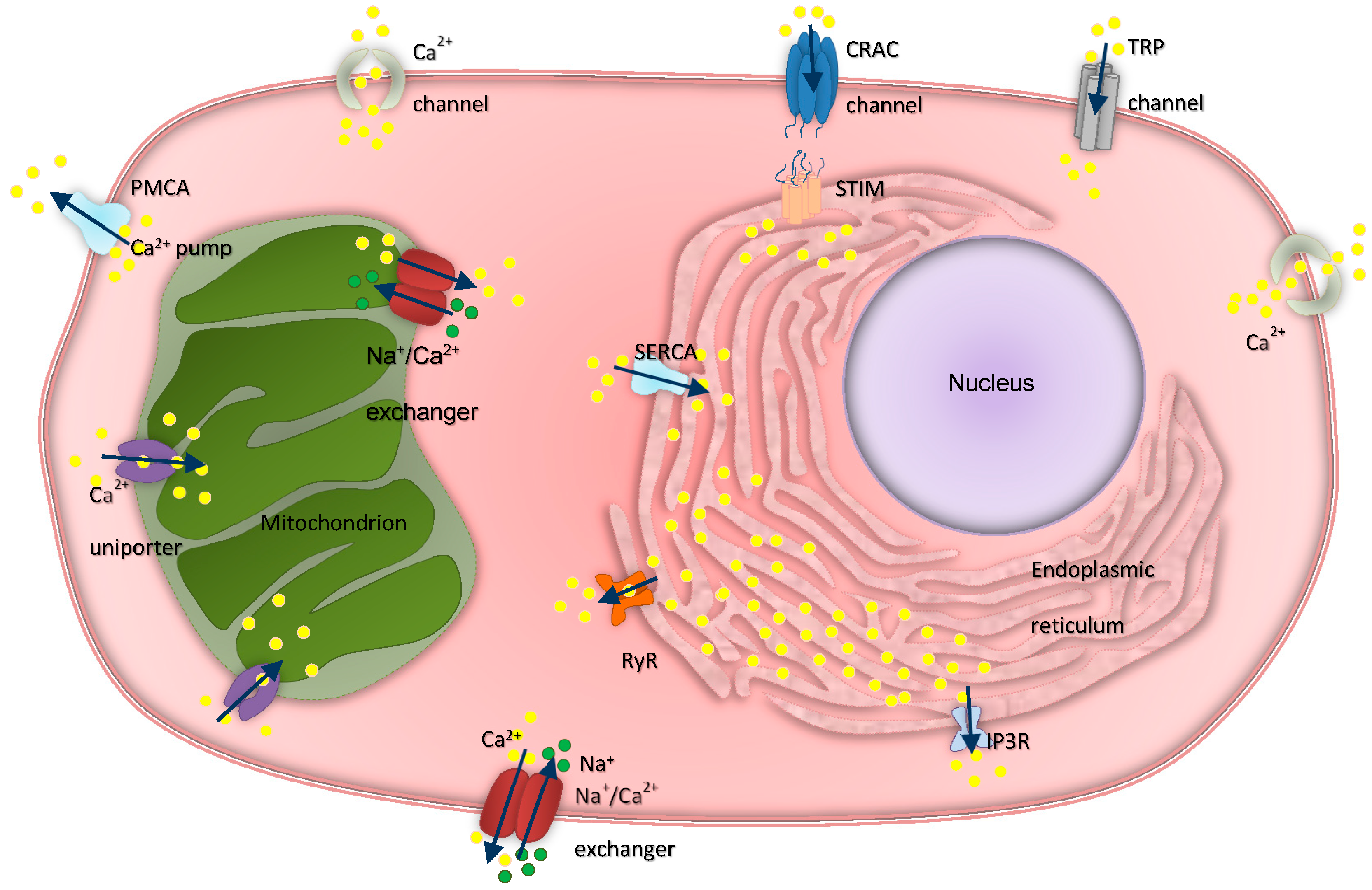
3. Intracellular Calcium Regulation in Ovarian Physiology and Cancer
3.1. Inositol Trisphosphate Receptors
3.2. Ryanodine Receptors
3.3. Transient Receptor Potential Channels and Calcium Release-Activated Channels
3.4. Calcium ATPases
3.5. Mitochondrial Calcium Channels
3.6. G-Protein-Coupled Receptors
3.7. Hormone Receptors
4. Contribution of Altered Calcium Signaling to the Development of Chemotherapy Resistance in Ovarian Cancer
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yang, X.; Zhu, S.; Li, L.; Zhang, L.; Xian, S.; Wang, Y.; Cheng, Y. Identification of differentially expressed genes and signaling pathways in ovarian cancer by integrated bioinformatics analysis. OncoTargets Ther. 2018, 11, 1457–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, M.; Li, X.; Hu, Y.-X.; Dong, H.; Gou, R.; Nie, X.; Liu, Q.; Ying-Ying, H.; Liu, J.-J.; Lin, B. Identification of molecular marker associated with ovarian cancer prognosis using bioinformatics analysis and experiments. J. Cell. Physiol. 2019, 234, 11023–11036. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decuypere, J.P.; Monaco, G.; Bultynck, G.; Missiaen, L.; De Smedt, H.; Parys, J.B. The IP3 receptor–mitochondria connection in apoptosis and autophagy. Biochim. Biophys. Acta. 2011, 1813, 1003–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerella, C.; Diederich, M.; Ghibelli, L. The Dual Role of Calcium as Messenger and Stressor in Cell Damage, Death, and Survival. Int. J. Cell Boil. 2010, 2010, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Karlstad, J.; Sun, Y.; Singh, B.B. Ca2+ Signaling: An Outlook on the Characterization of Ca2+ Channels and Their Importance in Cellular Functions. Adv. Exp. Med. Biol. 2012, 740, 143–157. [Google Scholar] [CrossRef] [Green Version]
- Csanády, L.; Mindell, J.A. The twain shall meet: Channels, transporters and things between. Meeting on Membrane Transport in Flux: The Ambiguous Interface Between Channels and Pumps. EMBO Rep. 2008, 9, 960–965. [Google Scholar] [CrossRef] [Green Version]
- Catterall, W.A.; Perez-Reyes, E.; Snutch, T.P.; Striessnig, J. International Union of Pharmacology. XLVIII. Nomenclature and Structure-Function Relationships of Voltage-Gated Calcium Channels. Pharmacol. Rev. 2005, 57, 411–425. [Google Scholar] [CrossRef]
- Yamakage, M.; Namiki, A. Calcium channels—Basic aspects of their structure, function and gene encoding; anesthetic action on the channels—A review. Can. J. Anaesth. 2002, 49, 151–164. [Google Scholar] [CrossRef] [Green Version]
- Striggow, F.; Ehrlich, B.E. Ligand-gated calcium channels inside and out. Curr. Opin. Cell Boil. 1996, 8, 490–495. [Google Scholar] [CrossRef]
- Gadsby, D.C. Ion channels versus ion pumps: The principal difference, in principle. Nat. Rev. Mol. Cell Boil. 2009, 10, 344–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, S.-K.; Hirabayashi, Y.; Polleux, F. Organelle-Specific Sensors for Monitoring Ca2+ Dynamics in Neurons. Front. Synaptic Neurosci. 2016, 8, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, M.C.X.; Kihara, A.H.; Goulart, V.A.; Tonelli, F.M.P.; Gomes, K.N.; Ulrich, H.; Resende, R.R. Calcium signaling and cell proliferation. Cell. Signal. 2015, 27, 2139–2149. [Google Scholar] [CrossRef]
- Missiaen, L.; Robberecht, W.; Bosch, L.V.D.; Callewaert, G.; Parys, J.; Wuytack, F.; Raeymaekers, L.; Nilius, B.; Eggermont, J.; Smedt, H. Abnormal intracellular Ca2+homeostasis and disease. Cell Calcium 2000, 28, 1–21. [Google Scholar] [CrossRef]
- Lorenzon, N.M.; Beam, K.G. Disease causing mutations of calcium channels. Channels 2008, 2, 163–179. [Google Scholar] [CrossRef] [Green Version]
- Supnet, C.; Bezprozvanny, I. The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium 2010, 47, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J. Calcium hypothesis of Alzheimer’s disease. Pflugers Arch. 2010, 459, 441–449. [Google Scholar] [CrossRef]
- Blayney, L.M.; Lai, F.A. Ryanodine receptor-mediated arrhythmias and sudden cardiac death. Pharmacol. Ther. 2009, 123, 151–177. [Google Scholar] [CrossRef] [Green Version]
- Gyorke, S. Molecular basis of catecholaminergic polymorphic ventricular tachycardia. Hear. Rhythm. 2009, 6, 123–129. [Google Scholar] [CrossRef]
- Foggia, L.; Hovnanian, A. Calcium pump disorders of the skin. Am. J. Med. Genet. 2004, 131c, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Monteith, G.; Davis, F.M.; Roberts-Thomson, S. Calcium Channels and Pumps in Cancer: Changes and Consequences*. J. Boil. Chem. 2012, 287, 31666–31673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prevarskaya, N.; Ouadid-Ahidouch, H.; Skryma, R.; Shuba, Y. Remodelling of Ca2+ transport in cancer: How it contributes to cancer hallmarks? Philos. Trans. R. Soc. B Boil. Sci. 2014, 369, 20130097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Targeting Ca2+transport in cancer: Close reality or long perspective? Expert Opin. Ther. Targets 2013, 17, 225–241. [Google Scholar] [CrossRef]
- Feske, S. Calcium signalling in lymphocyte activation and disease. Nat. Rev. Immunol. 2007, 7, 690–702. [Google Scholar] [CrossRef]
- Feske, S.; Picard, C.; Fischer, A. Immunodeficiency due to mutations in ORAI1 and STIM1. Clin. Immunol. 2010, 135, 169–182. [Google Scholar] [CrossRef] [Green Version]
- Shim, A.H.-R.; Tirado-Lee, L.; Prakriya, M. Structural and Functional Mechanisms of CRAC Channel Regulation. J. Mol. Boil. 2015, 427, 77–93. [Google Scholar] [CrossRef] [Green Version]
- Klemann, C.; Ammann, S.; Heizmann, M.; Fuchs, S.; Bode, S.; Heeg, M.; Fuchs, H.; Lehmberg, K.; Stadt, U.Z.; Roll, C.; et al. Hemophagocytic lymphohistiocytosis as presenting manifestation of profound combined immunodeficiency due to an ORAI1 mutation. J. Allergy Clin. Immunol. 2017, 140, 1721–1724. [Google Scholar] [CrossRef] [Green Version]
- Borowiec, A.-S.; Bidaux, G.; Pigat, N.; Goffin, V.; Bernichtein, S.; Capiod, T. Calcium channels, external calcium concentration and cell proliferation. Eur. J. Pharmacol. 2014, 739, 19–25. [Google Scholar] [CrossRef]
- Lee, J.M.; Davis, F.M.; Roberts-Thomson, S.; Monteith, G. Ion channels and transporters in cancer. 4. Remodeling of Ca2+ signaling in tumorigenesis: Role of Ca2+ transport. Am. J. Physiol. 2011, 301, C969–C976. [Google Scholar] [CrossRef] [Green Version]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion channels and the hallmarks of cancer. Trends Mol. Med. 2010, 16, 107–121. [Google Scholar] [CrossRef]
- Høyer-Hansen, M.; Jäättelä, M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007, 14, 1576–1582. [Google Scholar] [CrossRef]
- Giorgi, C.; Baldassari, F.; Bononi, A.; Bonora, M.; De Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; et al. Mitochondrial Ca2+ and apoptosis. Cell Calcium 2012, 52, 36–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, M.; Zhang, Y.; Jin, K.; Lu, Z.; Zeng, Z.; Xiong, W. Communication between mitochondria and other organelles: A brand-new perspective on mitochondria in cancer. Cell Biosci. 2019, 9, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Crisosto, C.; Bravo-Sagua, R.; Rodriguez-Peña, M.; Mera, C.; Castro, P.F.; Quest, A.F.; Rothermel, B.A.; Cifuentes, M.; Lavandero, S. ER-to-mitochondria miscommunication and metabolic diseases. Biochim. Biophys. Acta. 2015, 1852, 2096–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Min, K.-T. The Interface Between ER and Mitochondria: Molecular Compositions and Functions. Mol. Cells 2018, 41, 1000–1007. [Google Scholar]
- Wieckowski, M.R.; Giorgi, C.; Lebiedzinska, M.; Duszynski, J.; Pinton, P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 2009, 4, 1582–1590. [Google Scholar] [CrossRef]
- Giorgi, C.; Missiroli, S.; Patergnani, S.; Duszynski, J.; Wieckowski, M.R.; Pinton, P.; Wieckowski, M.R. Mitochondria-Associated Membranes: Composition, Molecular Mechanisms, and Physiopathological Implications. Antioxidants Redox Signal. 2015, 22, 995–1019. [Google Scholar] [CrossRef]
- Cárdenas, C.; Pinton, P.; Bultynck, G. Editorial: Inter-Organelle Calcium Communication in Cancer. Front. Oncol. 2018, 8, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm. Sin. B 2016, 7, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Predescu, D.-V.; Crețoiu, S.M.; Pavelescu, L.A.; Suciu, N.; Radu, B.M.; Voinea, S.-C.; Crețoiu, D. G Protein-Coupled Receptors (GPCRs)-Mediated Calcium Signaling in Ovarian Cancer: Focus on GPCRs activated by Neurotransmitters and Inflammation-Associated Molecules. Int. J. Mol. Sci. 2019, 20, 5568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Seas, A.; Kiyani, M.; Ji, K.S.Y.; Bell, H.N. A temporal examination of calcium signaling in cancer- from tumorigenesis, to immune evasion, and metastasis. Cell Biosci. 2018, 8, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Zhan, X. Signaling pathway network alterations in human ovarian cancers identified with quantitative mitochondrial proteomics. EPMA J. 2019, 10, 153–172. [Google Scholar] [CrossRef] [Green Version]
- Erickson, B.K.; Conner, M.G.; Landen, C.N. The role of the fallopian tube in the origin of ovarian cancer. Am. J. Obstet. Gynecol. 2013, 209, 409–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magoffin, D.A. Ovarian theca cell. Int. J. Biochem. Cell Boil. 2005, 37, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Goodman, H.M. Chapter 13—Hormonal Control of Reproduction in the Female: The Menstrual Cycle. In Basic Medical Endocrinology, 4th ed.; Goodman, H.M., Ed.; Academic Press: San Diego, CA, USA, 2009; pp. 257–275. [Google Scholar]
- Fujisawa, M.; Moh-Moh-Aung, A.; Zeng, Z.; Yoshimura, T.; Wani, Y.; Matsukawa, A. Ovarian stromal cells as a source of cancer-associated fibroblasts in human epithelial ovarian cancer: A histopathological study. PLoS ONE 2018, 13, e0205494. [Google Scholar] [CrossRef] [Green Version]
- Richards, J.S.; Pangas, S.A. The ovary: Basic biology and clinical implications. J. Clin. Investig. 2010, 120, 963–972. [Google Scholar] [CrossRef]
- Matz, M.; Coleman, M.P.; Sant, M.; Chirlaque, M.D.; Visser, O.; Gore, M.; Allemani, C.; Bouzbid, S.; Hamdi-Chérif, M.; Zaidi, Z.; et al. The histology of ovarian cancer: Worldwide distribution and implications for international survival comparisons (CONCORD-2). Gynecol. Oncol. 2017, 144, 405–413. [Google Scholar] [CrossRef]
- Brenton, J.D.; Stingl, J. Anatomy of an ovarian cancer. Nature 2013, 495, 183–184. [Google Scholar] [CrossRef]
- Flesken-Nikitin, A.; Hwang, C.-I.; Cheng, C.-Y.; Michurina, T.V.; Yenikolopov, G.; Nikitin, A.Y. Ovarian surface epithelium at the junction area contains a cancer-prone stem cell niche. Nature 2013, 495, 241–245. [Google Scholar] [CrossRef]
- Scully, R.E.; Sobin, L.H. Histologic typing of ovarian tumors. Arch. Pathol. Lab. Med. 1987, 111, 794–795. [Google Scholar]
- McCluggage, W.G. Morphological subtypes of ovarian carcinoma: A review with emphasis on new developments and pathogenesis. Pathology 2011, 43, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih, I.-M. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vo, C.; Carney, M.E. Ovarian Cancer Hormonal and Environmental Risk Effect. Obstet. Gynecol. Clin. North. Am. 2007, 34, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Dossus, L.; Allen, N.; Kaaks, R.; Bakken, K.; Lund, E.; Tjonneland, A.; Olsen, A.; Overvad, K.; Clavel-Chapelon, F.; Fournier, A.; et al. Reproductive risk factors and endometrial cancer: The European Prospective Investigation into Cancer and Nutrition. Int. J. Cancer 2010, 127, 442–451. [Google Scholar] [CrossRef]
- Fuh, K.; Shin, J.; Kapp, D.S.; Brooks, R.A.; Ueda, S.; Urban, R.R.; Chen, L.-M.; Chan, J.K. Survival differences of Asian and Caucasian epithelial ovarian cancer patients in the United States. Gynecol. Oncol. 2015, 136, 491–497. [Google Scholar] [CrossRef]
- Sugiyama, T.; Kamura, T.; Kigawa, J.; Terakawa, N.; Kikuchi, Y.; Kita, T.; Suzuki, M.; Sato, I.; Taguchi, K. Clinical characteristics of clear cell carcinoma of the ovary. Cancer 2000, 88, 2584–2589. [Google Scholar] [CrossRef]
- Kobayashi, C.; Kajihara, H.; Kanayama, S.; Noguchi, T.; Haruta, S.; Sado, T.; Yamada, Y.; Furukawa, N.; Yoshida, S.; Oi, H.; et al. Clear cell carcinoma of the ovary: Potential pathogenic mechanisms (Review). Oncol. Rep. 2010, 23, 1193–1203. [Google Scholar] [CrossRef]
- Uekuri, C.; Shigetomi, H.; Ono, S.; Sasaki, Y.; Matsuura, M.; Kobayashi, H. Toward an understanding of the pathophysiology of clear cell carcinoma of the ovary (Review). Oncol. Lett. 2013, 6, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Terada, T. Endometrioid adenocarcinoma of the ovary arising in atypical endometriosis. Int. J. Clin. Exp. Pathol. 2012, 5, 924–927. [Google Scholar]
- Jung, S.E.; Lee, J.M.; Rha, S.E.; Byun, J.Y.; Jung, J.I.; Hahn, S.T. CT and MR Imaging of Ovarian Tumors with Emphasis on Differential Diagnosis. Radiographics 2002, 22, 1305–1325. [Google Scholar] [CrossRef] [PubMed]
- DiPiro, J.T.; Talbert, R.L.; Yee, G.C.; Matzke, G.R.; Wells, B.G.; Posey, L.M.; Streetman, D.S.; Streetman, D.-A.D. Book Review: Pharmacotherapy: A Pathophysiologic Approach, 7th Edition. Ann. Pharmacother. 2009, 43, 395. [Google Scholar] [CrossRef]
- Kelemen, L.E.; Köbel, M. Mucinous carcinomas of the ovary and colorectum: Different organ, same dilemma. Lancet Oncol. 2011, 12, 1071–1080. [Google Scholar] [CrossRef]
- Patni, R. Squamous cell carcinoma arising in mature cystic teratoma of ovary. J. MidLife Health 2014, 5, 195–197. [Google Scholar] [CrossRef]
- Ichigo, S.; Takagi, H.; Matsunami, K.; Murase, T.; Ikeda, T.; Imai, A. Transitional cell carcinoma of the ovary (Review). Oncol. Lett. 2011, 3, 3–6. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.K.; Lu, K.H.; Malpica, A.; Bodurka, D.; Shvartsman, H.S.; Schmandt, R.E.; Thornton, A.D.; Deavers, M.T.; Silva, E.G.; Gershenson, D.M. Significantly Greater Expression of ER, PR, and ECAD in Advanced-Stage Low-Grade Ovarian Serous Carcinoma as Revealed by Immunohistochemical Analysis. Int. J. Gynecol. Pathol. 2007, 26, 404–409. [Google Scholar] [CrossRef]
- Kaldawy, A.; Segev, Y.; Lavie, O.; Auslender, R.; Sopik, V.; Narod, S.A. Low-grade serous ovarian cancer: A review. Gynecol. Oncol. 2016, 143, 433–438. [Google Scholar] [CrossRef]
- Gore, C.R.; Patvekar, M.M.; Kurade, S.J.; Kumar, H.; Pagaro, P.M. Malignant Mixed Mullerian Tumor of the Ovary. J. Obstet. Gynecol. India 2012, 64, 62–64. [Google Scholar] [CrossRef] [Green Version]
- Tafe, L.J.; Garg, K.; Chew, I.; Tornos, C.; Soslow, R.A. Endometrial and ovarian carcinomas with undifferentiated components: Clinically aggressive and frequently underrecognized neoplasms. Mod. Pathol. 2010, 23, 781–789. [Google Scholar] [CrossRef]
- Salcedo-Hernández, R.A.; Lino-Silva, L.S.; De León, D.C.; Perez-Montiel, D.; Luna-Ortiz, K. Ovarian undifferentiated carcinoma with voluminous mesenteric presentation. Int. J. Surg. Case Rep. 2012, 3, 551–554. [Google Scholar] [CrossRef] [Green Version]
- Bowtell, D.; Böhm, S.; Ahmed, A.A.; Aspuria, P.-J.; Bast, R.C.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Bowtell, D. The genesis and evolution of high-grade serous ovarian cancer. Nat. Rev. Cancer 2010, 10, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Park, E.Y.; Kim, O.; Schilder, J.M.; Coffey, D.M.; Cho, C.-H.; Bast, R.C. Cell Origins of High-Grade Serous Ovarian Cancer. Cancers 2018, 10, 433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisio, M.-A.; Fu, L.; Goyeneche, A.; Gao, Z.-H.; Telleria, C.M. High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. Int. J. Mol. Sci. 2019, 20, 952. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Cao, L.; Nguyen, D.; Lu, H. TP53 mutations in epithelial ovarian cancer. Transl. Cancer Res. 2016, 5, 650–663. [Google Scholar] [CrossRef]
- Kraggerud, S.M.; Hoei-Hansen, C.E.; Alagaratnam, S.; Skotheim, R.I.; Abeler, V.M.; De Meyts, E.R.; Lothe, R.A. Molecular characteristics of malignant ovarian germ cell tumors and comparison with testicular counterparts: Implications for pathogenesis. Endocr. Rev. 2013, 34, 339–376. [Google Scholar] [CrossRef]
- Gimelli, S.; Beri, S.; Drabkin, H.A.; Gambini, C.; Gregorio, A.; Fiorio, P.; Zuffardi, O.; Gemmill, R.M.; Giorda, R.; Gimelli, G. The tumor suppressor gene TRC8/RNF139 is disrupted by a constitutional balanced translocation t(8;22)(q24.13;q11.21) in a young girl with dysgerminoma. Mol. Cancer 2009, 8, 52. [Google Scholar] [CrossRef]
- Schultz, K.A.P.; Harris, A.K.; Schneider, D.T.; Young, R.H.; Brown, J.; Gershenson, D.M.; Dehner, L.P.; Hill, D.A.; Messinger, Y.H.; Frazier, A.L. Ovarian Sex Cord-Stromal Tumors. J. Oncol. Pr. 2016, 12, 940–946. [Google Scholar] [CrossRef] [Green Version]
- Horta, M.; Cunha, T.M. Sex cord-stromal tumors of the ovary: A comprehensive review and update for radiologists. Diagn. Interv. Radiol. 2015, 21, 277–286. [Google Scholar] [CrossRef]
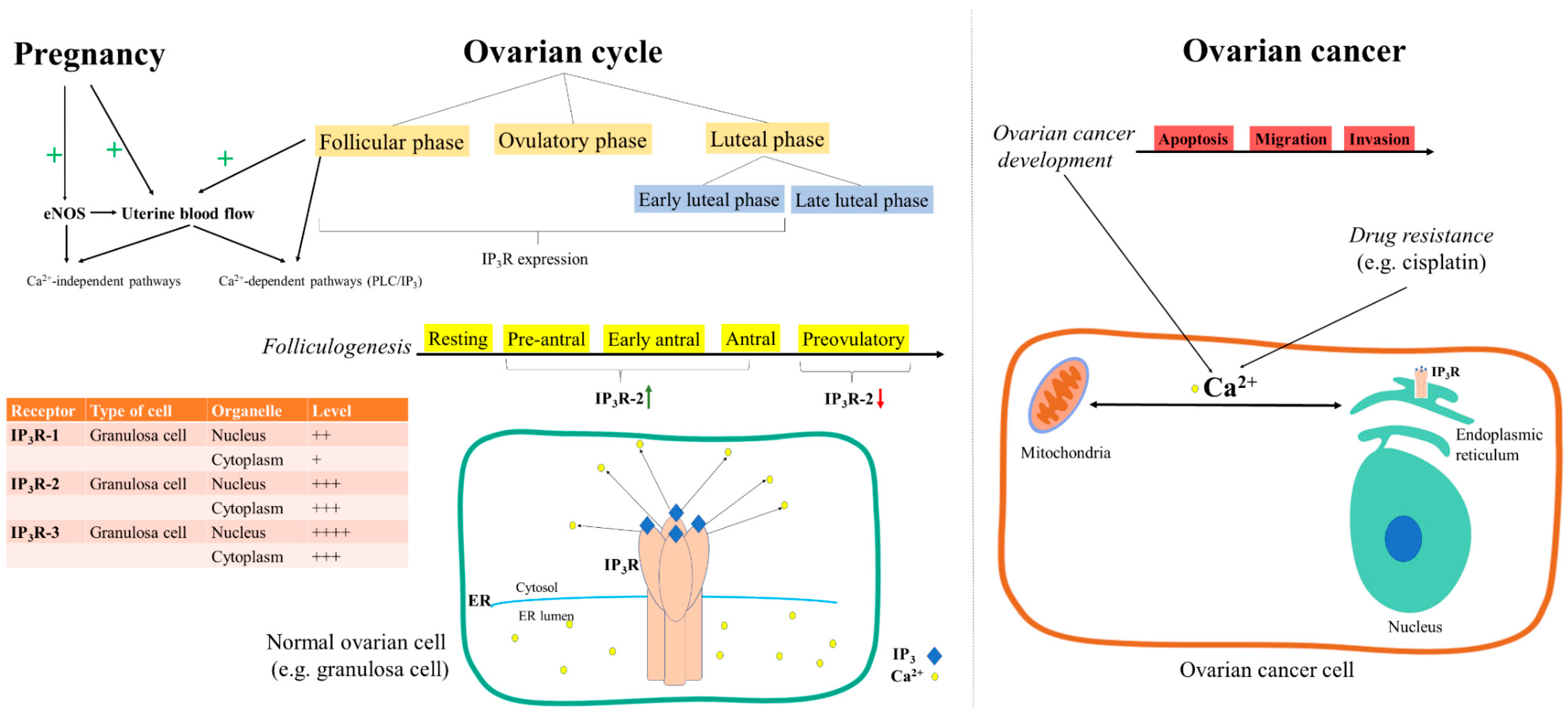
- Diaz-Munoz, M.; De La Rosa-Santander, P.; Juárez-Espinosa, A.B.; Arellano, R.O.; Tlalpan, V.M. Granulosa cells express three inositol 1,4,5-trisphosphate receptor isoforms: Cytoplasmic and nuclear Ca2+ mobilization. Reprod. Boil. Endocrinol. 2008, 6, 60. [Google Scholar] [CrossRef] [Green Version]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neher, E.; Sakaba, T. Multiple Roles of Calcium Ions in the Regulation of Neurotransmitter Release. Neuron 2008, 59, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanova, H.; Kerkhofs, M.; La Rovere, R.M.; Bultynck, G. Endoplasmic Reticulum–Mitochondrial Ca2+ Fluxes Underlying Cancer Cell Survival. Front. Oncol. 2017, 7, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Li, H. Effects of calcium channel on ovarian cancer cells. Oncol. Lett. 2017, 14, 6341–6344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frede, J.; Fraser, S.P.; Oskay-Özcelik, G.; Hong, Y.; Braicu, E.I.; Sehouli, J.; Gabra, H.; Djamgoz, M.B.A. Ovarian cancer: Ion channel and aquaporin expression as novel targets of clinical potential. Eur. J. Cancer 2013, 49, 2331–2344. [Google Scholar] [CrossRef]
- Li, W.; Zhang, S.-L.; Wang, N.; Zhang, B.-B.; Li, M. Blockade of T-Type Ca2+Channels Inhibits Human Ovarian Cancer Cell Proliferation. Cancer Investig. 2011, 29, 339–346. [Google Scholar] [CrossRef]
- Marchi, S.; Pinton, P. Alterations of calcium homeostasis in cancer cells. Curr. Opin. Pharmacol. 2016, 29, 1–6. [Google Scholar] [CrossRef]
- Di Capite., J.; Ng, S.W.; Parekh, A.B. Decoding of cytoplasmic Ca(2+) oscillations through the spatial signature drives gene expression. Curr. Biol. 2009, 19, 853–858. [Google Scholar] [CrossRef] [Green Version]
- De Koninck, P.; Schulman, H. Sensitivity of CaM Kinase II to the Frequency of Ca2+Oscillations. Science 1998, 279, 227–230. [Google Scholar] [CrossRef] [Green Version]
- Heemskerk, J.W.; Vis, P.; Feijge, M.A.; Hoyland, J.; Mason, W.T.; Sage, S.O. Roles of phospholipase C and Ca(2+)-ATPase in calcium responses of single, fibrinogen-bound platelets. J. Boil. Chem. 1993, 268, 356–363. [Google Scholar]
- Wakui, M.; Potter, B.V.L.; Petersen, O.H. Pulsatile intracellular calcium release does not depend on fluctuations in inositol trisphosphate concentration. Nature 1989, 339, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Tertyshnikova, S.; Fein, A. Dual Regulation of Calcium Mobilization by Inositol 1,4,5-Trisphosphate in a Living Cell. J. Gen. Physiol. 2000, 115, 481–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaji, R.; Bielmeier, C.; Harz, H.; Bates, J.A.; Stadler, C.; Hildebrand, A.; Classen, A.-K. Calcium spikes, waves and oscillations in a large, patterned epithelial tissue. Sci. Rep. 2017, 7, 42786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casarini, L.; Crépieux, P. Molecular Mechanisms of Action of FSH. Front. Endocrinol. 2019, 10, 305. [Google Scholar] [CrossRef]
- Selstam, G.; Rosberg, S.; Liljekvist, J.; Grönquist, L.; Perklev, T.; Ahrén, K. Differences in action of LH and FSH on the formation of cyclic amp in the prepubertal rat ovary. Eur. J. Endocrinol. 1976, 81, 150–164. [Google Scholar] [CrossRef]
- Flores, J.A.; Aguirre, C.; Sharma, O.P.; Veldhuis, J.D. Luteinizing Hormone (LH) Stimulates Both Intracellular Calcium Ion ([Ca2+]i) Mobilization and Transmembrane Cation Influx in Single Ovarian (Granulosa) Cells: Recruitment as a Cellular Mechanism of LH-[Ca2+]i Dose Response*. Endocrinology 1998, 139, 3606–3612. [Google Scholar] [CrossRef]
- Dziegielewska, B.; Casarez, E.V.; Yang, W.Z.; Gray, L.S.; Dziegielewski, J.; Slack-Davis, J.K. T-Type Ca2+ Channel Inhibition Sensitizes Ovarian Cancer to Carboplatin. Mol. Cancer Ther. 2016, 15, 460–470. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Rubaiy, H.N.; Chen, G.; Hallett, T.; Zaibi, N.; Zeng, B.; Saurabh, R.; Xu, S.-Z. Mibefradil, a T-type Ca2+ channel blocker also blocks Orai channels by action at the extracellular surface. Br. J. Pharmacol. 2019, 176, 3845–3856. [Google Scholar] [CrossRef]
- Lee, H.; Kim, J.W.; Kim, D.K.; Choi, D.K.; Lee, S.; Yu, J.H.; Kwon, O.-B.; Lee, J.; Lee, D.; Kim, J.H.; et al. Calcium Channels as Novel Therapeutic Targets for Ovarian Cancer Stem Cells. Int. J. Mol. Sci. 2020, 21, 2327. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, E.J.; Rutter, G.A. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim. Biophys. Acta. 2009, 1787, 1324–1333. [Google Scholar] [CrossRef] [Green Version]
- Monteith, G.; Prevarskaya, N.; Roberts-Thomson, S. The calcium–cancer signalling nexus. Nat. Rev. Cancer 2017, 17, 373–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foskett, J.K.; White, C.; Cheung, K.-H.; Mak, N.-O.D. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007, 87, 593–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermassen, E.; Parys, J.; Mauger, J.-P. Subcellular distribution of the inositol 1,4,5-trisphosphate receptors: Functional relevance and molecular determinants. Boil. Cell 2004, 96, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Rezuchova, I.; Hudecova, S.; Soltysova, A.; Matuskova, M.; Durinikova, E.; Chovancova, B.; Zuzcak, M.; Cihova, M.; Burikova, M.; Penesova, A.; et al. Type 3 inositol 1,4,5-trisphosphate receptor has antiapoptotic and proliferative role in cancer cells. Cell Death Dis. 2019, 10, 186. [Google Scholar] [CrossRef] [Green Version]
- Yi, F.-X.; Magness, R.R.; Bird, I.M. Simultaneous imaging of [Ca2+]i and intracellular NO production in freshly isolated uterine artery endothelial cells: Effects of ovarian cycle and pregnancy. Am. J. Physiol. Integr. Comp. Physiol. 2005, 288, R140–R148. [Google Scholar] [CrossRef]
- Parrott, J.A.; Skinner, M.K. Developmental and hormonal regulation of hepatocyte growth factor expression and action in the bovine ovarian follicle. Boil. Reprod. 1998, 59, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Lail-Trecker, M.R.; Peluso, C.E.; Peluso, J.J. Hepatocyte Growth Factor Disrupts Cell Contact and Stimulates an Increase in Type 3 Inositol Triphosphate Receptor Expression, Intracellular Calcium Levels, and Apoptosis of Rat Ovarian Surface Epithelial Cells. Endocrine 2000, 12, 303–314. [Google Scholar] [CrossRef]
- Steffl, M.; Schweiger, M.; Amselgruber, W.M. Oestrous cycle-regulated expression of inositol 1,4,5-trisphosphate receptor type 2 in the pig ovary. Acta Histochem. 2004, 106, 137–144. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, C.; Su, J.; Xie, Q.; Ma, L.; Zeng, L.; Yu, Y.; Liu, S.; Li, S.; Li, Z.; et al. Tolerance to endoplasmic reticulum stress mediates cisplatin resistance in human ovarian cancer cells by maintaining endoplasmic reticulum and mitochondrial homeostasis. Oncol. Rep. 2015, 34, 3051–3060. [Google Scholar] [CrossRef] [Green Version]
- Kerkhofs, M.; Bittremieux, M.; Morciano, G.; Giorgi, C.; Pinton, P.; Parys, J.; Bultynck, G. Emerging molecular mechanisms in chemotherapy: Ca2+ signaling at the mitochondria-associated endoplasmic reticulum membranes. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Sneyers, F.; Rosa, N.; Bultynck, G. Type 3 IP3 receptors driving oncogenesis. Cell Calcium 2020, 86, 102141. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.Y. Role of Type 1 Inositol 1,4,5-triphosphate Receptors in Mammalian Oocytes. Dev. Reprod. 2019, 23, 1–9. [Google Scholar] [CrossRef]
- Ando, H.; Hirose, M.; Mikoshiba, K. Aberrant IP3 receptor activities revealed by comprehensive analysis of pathological mutations causing spinocerebellar ataxia 29. Proc. Natl. Acad. Sci. USA 2018, 115, 12259–12264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Andruska, N.; Lambrecht, M.J.; He, S.; Parissenti, A.; Hergenrother, P.J.; Nelson, E.R.; Shapiro, D.J. Targeting multidrug-resistant ovarian cancer through estrogen receptor α dependent ATP depletion caused by hyperactivation of the unfolded protein response. Oncotarget 2016, 9, 14741–14753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fill, M.; Copello, J.A. Ryanodine Receptor Calcium Release Channels. Physiol. Rev. 2002, 82, 893–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meissner, G. The structural basis of ryanodine receptor ion channel function. J. Gen. Physiol. 2017, 149, 1065–1089. [Google Scholar] [CrossRef] [PubMed]
- Tribe, R. Unravelling the role of the ryanodine receptor type 3 in smooth muscle. J. Physiol. 2002, 538, 673. [Google Scholar] [CrossRef]
- Bhat, M.; Zhao, J.; Takeshima, H.; Ma, J. Functional calcium release channel formed by the carboxyl-terminal portion of ryanodine receptor. Biophys. J. 1997, 73, 1329–1336. [Google Scholar] [CrossRef] [Green Version]
- Bhat, M.B.; Hayek, S.M.; Zhao, J.; Zang, W.; Takeshima, H.; Gil Wier, W.; Ma, J. Expression and Functional Characterization of the Cardiac Muscle Ryanodine Receptor Ca2+ Release Channel in Chinese Hamster Ovary Cells. Biophys. J. 1999, 77, 808–816. [Google Scholar] [CrossRef] [Green Version]
- Imagawa, T. Effects of saponin on contractility, intracellular Ca2+ and fluidity of plasma membrane in cultured heart cells. J. Mol. Cell. Cardiol. 1992, 24, 206. [Google Scholar] [CrossRef]
- Pan, Z.; Damron, D.; Nieminen, A.-L.; Bhat, M.B.; Ma, J. Depletion of Intracellular Ca2+by Caffeine and Ryanodine Induces Apoptosis of Chinese Hamster Ovary Cells Transfected with Ryanodine Receptor. J. Boil. Chem. 2000, 275, 19978–19984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, S.S.; Lamb, H.K.; Morgan, J.M.; Dunlop, W.; Gillespie, J.I. Differential expression of ryanodine receptor RyR2 mRNA in the non-pregnant and pregnant human myometrium. Biochem. J. 1997, 322, 777–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulhunty, A.F.; Gage, P.; Curtis, S.; Chelvanayagam, G.; Board, P. The Glutathione Transferase Structural Family Includes a Nuclear Chloride Channel and a Ryanodine Receptor Calcium Release Channel Modulator. J. Boil. Chem. 2000, 276, 3319–3323. [Google Scholar] [CrossRef] [Green Version]
- Morari, E.C.; Lima, A.B.C.; Bufalo, N.E.; Leite, J.L.; Granja, F.; Ward, L.S. Role of glutathione-S-transferase and codon 72 of P53 genotypes in epithelial ovarian cancer patients. J. Cancer Res. Clin. Oncol. 2006, 132, 521–528. [Google Scholar] [CrossRef]
- Mertens-Walker, I.; Bolitho, C.; Baxter, R.C.; Marsh, D.J. Gonadotropin-induced ovarian cancer cell migration and proliferation require extracellular signal-regulated kinase 1/2 activation regulated by calcium and protein kinase Cδ. Endoc.-Relat. Cancer 2010, 17, 335–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, C.S.; Yeung, T.-L.; Yip, K.-P.; Pradeep, S.; Balasubramanian, L.; Liu, J.; Wong, K.-K.; Mangala, L.S.; Armaiz-Pena, G.N.; Lopez-Berestein, G.; et al. Calcium-dependent FAK/CREB/TNNC1 signalling mediates the effect of stromal MFAP5 on ovarian cancer metastatic potential. Nat. Commun. 2014, 5, 5092. [Google Scholar] [CrossRef] [PubMed]
- Andruska, N.D.; Zheng, X.; Yang, X.; Mao, C.; Cherian, M.M.; Mahapatra, L.; Helferich, W.G.; Shapiro, D.J. Estrogen receptor α inhibitor activates the unfolded protein response, blocks protein synthesis, and induces tumor regression. Proc. Natl. Acad. Sci. USA 2015, 112, 4737–4742. [Google Scholar] [CrossRef] [Green Version]
- Clapham, D.E.; Runnels, L.W.; Strübing, C. The trp ion channel family. Nat. Rev. Neurosci. 2001, 2, 387–396. [Google Scholar] [CrossRef]
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal structure of the calcium release-activated calcium channel Orai. Science 2012, 338, 1308–1313. [Google Scholar] [CrossRef] [Green Version]
- Venkatachalam, K.; Montell, C. TRP channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef] [Green Version]
- Tóth, B.I.; Nilius, B. Chapter 2—Transient Receptor Potential Dysfunctions in Hereditary Diseases: TRP Channelopathies and Beyond. In TRP Channels as Therapeutic Targets; Szallasi, A., Ed.; Academic Press: Boston, MA, USA, 2015; pp. 13–33. [Google Scholar]
- Vaca, L.; Sampieri, A. Calmodulin Modulates the Delay Period between Release of Calcium from Internal Stores and Activation of Calcium Influx via Endogenous TRP1 Channels. J. Boil. Chem. 2002, 277, 42178–42187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gailly, P.; Schoor, M.C.-V. Involvement of trp- 2 protein in store-operated influx of calcium in fibroblasts. Cell Calcium 2001, 30, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Hogan, P.G.; Lewis, R.S.; Rao, A. Molecular Basis of Calcium Signaling in Lymphocytes: STIM and ORAI. Annu. Rev. Immunol. 2010, 28, 491–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.-H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Huang, Y.-C.; Xiu, H.-H.; Shan, Z.-M.; Xu, K.-Q. Altered expression of stromal interaction molecule (STIM)-calcium release-activated calcium channel protein (ORAI) and inositol 1,4,5-trisphosphate receptors (IP3Rs) in cancer: Will they become a new battlefield for oncotherapy? Chin. J. Cancer 2016, 35, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, N.; Lindemann, O.; Schwab, A. TRP channels and STIM/ORAI proteins: Sensors and effectors of cancer and stroma cell migration. Br. J. Pharmacol. 2014, 171, 5524–5540. [Google Scholar] [CrossRef] [Green Version]
- Schwab, A.; Fabian, A.; Hanley, P.J.; Stock, C. Role of Ion Channels and Transporters in Cell Migration. Physiol. Rev. 2012, 92, 1865–1913. [Google Scholar] [CrossRef]
- Chantome, A.; Potier-Cartereau, M.; Clarysse, L.; Fromont, G.; Marionneau-Lambot, S.; Gueguinou, M.; Pages, J.-C.; Collin, C.; Oullier, T.; Girault, A.; et al. Pivotal Role of the Lipid Raft SK3-Orai1 Complex in Human Cancer Cell Migration and Bone Metastases. Cancer Res. 2013, 73, 4852–4861. [Google Scholar] [CrossRef] [Green Version]
- Hammadi, M.; Chopin, V.; Matifat, F.; Dhennin-Duthille, I.; Chasseraud, M.; Sevestre, H.; Ouadid-Ahidouch, H. Human ether à-gogo K+ channel 1 (hEag1) regulates MDA-MB-231 breast cancer cell migration through Orai1-dependent calcium entry. J. Cell. Physiol. 2012, 227, 3837–3846. [Google Scholar] [CrossRef]
- Liu, X.; Zou, J.; Su, J.; Lu, Y.; Zhang, J.; Li, L.; Yin, F. Downregulation of transient receptor potential cation channel, subfamily C, member 1 contributes to drug resistance and high histological grade in ovarian cancer. Int. J. Oncol. 2015, 48, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.L.; Cao, Q.; Zhou, K.C.; Feng, Y.J.; Wang, Y.-Z. Transient receptor potential channel C3 contributes to the progression of human ovarian cancer. Oncogene 2009, 28, 1320–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezzerides, V.J.; Ramsey, I.S.; Kotecha, S.; Greka, A.; Clapham, D.E. Rapid vesicular translocation and insertion of TRP channels. Nature 2004, 6, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Maihle, N.J.; Baron, A.; Barrette, B.A.; Boardman, C.H.; Christensen, T.A.; Cora, E.M.; Faupel-Badger, J.M.; Greenwood, T.; Juneja, S.C.; Lafky, J.M.; et al. EGF/ErbB receptor family in ovarian cancer. Cancer Treat. Res. 2002, 107, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Nicosia, S.V.; Bai, W.; Cheng, J.Q.; Coppola, M.; Kruk, P.A. Oncogenic pathways implicated in ovarian epithelial cancer. Hematol. Clin. North. Am. 2003, 17, 927–943. [Google Scholar] [CrossRef]
- Psyrri, A.; Kassar, M.; Yu, Z.; Bamias, A.; Weinberger, P.M.; Markakis, S.; Kowalski, D.; Camp, R.L.; Rimm, D.L.; Dimopoulos, M.A. Effect of Epidermal Growth Factor Receptor Expression Level on Survival in Patients with Epithelial Ovarian Cancer. Clin. Cancer Res. 2005, 11, 8637–8643. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, S.; Liu, G.; Liu, G.; Yang, W.; Honisch, S.; Pantelakos, S.; Stournaras, C.; Hönig, A.; Lang, F. Enhanced Orai1 and STIM1 expression as well as store operated Ca2+ entry in therapy resistant ovary carcinoma cells. Oncotarget 2014, 5, 4799–4810. [Google Scholar] [CrossRef] [Green Version]
- Vashisht, A.; Trebak, M.; Motiani, R.K. STIM and Orai proteins as novel targets for cancer therapy. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. Am. J. Physiol. 2015, 309, C457–C469. [Google Scholar] [CrossRef] [Green Version]
- Pla, A.F.; Kondratska, K.; Prevarskaya, N. STIM and ORAI proteins: Crucial roles in hallmarks of cancer. Am. J. Physiol. 2016, 310, C509–C519. [Google Scholar] [CrossRef]
- Williams, C.J.; Erickson, G.F. Morphology and Physiology of the Ovary. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., Dungan, K., Grossman, A., Hershman, J.M., Kaltsas, G., Koch, C., Kopp, P., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Sun, Z.; Zhang, H.; Wang, X.; Wang, Q.-C.; Zhang, C.; Wang, J.-Q.; Wang, Y.-H.; An, C.-Q.; Yang, K.-Y.; Wang, Y.; et al. TMCO1 is essential for ovarian follicle development by regulating ER Ca2+ store of granulosa cells. Cell Death Differ. 2018, 25, 1686–1701. [Google Scholar] [CrossRef] [Green Version]
- Periasamy, M.; Kalyanasundaram, A. SERCA pump isoforms: Their role in calcium transport and disease. Muscle Nerve 2007, 35, 430–442. [Google Scholar] [CrossRef]
- Huang, N.; Yu, Y.; Qiao, J. Dual role for the unfolded protein response in the ovary: Adaption and apoptosis. Protein Cell 2016, 8, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Boil. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Peluso, J.J. Basic fibroblast growth factor (bFGF) regulation of the plasma membrane calcium ATPase (PMCA) as part of an anti-apoptotic mechanism of action. Biochem. Pharmacol. 2003, 66, 1363–1369. [Google Scholar] [CrossRef]
- Hegedũs, L.; Garay, T.; Molnár, E.; Varga, K.; Bilecz, Á.; Török, S.; Padányi, R.; Pászty, K.; Wolf, M.; Grusch, M.; et al. The plasma membrane C a 2+ pump PMCA 4b inhibits the migratory and metastatic activity of BRAF mutant melanoma cells. Int. J. Cancer 2016, 140, 2758–2770. [Google Scholar] [CrossRef] [Green Version]
- Solár, P.; Sytkowski, A.J. Differentially expressed genes associated with cisplatin resistance in human ovarian adenocarcinoma cell line A2780. Cancer Lett. 2011, 309, 11–18. [Google Scholar] [CrossRef]
- Stafford, N.; Wilson, C.; Oceandy, D.; Neyses, L.; Cartwright, E.J. The Plasma Membrane Calcium ATPases and Their Role as Major New Players in Human Disease. Physiol. Rev. 2017, 97, 1089–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunelle, J.K.; Letai, A. Control of mitochondrial apoptosis by the Bcl-2 family. J. Cell Sci. 2009, 122, 437–441. [Google Scholar] [CrossRef] [Green Version]
- Wilson, B.E.; Mochon, E.; Boxer, L.M. Induction of bcl-2 expression by phosphorylated CREB proteins during B-cell activation and rescue from apoptosis. Mol. Cell. Boil. 1996, 16, 5546–5556. [Google Scholar] [CrossRef] [Green Version]
- Schuh, R.A.; Kristian, T.; Fiskum, G. Calcium-dependent dephosphorylation of brain mitochondrial calcium/cAMP response element binding protein (CREB). J. Neurochem. 2005, 92, 388–394. [Google Scholar] [CrossRef] [Green Version]
- Barylyak, R.V.; Iefremova, U.P.; Onufrovych, O.; Melnyk, O.V.; Vorobets, D.Z.; Vorobets, Z.D. Characterization of Ca2+,Mg2+-ATPase of blood lymphocytes in women with ovarian cancer. Regul. Mech. Biosyst. 2018, 9, 85–89. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.-A.; Kim, B.; Dhanasekaran, D.N.; Tsang, B.K.; Song, Y.S. Curcumin induces apoptosis by inhibiting sarco/endoplasmic reticulum Ca2+ ATPase activity in ovarian cancer cells. Cancer Lett. 2016, 371, 30–37. [Google Scholar] [CrossRef]
- Nakayama, K.; Kanzaki, A.; Terada, K.; Mutoh, M.; Ogawa, K.; Sugiyama, T.; Takenoshita, S.; Itoh, K.; Yaegashi, N.; Miyazaki, K.; et al. Prognostic value of the Cu-transporting ATPase in ovarian carcinoma patients receiving cisplatin-based chemotherapy. Clin. Cancer Res. 2004, 10, 2804–2811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultze-Mosgau, S.; Erbe, M.; Keilholz, L.; Radespiel-Troger, M.; Wiltfang, J.; Minge, N.; Neukam, F.W. Histomorphometric analysis of irradiated recipient vessels and transplant vessels of free flaps in patients undergoing reconstruction after ablative surgery. Int. J. Oral Maxillofac. Surg. 2000, 29, 112–118. [Google Scholar] [CrossRef]
- Al-Bahlani, S.; Fraser, M.; Wong, A.Y.; Sayan, B.S.; Bergeron, R.; Melino, G.; Tsang, B.K. P73 regulates cisplatin-induced apoptosis in ovarian cancer cells via a calcium/calpain-dependent mechanism. Oncogene 2011, 30, 4219–4230. [Google Scholar] [CrossRef] [PubMed]
- Torner, H.; Brüssow, K.-P.; Alm, H.; Rátky, J.; Pohland, R.; Tuchscherer, A.; Kanitz, W. Mitochondrial aggregation patterns and activity in porcine oocytes and apoptosis in surrounding cumulus cells depends on the stage of pre-ovulatory maturation. Theriogenology 2004, 61, 1675–1689. [Google Scholar] [CrossRef]
- Wang, L.; Wang, D.-H.; Zou, X.-Y.; Xu, C.-M. Mitochondrial functions on oocytes and preimplantation embryos. J. Zhejiang Univ. Sci. B 2009, 10, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Krisher, R. The effect of oocyte quality on development. J. Anim. Sci. 2004, 82, 82. [Google Scholar]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [Green Version]
- Patron, M.; Checchetto, V.; Raffaello, A.; Teardo, E.; Reane, D.V.; Mantoan, M.; Granatiero, V.; Szabò, I.; De Stefani, D.; Rizzuto, R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 2014, 53, 726–737. [Google Scholar] [CrossRef] [Green Version]
- Marchi, S.; Pinton, P. The mitochondrial calcium uniporter complex: Molecular components, structure and physiopathological implications. J. Physiol. 2014, 592, 829–839. [Google Scholar] [CrossRef]
- Matesanz-Isabel, J.; Arias-Del-Val, J.; Alvarez-Illera, P.; Fonteriz, R.I.; Montero, M.; Alvarez, J. Functional roles of MICU1 and MICU2 in mitochondrial Ca 2+ uptake. Biochim. Biophys. Acta. 2016, 1858, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Gunter, T.E.; Yule, D.I.; Gunter, K.K.; Eliseev, R.A.; Salter, J.D. Calcium and mitochondria. FEBS Lett. 2004, 567, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Bolinches-Amorãs, A.; Mollã, B.; Pla-Martãn, D.; Palau, F.; Gonzã¡lez-Cabo, P. Mitochondrial dysfunction induced by frataxin deficiency is associated with cellular senescence and abnormal calcium metabolism. Front. Cell. Neurosci. 2014, 8, 124. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, D.V.; Wiley, C.D.; Velarde, M.C. Mitochondrial effectors of cellular senescence: Beyond the free radical theory of aging. Aging Cell 2014, 14, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallilankaraman, K.; Doonan, P.; Cárdenas, C.; Chandramoorthy, H.C.; Müller, M.; Miller, R.; Hoffman, N.E.; Gandhirajan, R.K.; Molgó, J.; Birnbaum, M.J.; et al. MICU1 Is an Essential Gatekeeper for MCU-Mediated Mitochondrial Ca2+ Uptake that Regulates Cell Survival. Cell 2012, 151, 630–644. [Google Scholar] [CrossRef] [Green Version]
- Csordás, G.; Golenár, T.; Seifert, E.L.; Kamer, K.J.; Sancak, Y.; Perocchi, F.; Moffat, C.; Weaver, D.; Perez, S.D.L.F.; Bogorad, R.; et al. MICU1 Controls Both the Threshold and Cooperative Activation of the Mitochondrial Ca2+ Uniporter. Cell Metab. 2013, 17, 976–987. [Google Scholar] [CrossRef] [Green Version]
- Wiel, C.; Lallet-Daher, H.; Gitenay, D.; Gras, B.; Le Calve, B.; Augert, A.; Ferrand, M.; Prevarskaya, N.; Simonnet, H.; Vindrieux, D.; et al. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat. Commun. 2014, 5, 3792. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, P.K.; Mustafi, S.B.; Xiong, X.; Dwivedi, S.K.D.; Nesin, V.; Saha, S.; Zhang, M.; Dhanasekaran, D.; Jayaraman, M.; Mannel, R.; et al. MICU1 drives glycolysis and chemoresistance in ovarian cancer. Nat. Commun. 2017, 8, 14634. [Google Scholar] [CrossRef]
- Arvizo, R.R.; Moyano, D.F.; Saha, S.; Thompson, M.A.; Bhattacharya, R.; Rotello, V.M.; Prakash, Y.S.; Mukherjee, P. Probing Novel Roles of the Mitochondrial Uniporter in Ovarian Cancer Cells Using Nanoparticles. J. Boil. Chem. 2013, 288, 17610–17618. [Google Scholar] [CrossRef] [Green Version]
- Lemasters, J.J.; Qian, T.; He, L.; Kim, J.-S.; Elmore, S.P.; Cascio, W.E.; Brenner, D.A. Role of Mitochondrial Inner Membrane Permeabilization in Necrotic Cell Death, Apoptosis, and Autophagy. Antioxidants Redox Signal. 2002, 4, 769–781. [Google Scholar] [CrossRef]
- Danese, A.; Patergnani, S.; Bonora, M.; Wieckowski, M.R.; Previati, M.; Giorgi, C.; Pinton, P. Calcium regulates cell death in cancer: Roles of the mitochondria and mitochondria-associated membranes (MAMs). Biochim. Biophys. Acta. 2017, 1858, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Bittremieux, M.; Missiroli, S.; Morganti, C.; Patergnani, S.; Sbano, L.; Rimessi, A.; Kerkhofs, M.; Parys, J.; Bultynck, G.; et al. Endoplasmic Reticulum-Mitochondria Communication Through Ca2+ Signaling: The Importance of Mitochondria-Associated Membranes (MAMs). Adv. Exp. Med. Biol. 2017, 997, 49–67. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.N.; McDonald, P.H. GPCRs: Emerging anti-cancer drug targets. Cell. Signal. 2018, 41, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Oldham, W.M.; Hamm, H.E. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Boil. 2008, 9, 60–71. [Google Scholar] [CrossRef]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef]
- Lundstrom, K. Structural genomics of GPCRs. Trends Biotechnol. 2005, 23, 103–108. [Google Scholar] [CrossRef]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Boil. 2002, 3, 639–650. [Google Scholar] [CrossRef]
- Lappano, R.; Maggiolini, M. GPCRs and cancer. Acta Pharmacol. Sin. 2012, 33, 351–362. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Madden, N.E.; Wong, A.S.T.; Chow, B.K.C.; Lee, L.T.O. The Role of Endocrine G Protein-Coupled Receptors in Ovarian Cancer Progression. Front. Endocrinol. 2017, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Lenhard, M.S.; Tereza, L.; Heublein, S.; Ditsch, N.; Himsl, I.; Mayr, D.; Friese, K.; Jeschke, U. Steroid hormone receptor expression in ovarian cancer: Progesterone receptor B as prognostic marker for patient survival. BMC Cancer 2012, 12, 553. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Sun, Y.; Gao, D. Role of the nervous system in cancer metastasis. Oncol. Lett. 2013, 5, 1101–1111. [Google Scholar] [CrossRef] [Green Version]
- Murata, M. Inflammation and cancer. Environ. Heal. Prev. Med. 2018, 23, 50. [Google Scholar] [CrossRef] [Green Version]
- Park, K.S.; Kim, M.-K.; Lee, H.Y.; Kim, S.D.; Lee, S.Y.; Kim, J.M.; Ryu, S.H.; Bae, Y.-S. S1P stimulates chemotactic migration and invasion in OVCAR3 ovarian cancer cells. Biochem. Biophys. Res. Commun. 2007, 356, 239–244. [Google Scholar] [CrossRef]
- Park, K.S.; Lee, H.Y.; Lee, S.Y.; Kim, M.-K.; Kim, S.D.; Kim, J.M.; Yun, J.; Im, D.-S.; Bae, Y.-S. Lysophosphatidylethanolamine stimulates chemotactic migration and cellular invasion in SK-OV3 human ovarian cancer cells: Involvement of pertussis toxin-sensitive G-protein coupled receptor. FEBS Lett. 2007, 581, 4411–4416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batra, S.; Fadeel, I. Release of intracellular calcium and stimulation of cell growth by ATP and histamine in human ovarian cancer cells (SKOV-3). Cancer Lett. 1994, 77, 57–63. [Google Scholar] [CrossRef]
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerasimova, T.; Thanasoula, M.N.; Zattas, D.; Seli, E.; Sakkas, D.; Lalioti, M.D. Identification and in vitro characterization of follicle stimulating hormone (FSH) receptor variants associated with abnormal ovarian response to FSH. J. Clin. Endocrinol. Metab. 2010, 95, 529–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Liu, H.; Chen, X.; Chen, P.-H.; Fischer, D.; Sriraman, V.; Yu, H.N.; Arkinstall, S.; He, X. Structure of follicle-stimulating hormone in complex with the entire ectodomain of its receptor. Proc. Natl. Acad. Sci. USA 2012, 109, 12491–12496. [Google Scholar] [CrossRef] [Green Version]
- Khor, S.; Lyu, Q.; Kuang, Y.; Lu, X. Novel FSHR variants causing female resistant ovary syndrome. Mol. Genet. Genom. Med. 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- DeRoo, B.J.; Korach, K.S. Estrogen receptors and human disease. J. Clin. Investig. 2006, 116, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Burns, K.A.; Korach, K.S. Estrogen receptors and human disease: An update. Arch. Toxicol. 2012, 86, 1491–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulac-Jericevic, B.; Mullinax, R.A.; DeMayo, F.J.; Lydon, J.P.; Conneely, O.M. Subgroup of Reproductive Functions of Progesterone Mediated by Progesterone Receptor-B Isoform. Science 2000, 289, 1751–1754. [Google Scholar] [CrossRef] [PubMed]
- Attia, G.R.; Zeitoun, K.; Edwards, D.; Johns, A.; Carr, B.R.; Bulun, S.E. Progesterone Receptor Isoform A But Not B Is Expressed in Endometriosis 1. J. Clin. Endocrinol. Metab. 2000, 85, 2897–2902. [Google Scholar] [CrossRef] [Green Version]
- Kjaer, S.K.; Christensen, I.J.; Jacobs, I.J.; Gayther, S.; Christensen, L.; Høgdall, C.; Blaakaer, J.; Høgdall, E. Prognostic value of estrogen receptor and progesterone receptor tumor expression in Danish ovarian cancer patients: From the ’MALOVA’ ovarian cancer study. Oncol. Rep. 2007, 18, 1051–1059. [Google Scholar] [CrossRef] [Green Version]
- Jönsson, J.-M.; Arildsen, N.; Malander, S.; Måsbäck, A.; Hartman, L.; Nilbert, M.; Hedenfalk, I. Sex Steroid Hormone Receptor Expression Affects Ovarian Cancer Survival. Transl. Oncol. 2015, 8, 424–433. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Wen, H.; Bi, R.; Ju, X.; Chen, X.; Yang, W.; Wu, X. A clinically applicable molecular classification for high-grade serous ovarian cancer based on hormone receptor expression. Sci. Rep. 2016, 6, 25408. [Google Scholar] [CrossRef] [Green Version]
- Chuffa, L.G.A.; Lupi-Júnior, L.A.; Costa, A.B.; de Arruda Amorim, J.P.; Seiva, F.R.F. The role of sex hormones and steroid receptors on female reproductive cancers. Steroids 2017, 118, 93–108. [Google Scholar] [CrossRef] [Green Version]
- Shen, F.; Zhang, X.; Zhang, Y.; Ding, J.; Chen, Q. Hormone receptors expression in ovarian cancer taking into account menopausal status: A retrospective study in Chinese population. Oncotarget 2017, 8, 84019–84027. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-H.; Cheung, L.W.T.; Wong, A.S.T.; Leung, P.C.K. Estrogen Regulates Snail and Slug in the Down-Regulation of E-Cadherin and Induces Metastatic Potential of Ovarian Cancer Cells through Estrogen Receptor α. Mol. Endocrinol. 2008, 22, 2085–2098. [Google Scholar] [CrossRef]
- Prat, J. Ovarian carcinomas: Five distinct diseases with different origins, genetic alterations, and clinicopathological features. Virchows Arch. 2012, 460, 237–249. [Google Scholar] [CrossRef]
- Gomora, M.J.; Morales-Vasquez, F.; Pedernera, E.; Perez-Montiel, D.; López-Basave, H.; Villa, A.R.; Hernandez-Martinez, A.; Mena, E.; Mendez, C. Sexual steroid hormone receptors profiles of ovarian carcinoma in Mexican women. Endocr. Connect. 2018, 7, 1006–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Zhao, M.; Dai, X.; Tong, M.; Wei, J.; Chen, Q. The prevalence of endometrial cancer in pre- and postmenopausal Chinese women. Menopause 2016, 23, 884–887. [Google Scholar] [CrossRef]
- Mironov, S.; Akin, O.; Pandit-Taskar, N.; Hann, L.E. Ovarian Cancer. Radiol. Clin. North. Am. 2007, 45, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Darb-Esfahani, S.; Wirtz, R.M.; Sinn, B.V.; Budczies, J.; Noske, A.; Weichert, W.; Faggad, A.; Scharff, S.; Sehouli, J.; Oskay-Ozcelik, G.; et al. Estrogen receptor 1 mRNA is a prognostic factor in ovarian carcinoma: Determination by kinetic PCR in formalin-fixed paraffin-embedded tissue. Endocr. Relat. Cancer 2009, 16, 1229–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.F.; Hirsch, M.S.; Lee, H.; Matulonis, U. Prognosis and hormone receptor status in older and younger patients with advanced-stage papillary serous ovarian carcinoma. Gynecol. Oncol. 2009, 115, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Sinn, B.V.; Darb-Esfahani, S.; Wirtz, R.M.; Budczies, J.; Sehouli, J.; Chekerov, R.; Dietel, M.; Denkert, C. Evaluation of a hormone receptor-positive ovarian carcinoma subtype with a favourable prognosis by determination of progesterone receptor and oestrogen receptor 1 mRNA expression in formalin-fixed paraffin-embedded tissue. Histopathology 2011, 59, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Tkalia, I.G.; Vorobyova, L.I.; Svintsitsky, V.S.; Nespryadko, S.V.; Goncharuk, I.V.; Lukyanova, N.Y.; Chekhun, V.F. Clinical significance of hormonal receptor status of malignant ovarian tumors. Exp. Oncol. 2014, 36, 125–133. [Google Scholar] [PubMed]
- Zhao, M.; Shen, F.; Yin, Y.X.; Yang, Y.Y.; Xiang, D.J.; Chen, Q. Increased Expression of Heat Shock Protein 27 Correlates With Peritoneal Metastasis in Epithelial Ovarian Cancer. Reprod. Sci. 2012, 19, 748–753. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Combe, P. Recurrent ovarian cancer. Ann. Oncol. 2016, 27, i63–i65. [Google Scholar] [CrossRef]
- Burger, R.A.; Brady, M.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H.; Mannel, R.S.; Homesley, H.D.; Fowler, J.; Greer, B.E.; et al. Incorporation of Bevacizumab in the Primary Treatment of Ovarian Cancer. New Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef] [Green Version]
- Schwab, M. Encyclopedia of Cancer; Springer Science & Business Media: Berlin, Germany, 2008. [Google Scholar]
- Florea, A.-M.; Büsselberg, D. Anti-cancer drugs interfere with intracellular calcium signaling. NeuroToxicology 2009, 30, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Ferri, K.F.; Kroemer, G. Organelle-specific initiation of cell death pathways. Nature 2001, 3, E255–E263. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Xu, Y.; Su, J.; Yu, H.; Kang, J.; Li, H.; Li, X.; Xie, Q.; Yu, C.; Sun, L.; et al. Autophagic flux promotes cisplatin resistance in human ovarian carcinoma cells through ATP-mediated lysosomal function. Int. J. Oncol. 2015, 47, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Kucukkaya, B.; Basoglu, H.; Erdag, D.; Akbas, F.; Susgun, S.; Yalcintepe, L. Calcium homeostasis in cisplatin resistant epithelial ovarian cancer. Gen. Physiol. Biophys. 2019, 38, 353–363. [Google Scholar] [CrossRef]
- Büsselberg, D.; Florea, A.-M. Targeting Intracellular Calcium Signaling ([Ca2+]i) to Overcome Acquired Multidrug Resistance of Cancer Cells: A Mini-Overview. Cancers 2017, 9, 48. [Google Scholar] [CrossRef] [Green Version]
- Xie, Q.; Su, J.; Jiao, B.; Shen, L.; Ma, L.; Qu, X.; Yu, C.; Jiang, X.; Xu, Y.; Sun, L. ABT737 reverses cisplatin resistance by regulating ER-mitochondria Ca2+ signal transduction in human ovarian cancer cells. Int. J. Oncol. 2016, 49, 2507–2519. [Google Scholar] [CrossRef] [Green Version]
- Varghese, E.; Büsselberg, D. Auranofin, an Anti-Rheumatic Gold Compound, Modulates Apoptosis by Elevating the Intracellular Calcium Concentration ([Ca2+]i) in MCF-7 Breast Cancer Cells. Cancers 2014, 6, 2243–2258. [Google Scholar] [CrossRef] [Green Version]
- Marzo, T.; Massai, L.; Pratesi, A.; Stefanini, M.; Cirri, D.; Magherini, F.; Becatti, M.; Landini, I.; Nobili, S.; Mini, E.; et al. Replacement of the Thiosugar of Auranofin with Iodide Enhances the Anticancer Potency in a Mouse Model of Ovarian Cancer. ACS Med. Chem. Lett. 2019, 10, 656–660. [Google Scholar] [CrossRef]
- Zadran, S.; Remacle, F.; Levine, R.D. miRNA and mRNA cancer signatures determined by analysis of expression levels in large cohorts of patients. Proc. Natl. Acad. Sci. USA 2013, 110, 19160–19165. [Google Scholar] [CrossRef] [Green Version]
- Pillozzi, S.; D’Amico, M.; Bartoli, G.; Gasparoli, L.; Petroni, G.; Crociani, O.; Marzo, T.; Guerriero, A.; Messori, L.; Severi, M.; et al. The combined activation of KCa3.1 and inhibition of Kv11.1/hERG1 currents contribute to overcome Cisplatin resistance in colorectal cancer cells. Br. J. Cancer 2017, 118, 200–212. [Google Scholar] [CrossRef] [Green Version]
- Bonnefond, M.-L.; Lambert, B.; Giffard, F.; Abeilard, E.; Brotin, E.; Louis, M.-H.; Gueye, M.S.; Gauduchon, P.; Poulain, L.; N’Diaye, M. Calcium signals inhibition sensitizes ovarian carcinoma cells to anti-Bcl-xL strategies through Mcl-1 down-regulation. Apoptosis 2015, 20, 535–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Histological Subtype | Distinguishing Features |
|---|---|---|
| Type I epithelial ovarian cancers | Clear-cell carcinoma |
|
| Endometrioid carcinoma | ||
| Mucinous carcinoma | ||
| Squamous carcinoma |
| |
| Transitional cell carcinoma |
| |
| Low-grade serous carcinoma (LGSC) | ||
| Type II epithelial ovarian cancers | Mixed mesodermal tumor |
|
| Undifferentiated carcinoma | ||
| High-grade serous carcinoma (HGSC) |
| |
| Germ cell tumors | Germ cell tumors |
|
| Sex cord-stromal tumors | Sex cord-stromal tumors |
| TRP Family | TRP Members | Associated Genetic Disorder |
|---|---|---|
| TRPC | TRPC1, mTRPC2, TRPC3/4/5/6/7 | Focal segmental glomerulosclerosis (TRPC6) |
| TRPV | TRPV1/2/3/4/5/6 | Olmsted syndrome (TRPV3) |
| Type 3 brachyolmia (TRPV4) | ||
| Hereditary motor and sensory neuropathy type 2 (TRPV4) | ||
| Congenital distal spinal muscular atrophy (TRPV4) | ||
| Spondyloepiphyseal dysplasia Maroteaux type (TRPV4) | ||
| TRPM | TRPM1/2/3/4/5/6/7/8 | Autosomal recessive congenital stationary night blindness (TRPM1) |
| Progressive familial heart block type IB (TRPM4) | ||
| Primary hypomagnesemia with secondary hypocalcemia (TRPM6) | ||
| TRPML | TRPML1/2/3 | Mucolipidosis type IV (TRPML1) |
| TRPP | TRPP1-PKD2, TRPP2-PKD2-L1, TRPP3-PKD2-L2 | Autosomal dominant polycystic kidney disease (TRPP1-PKD2) |
| TRPA1 | TRPA1 | Familial episodic pain syndrome |
| GPCR | Stimuli | Calcium Effect | Type of Cells | Effect | Reference |
|---|---|---|---|---|---|
| Lipid Receptor | S1P1/3 | Increased intracellular Ca2+ | OVCAR-3 cells | Chemotactic migration Cellular invasion | Park et al. [196] |
| Lipid Receptor | LPE | PTX-sensitive G-protein-dependent Ca2+ increase | SKOV-3 cells OVOCAR 3 cells | Chemotactic migration | Park et al. [197] |
| Biogenic amines | ATP Histamine | Increased intracellular Ca2+ | SKOV-3 cells | Cellular proliferation | Batra et al. [198] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caravia, L.; Staicu, C.E.; Radu, B.M.; Condrat, C.E.; Crețoiu, D.; Bacalbașa, N.; Suciu, N.; Crețoiu, S.M.; Voinea, S.C. Altered Organelle Calcium Transport in Ovarian Physiology and Cancer. Cancers 2020, 12, 2232. https://doi.org/10.3390/cancers12082232
Caravia L, Staicu CE, Radu BM, Condrat CE, Crețoiu D, Bacalbașa N, Suciu N, Crețoiu SM, Voinea SC. Altered Organelle Calcium Transport in Ovarian Physiology and Cancer. Cancers. 2020; 12(8):2232. https://doi.org/10.3390/cancers12082232
Chicago/Turabian StyleCaravia, Laura, Cristina Elena Staicu, Beatrice Mihaela Radu, Carmen Elena Condrat, Dragoș Crețoiu, Nicolae Bacalbașa, Nicolae Suciu, Sanda Maria Crețoiu, and Silviu Cristian Voinea. 2020. "Altered Organelle Calcium Transport in Ovarian Physiology and Cancer" Cancers 12, no. 8: 2232. https://doi.org/10.3390/cancers12082232
APA StyleCaravia, L., Staicu, C. E., Radu, B. M., Condrat, C. E., Crețoiu, D., Bacalbașa, N., Suciu, N., Crețoiu, S. M., & Voinea, S. C. (2020). Altered Organelle Calcium Transport in Ovarian Physiology and Cancer. Cancers, 12(8), 2232. https://doi.org/10.3390/cancers12082232