Systematic Analysis of Aberrant Biochemical Networks and Potential Drug Vulnerabilities Induced by Tumor Suppressor Loss in Malignant Pleural Mesothelioma

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
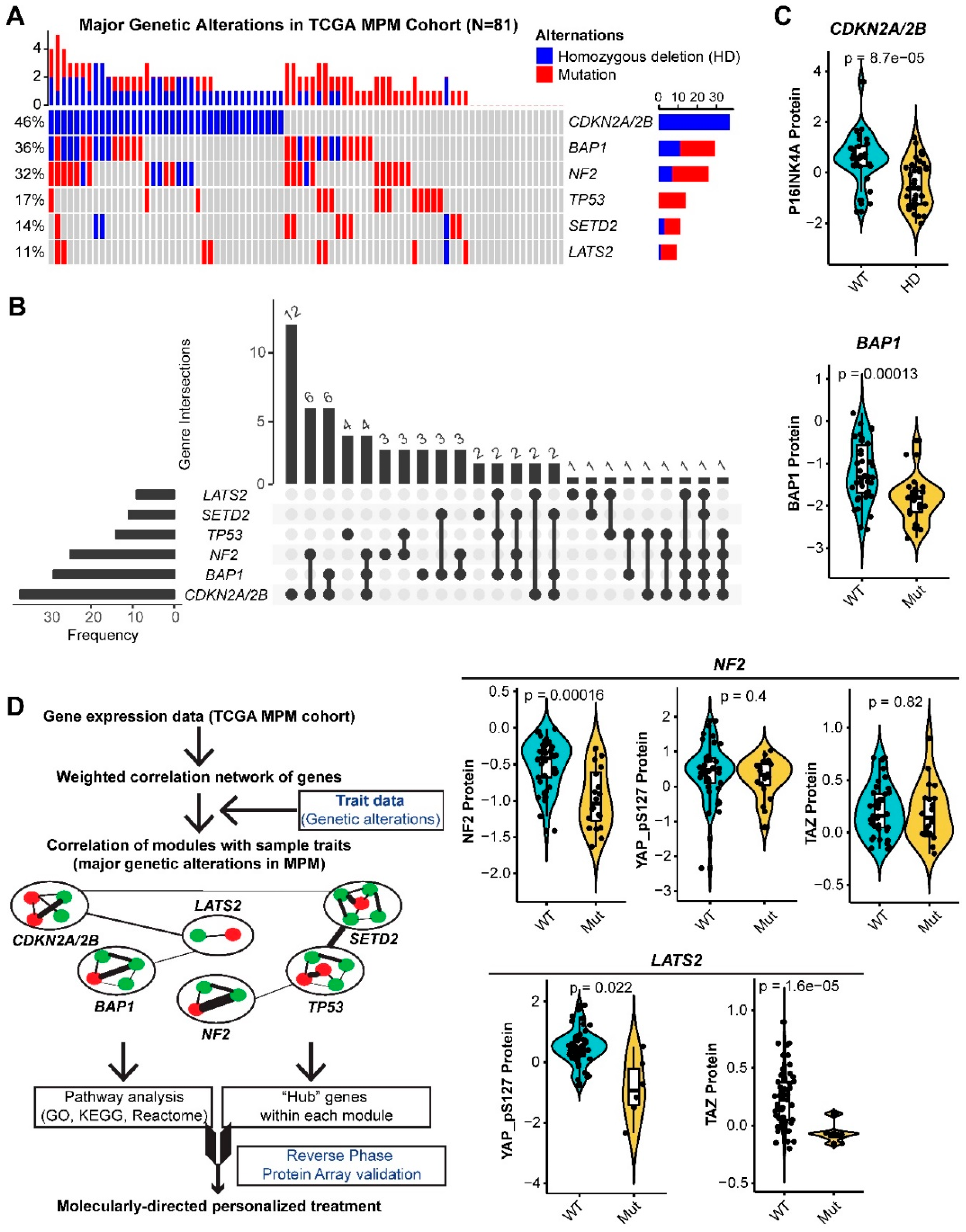
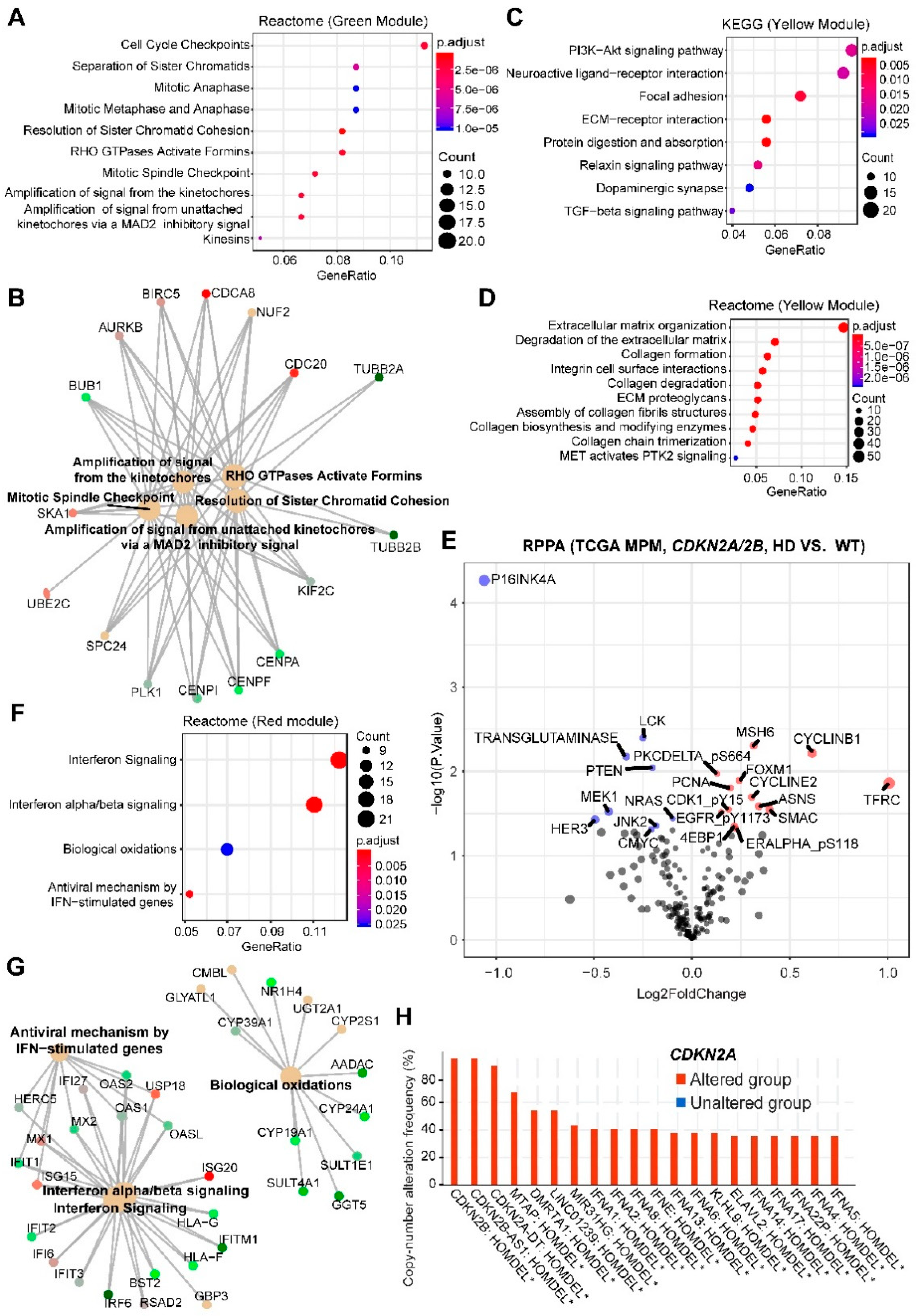
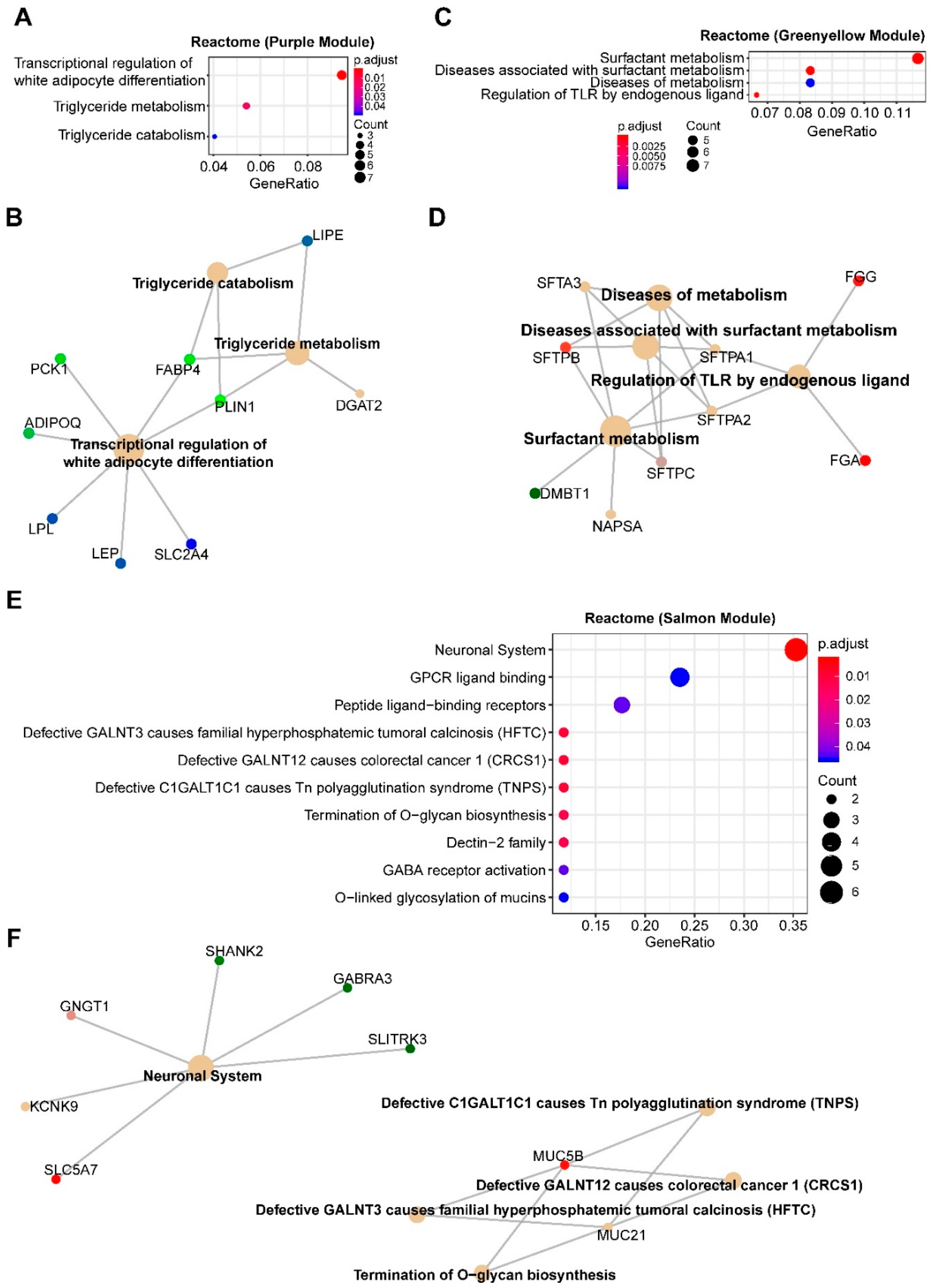
2.1. Systematic Analysis of Rewired Biochemical Networks and Therapeutic Vulnerabilities Enabled by Tumor Suppressor Loss in MPM
2.2. CDKN2A/2B
2.3. NF2
2.4. TP53
2.5. SETD2
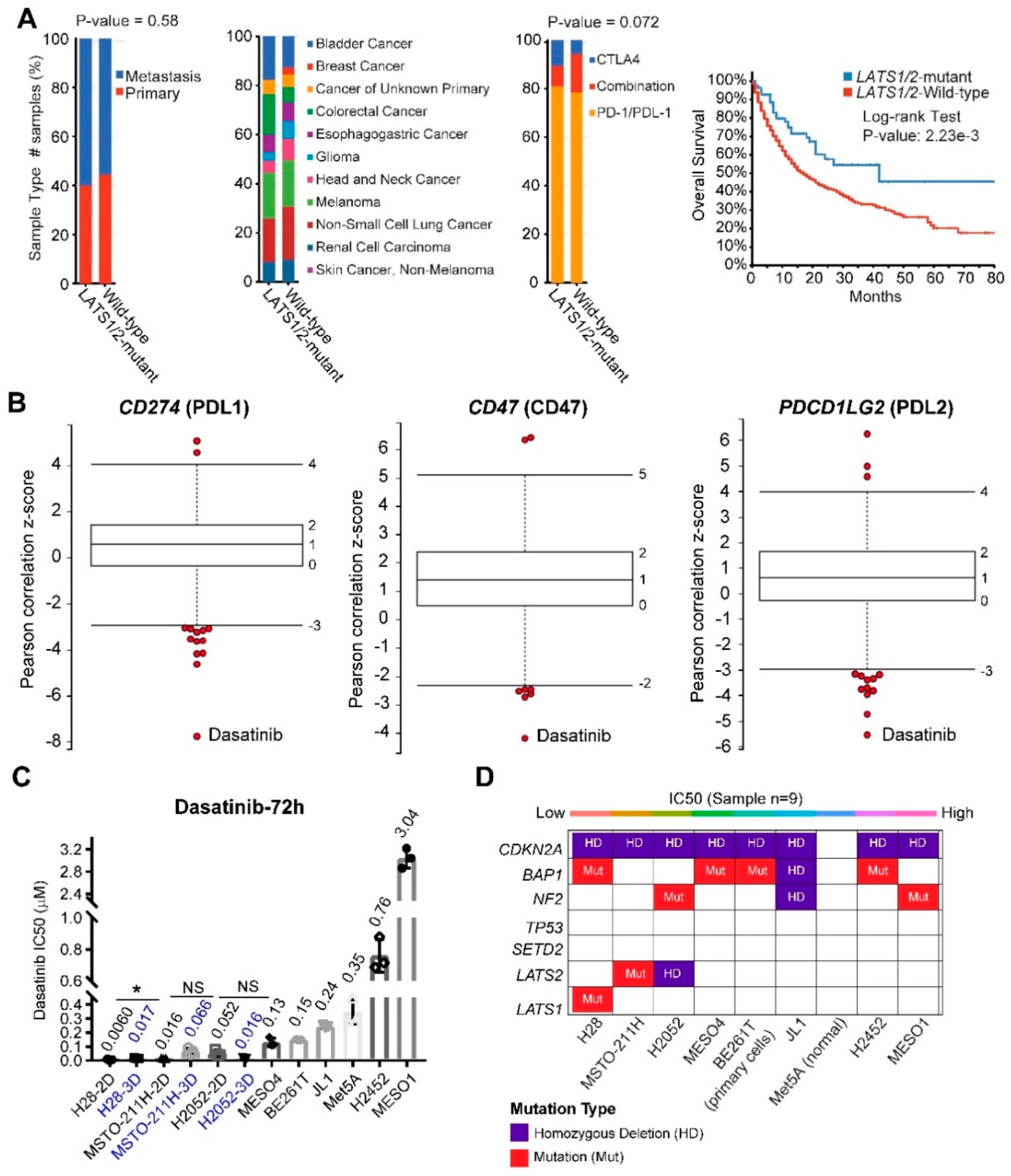
2.6. LATS2
3. Discussion
3.1. CDKN2A/2B and NF2
3.2. BAP1
3.3. TP53
3.4. SETD2
3.5. LATS2
4. Materials and Methods
4.1. WGCNA and Function Enrichment Analyses
4.2. Cell Viability Assay
4.3. Public Databases
4.4. Survival Analysis
4.5. ECM Gene Signature
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mutti, L.; Peikert, T.; Robinson, B.W.S.; Scherpereel, A.; Tsao, A.S.; De Perrot, M.; Woodard, G.A.; Jablons, D.M.; Wiens, J.; Hirsch, F.R.; et al. Scientific Advances and New Frontiers in Mesothelioma Therapeutics. J. Thorac. Oncol. 2018, 13, 1269–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffié, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C.; et al. Phase III Study of Pemetrexed in Combination with Cisplatin Versus Cisplatin Alone in Patients with Malignant Pleural Mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644. [Google Scholar] [CrossRef] [PubMed]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat. Genet. 2016, 48, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A.; et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discov. 2018, 8, 1548–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, G.; Chmielecki, J.; Goparaju, C.; Heguy, A.; Dolgalev, I.; Carbone, M.; Seepo, S.; Meyerson, M.; Pass, H.I. Whole-Exome Sequencing Reveals Frequent Genetic Alterations in BAP1, NF2, CDKN2A, and CUL1 in Malignant Pleural Mesothelioma. Cancer Res. 2014, 75, 264–269. [Google Scholar] [CrossRef] [Green Version]
- Dudek, A.Z.; Pang, H.; Kratzke, R.A.; Otterson, G.A.; Hodgson, L.; Vokes, E.E.; Kindler, H.L.; Cancer and Leukemia Group B. Phase II Study of Dasatinib in Patients with Previously Treated Malignant Mesothelioma (Cancer and Leukemia Group B 30601): A Brief Report. J. Thorac. Oncol. 2012, 7, 755–759. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.-H.I.; Moon, J.; Garland, L.L.; Mack, P.C.; Testa, J.R.; Tsao, A.S.; Wozniak, A.J.; Gandara, D.R. SWOG S0722: Phase II study of mTOR inhibitor everolimus (RAD001) in advanced malignant pleural mesothelioma (MPM). J. Thorac. Oncol. 2015, 10, 387–391. [Google Scholar] [CrossRef] [Green Version]
- Govindan, R. Gefitinib in Patients with Malignant Mesothelioma: A Phase II Study by the Cancer and Leukemia Group B. Clin. Cancer Res. 2005, 11, 2300–2304. [Google Scholar] [CrossRef] [Green Version]
- Garland, L.L.; Rankin, C.; Gandara, D.R.; Rivkin, S.E.; Scott, K.M.; Nagle, R.B.; Klein-Szanto, A.J.; Testa, J.R.; Altomare, D.A.; Borden, E.C. Phase II Study of Erlotinib in Patients with Malignant Pleural Mesothelioma: A Southwest Oncology Group Study. J. Clin. Oncol. 2007, 25, 2406–2413. [Google Scholar] [CrossRef]
- Ding, H.; Zhao, J.; Zhang, Y.; Yu, J.; Liu, M.; Li, X.; Xu, L.; Lin, M.; Liu, C.; He, Z.; et al. Systematic Analysis of Drug Vulnerabilities Conferred by Tumor Suppressor Loss. Cell Rep. 2019, 27, 3331–3344.e6. [Google Scholar] [CrossRef] [Green Version]
- Rees, M.G.; Seashore-Ludlow, B.; Cheah, J.H.; Adams, D.J.; Price, E.V.; Gill, S.; Javaid, S.; Coletti, M.E.; Jones, V.L.; Bodycombe, N.E.; et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Methods 2015, 12, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.; Choi, B.Y.; Lee, M.-H.; Bode, A.M.; Surh, Y.-J. Implications of Genetic and Epigenetic Alterations of CDKN2A (p16(INK4a)) in Cancer. EBioMedicine 2016, 8, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Delaunay, T.; Achard, C.; Boisgerault, N.; Grard, M.; Petithomme, T.; Chatelain, C.; Dutoit, S.; Blanquart, C.; Royer, P.-J.; Minvielle, S.; et al. Frequent Homozygous Deletions of Type I Interferon Genes in Pleural Mesothelioma Confer Sensitivity to Oncolytic Measles Virus. J. Thorac. Oncol. 2020, 15, 827–842. [Google Scholar] [CrossRef] [PubMed]
- Musa, J.; Aynaud, M.-M.; Mirabeau, O.; Delattre, O.; Grünewald, T.G.P. MYBL2 (B-Myb): A central regulator of cell proliferation, cell survival and differentiation involved in tumorigenesis. Cell Death Dis. 2017, 8, e2895. [Google Scholar] [CrossRef]
- Mushtaq, M.U.; Papadas, A.; Pagenkopf, A.; Flietner, E.; Morrow, Z.; Chaudhary, S.G.; Asimakopoulos, F. Tumor matrix remodeling and novel immunotherapies: The promise of matrix-derived immune biomarkers. J. Immunother. Cancer 2018, 6, 65. [Google Scholar] [CrossRef]
- Chakravarthy, A.; Khan, L.; Bensler, N.P.; Bose, P.; De Carvalho, D.D. TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat. Commun. 2018, 9, 4692. [Google Scholar] [CrossRef]
- Bommhardt, U.; Schraven, B.; Simeoni, L. Beyond TCR Signaling: Emerging Functions of Lck in Cancer and Immunotherapy. Int. J. Mol. Sci. 2019, 20, 3500. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Pak, H.; Hammond-Martel, I.; Ghram, M.; Rodrigue, A.; Daou, S.; Barbour, H.; Corbeil, L.; Hébert, J.; Drobetsky, E.; et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 2013, 111, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Xu, D.; Gao, Y.; Schmid, R.A.; Peng, R.-W. The Association of BAP1 Loss-of-Function with the Defect in Homologous Recombination Repair and Sensitivity to PARP-Targeted Therapy. J. Thorac. Oncol. 2020, 15, e88–e90. [Google Scholar] [CrossRef]
- Yang, H. Co-Occurring LKB1 Deficiency Determinates the Susceptibility to ERK-Targeted Therapy in RAS-Mutant Lung Cancer. J. Thorac. Oncol. 2020, 15, e58–e59. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.-H.; Gallagher, R.; Shetler, J.; Skele, K.; Altomare, D.A.; Pestell, R.G.; Jhanwar, S.; Testa, J.R. The NF2 Tumor Suppressor Gene Product, Merlin, Inhibits Cell Proliferation and Cell Cycle Progression by Repressing Cyclin D1 Expression. Mol. Cell. Biol. 2005, 25, 2384–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Bollam, S.R.; White, S.M.; Laughlin, S.Z.; Graham, G.T.; Wadhwa, M.; Chen, H.; Nguyen, C.; Vitte, J.; Giovannini, M.; et al. Rac1-Mediated DNA Damage and Inflammation Promote Nf2 Tumorigenesis but Also Limit Cell-Cycle Progression. Dev. Cell 2016, 39, 452–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Lago, M.A.; Okada, T.; Murillo, M.M.; Socci, N.; Giancotti, F.G. Loss of the Tumor Suppressor Gene NF2, Encoding Merlin, Constitutively Activates Integrin-Dependent mTORC1 Signaling. Mol. Cell. Biol. 2009, 29, 4235–4249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishay-Ronen, D.; Diepenbruck, M.; Kalathur, R.K.R.; Sugiyama, N.; Tiede, S.; Ivanek, R.; Bantug, G.; Morini, M.F.; Wang, J.; Hess, C.; et al. Gain Fat—Lose Metastasis: Converting Invasive Breast Cancer Cells into Adipocytes Inhibits Cancer Metastasis. Cancer Cell 2019, 35, 17–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Duns, G.; Westers, H.; Sijmons, R.; Berg, A.V.D.; Kok, K. SETD2: An epigenetic modifier with tumor suppressor functionality. Oncotarget 2016, 7, 50719–50734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viaene, A.N.; Santi, M.; Rosenbaum, J.; Li, M.M.; Surrey, L.F.; Nasrallah, M.L.P. SETD2 mutations in primary central nervous system tumors. Acta Neuropathol. Commun. 2018, 6, 123. [Google Scholar] [CrossRef]
- Moroishi, T.; Hayashi, T.; Pan, W.-W.; Fujita, Y.; Holt, M.V.; Qin, J.; Carson, D.A.; Guan, K.-L. The Hippo Pathway Kinases LATS1/2 Suppress Cancer Immunity. Cell 2016, 167, 1525–1539.e7. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Liu, C.; Peng, W.; Lizée, G.; Overwijk, W.W.; Liu, Y.; Woodman, S.E.; Hwu, P. Antitumor T-cell responses contribute to the effects of dasatinib on c-KIT mutant murine mastocytoma and are potentiated by anti-OX40. Blood 2012, 120, 4533–4543. [Google Scholar] [CrossRef] [Green Version]
- Tu, M.M.; Lee, F.Y.F.; Jones, R.T.; Kimball, A.K.; Saravia, E.; Graziano, R.F.; Coleman, B.; Menard, K.; Yan, J.; Michaud, E.; et al. Targeting DDR2 enhances tumor response to anti–PD-1 immunotherapy. Sci. Adv. 2019, 5, eaav2437. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Minikes, A.; Gao, M.; Bian, H.; Li, Y.; Stockwell, B.R.; Chen, Z.-N.; Jiang, X. Intercellular interaction dictates cancer cell ferroptosis via Merlin-YAP signalling. Nature 2019, 572, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liang, S.-Q.; Schmid, R.A.; Peng, R.-W. New Horizons in KRAS-Mutant Lung Cancer: Dawn after Darkness. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausser, J.; Alon, U. Tumour heterogeneity and the evolutionary trade-offs of cancer. Nat. Rev. Cancer 2020, 20, 247–257. [Google Scholar] [CrossRef]
- Zhou, S.; Liu, L.; Li, H.; Eilers, G.; Kuang, Y.; Shi, S.; Yan, Z.; Li, X.; Corson, J.M.; Meng, F.; et al. Multipoint targeting of the PI3K/mTOR pathway in mesothelioma. Br. J. Cancer 2014, 110, 2479–2488. [Google Scholar] [CrossRef] [Green Version]
- Bonelli, M.; Digiacomo, G.; Fumarola, C.; Alfieri, R.; Quaini, F.; Falco, A.; Madeddu, D.; La Monica, S.; Cretella, D.; Ravelli, A.; et al. Combined Inhibition of CDK4/6 and PI3K/AKT/mTOR Pathways Induces a Synergistic Anti-Tumor Effect in Malignant Pleural Mesothelioma Cells. Neoplasia 2017, 19, 637–648. [Google Scholar] [CrossRef]
- Altomare, D.A.; You, H.; Xiao, G.-H.; Ramos-Nino, M.E.; Skele, K.L.; De Rienzo, A.; Jhanwar, S.C.; Mossman, B.T.; Kane, A.B.; Testa, J.R. Human and mouse mesotheliomas exhibit elevated AKT/PKB activity, which can be targeted pharmacologically to inhibit tumor cell growth. Oncogene 2005, 24, 6080–6089. [Google Scholar] [CrossRef] [Green Version]
- Yamaji, M.; Ota, A.; Wahiduzzaman, M.; Karnan, S.; Hyodo, T.; Konishi, H.; Tsuzuki, S.; Hosokawa, Y.; Haniuda, M. Novel ATP-competitive Akt inhibitor afuresertib suppresses the proliferation of malignant pleural mesothelioma cells. Cancer Med. 2017, 6, 2646–2659. [Google Scholar] [CrossRef] [Green Version]
- Sobhani, N.; Corona, S.P.; Zanconati, F.; Generali, D. Cyclin dependent kinase 4 and 6 inhibitors as novel therapeutic agents for targeted treatment of malignant mesothelioma. Genes Cancer 2017, 8, 495–496. [Google Scholar] [CrossRef] [Green Version]
- Pease, D.F.; Kratzke, R.A. Oncolytic Viral Therapy for Mesothelioma. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef]
- Carbone, M.; Yang, H.; Pass, H.I.; Krausz, T.; Testa, J.R.; Gaudino, G. BAP1 and Cancer. Nat. Rev. Cancer 2013, 13, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Rathkey, D.; Khanal, M.; Murai, J.; Zhang, J.; Sengupta, M.; Jiang, Q.; Morrow, B.; Evans, C.N.; Chari, R.; Fetsch, P.; et al. Sensitivity of Mesothelioma Cells to PARP Inhibitors Is Not Dependent on BAP1 but Is Enhanced by Temozolomide in Cells With High-Schlafen 11 and Low-O6-methylguanine-DNA Methyltransferase Expression. J. Thorac. Oncol. 2020, 15, 843–859. [Google Scholar] [CrossRef] [PubMed]
- Guazzelli, A.; Meysami, P.; Bakker, E.; Demonacos, C.; Giordano, A.; Demonacos, C.; Mutti, L. BAP1 Status Determines the Sensitivity of Malignant Mesothelioma Cells to Gemcitabine Treatment. Int. J. Mol. Sci. 2019, 20, 429. [Google Scholar] [CrossRef] [Green Version]
- Okonska, A.; Bühler, S.; Rao, V.; Ronner, M.; Blijlevens, M.; Van Der Meulen-Muileman, I.H.; De Menezes, R.X.; Wipplinger, M.; Oehl, K.; Smit, E.F.; et al. Functional Genomic Screen in Mesothelioma Reveals that Loss of Function of BRCA1-Associated Protein 1 Induces Chemoresistance to Ribonucleotide Reductase Inhibition. Mol. Cancer Ther. 2019, 19, 552–563. [Google Scholar] [CrossRef] [Green Version]
- Sen, N.; Satija, Y.K.; Das, S. PGC-1α, a Key Modulator of p53, Promotes Cell Survival upon Metabolic Stress. Mol. Cell 2011, 44, 621–634. [Google Scholar] [CrossRef] [Green Version]
- Assaily, W.; Rubinger, D.A.; Wheaton, K.; Lin, Y.; Ma, W.; Xuan, W.; Brown-Endres, L.; Tsuchihara, K.; Mak, T.W.; Benchimol, S. ROS-Mediated p53 Induction of Lpin1 Regulates Fatty Acid Oxidation in Response to Nutritional Stress. Mol. Cell 2011, 44, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, I.; Rotter, V. Regulation of lipid metabolism by p53—Fighting two villains with one sword. Trends Endocrinol. Metab. 2012, 23, 567–575. [Google Scholar] [CrossRef]
- Lacroix, M.; Riscal, R.; Arena, G.; Linares, L.K.; Le Cam, L. Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Mol. Metab. 2020, 33, 2–22. [Google Scholar] [CrossRef]
- Tian, K.; Bakker, E.; Hussain, M.; Guazzelli, A.; Alhebshi, H.; Meysami, P.; Demonacos, C.; Schwartz, J.-M.; Mutti, L.; Krstic-Demonacos, M. p53 modeling as a route to mesothelioma patients stratification and novel therapeutic identification. J. Transl. Med. 2018, 16, 282. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Simon, R. Identification of potential synthetic lethal genes to p53 using a computational biology approach. BMC Med. Genom. 2013, 6, 30. [Google Scholar] [CrossRef] [Green Version]
- Aning, O.A.; Cheok, C.F. Drugging in the absence of p53. J. Mol. Cell Biol. 2019, 11, 255–264. [Google Scholar] [CrossRef]
- Mairinger, F.D.; Walter, R.F.; Ting, S.; Vollbrecht, C.; Kollmeier, J.; Griff, S.; Hager, T.; Mairinger, T.; Christoph, D.C.; Theegarten, D.; et al. Mdm2 protein expression is strongly associated with survival in malignant pleural mesothelioma. Future Oncol. 2014, 10, 995–1005. [Google Scholar] [CrossRef]
- Di Marzo, D.; Forte, I.M.; Indovina, P.; Di Gennaro, E.; Rizzo, V.; Giorgi, F.; Mattioli, E.; Ianuzzi, C.A.; Budillon, A.; Giordano, A.; et al. Pharmacological targeting of p53 through RITA is an effective antitumoral strategy for malignant pleural mesothelioma. Cell Cycle 2013, 13, 652–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, R.F.; Werner, R.; Wessolly, M.; Mairinger, E.; Borchert, S.; Schmeller, J.; Kollmeier, J.; Mairinger, T.; Hager, T.; Bankfalvi, A.; et al. Inhibition of MDM2 via Nutlin-3A: A Potential Therapeutic Approach for Pleural Mesotheliomas with MDM2-Induced Inactivation of Wild-Type P53. J. Oncol. 2018, 2018, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Urso, L.; Cavallari, I.; Silic-Benussi, M.; Biasini, L.; Zago, G.; Calabrese, F.; Conte, P.; Ciminale, V.; Pasello, G. Synergistic targeting of malignant pleural mesothelioma cells by MDM2 inhibitors and TRAIL agonists. Oncotarget 2017, 8, 44232–44241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Liang, S.-Q.; Yang, H.; Bruggmann, R.; Berezowska, S.; Yang, Z.; Marti, T.M.; Hall, S.R.R.; Gao, Y.; Kocher, G.J.; et al. CRISPR Screening Identifies WEE1 as a Combination Target for Standard Chemotherapy in Malignant Pleural Mesothelioma. Mol. Cancer Ther. 2019, 19, 661–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indovina, P.; Marcelli, E.; Di Marzo, D.; Casini, N.; Forte, I.M.; Giorgi, F.; Alfano, L.; Pentimalli, F.; Giordano, A. Abrogating G2/M checkpoint through WEE1 inhibition in combination with chemotherapy as a promising therapeutic approach for mesothelioma. Cancer Biol. Ther. 2014, 15, 380–388. [Google Scholar] [CrossRef] [Green Version]
- Destro, A.; Ceresoli, G.; Falleni, M.; Zucali, P.; Morenghi, E.; Bianchi, P.; Pellegrini, C.; Cordani, N.; Vaira, V.; Alloisio, M.; et al. EGFR overexpression in malignant pleural mesothelioma. Lung Cancer 2006, 51, 207–215. [Google Scholar] [CrossRef]
- Mezzapelle, R.; Miglio, U.; Rena, O.; Paganotti, A.; Allegrini, S.; Antona, J.; Molinari, F.; Frattini, M.; Monga, G.; Alabiso, O.; et al. Mutation analysis of the EGFR gene and downstream signalling pathway in histologic samples of malignant pleural mesothelioma. Br. J. Cancer 2013, 108, 1743–1749. [Google Scholar] [CrossRef] [Green Version]
- Horvai, A.E.; Li, L.; Xu, Z.; Kramer, M.J.; Jablons, D.; Treseler, P.A. Malignant mesothelioma does not demonstrate overexpression or gene amplification despite cytoplasmic immunohistochemical staining for c-Erb-B2. Arch. Pathol. Lab. Med. 2003, 127, 465–469. [Google Scholar]
- Klampatsa, A.; Achkova, D.Y.; Davies, D.M.; Parente-Pereira, A.C.; Woodman, N.; Rosekilly, J.; Osborne, G.; Thayaparan, T.; Bille, A.; Sheaf, M.; et al. Intracavitary ‘T4 immunotherapy’ of malignant mesothelioma using pan-ErbB re-targeted CAR T-cells. Cancer Lett. 2017, 393, 52–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toma, S.; Colucci, L.; Scarabelli, L.; Scaramuccia, A.; Emionite, L.; Betta, P.G.; Mutti, L. Synergistic effect of the anti-HER-2/neu antibody and cisplatin in immortalized and primary mesothelioma cell lines. J. Cell. Physiol. 2002, 193, 37–41. [Google Scholar] [CrossRef]
- Popat, S.; Curioni-Fontecedro, A.; Polydoropoulou, V.; Shah, R.; O’Brien, M.; Pope, A.; Fisher, P.; Spicer, J.; Roy, A.; Gilligan, D.; et al. A multicentre randomized phase III trial comparing pembrolizumab (P) vs single agent chemotherapy (CT) for advanced pre-treated malignant pleural mesothelioma (MPM): Results from the European Thoracic Oncology Platform (ETOP 9-15) PROMISE-meso trial. Ann. Oncol. 2019, 30, v931. [Google Scholar] [CrossRef]
- Maio, M.; Scherpereel, A.; Calabrò, L.; Aerts, J.; Perez, S.C.; Bearz, A.; Nackaerts, K.; A Fennell, D.; Kowalski, D.; Tsao, A.S.; et al. Tremelimumab as second-line or third-line treatment in relapsed malignant mesothelioma (DETERMINE): A multicentre, international, randomised, double-blind, placebo-controlled phase 2b trial. Lancet Oncol. 2017, 18, 1261–1273. [Google Scholar] [CrossRef]
- Tsao, A.S.; Lin, H.; Carter, B.W.; Lee, J.J.; Rice, D.; Vaporcyan, A.; Swisher, S.; Mehran, R.; Heymach, J.; Nilsson, M.; et al. Biomarker-Integrated Neoadjuvant Dasatinib Trial in Resectable Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2018, 13, 246–257. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Xu, D.; Yang, H.; Berezowska, S.; Gao, Y.; Liang, S.-Q.; Marti, T.M.; Hall, S.R.R.; Dorn, P.; Kocher, G.J.; Schmid, R.A.; et al. Endoplasmic Reticulum Stress Signaling as a Therapeutic Target in Malignant Pleural Mesothelioma. Cancers 2019, 11, 1502. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Liang, S.-Q.; Xu, D.; Yang, Z.; Marti, T.M.; Gao, Y.; Kocher, G.J.; Zhao, H.; Schmid, R.A.; Peng, R.-W. HSP90/AXL/eIF4E-regulated unfolded protein response as an acquired vulnerability in drug-resistant KRAS-mutant lung cancer. Oncogenesis 2019, 8, 45. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Lu, Y.; Akbani, R.; Ju, Z.; Roebuck, P.L.; Liu, W.; Yang, J.-Y.; Broom, B.M.; Verhaak, R.G.W.; Kane, D.W.; et al. TCPA: A resource for cancer functional proteomics data. Nat. Methods 2013, 10, 1046–1047. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Zhao, L.; Yao, F.; Gao, Y.; Marti, T.M.; Schmid, R.A.; Peng, R.-W. Integrative Pharmacogenomic Profiling Identifies Novel Cancer Drugs and Gene Networks Modulating Ferroptosis Sensitivity in Pan-Cancer. 2020. Available online: https://doi.org/10.21203/rs.3.rs-34574/v1 (accessed on 3 July 2020).
- Alonso-López, D.; Campos-Laborie, F.J.; A Gutiérrez, M.; Lambourne, L.; Calderwood, M.A.; Vidal, M.; Rivas, J.D. APID database: Redefining protein–protein interaction experimental evidences and binary interactomes. Database 2019, 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Xu, D.; Yang, Z.; Yao, F.; Zhao, H.; Schmid, R.A.; Peng, R.-W. Systematic Analysis of Aberrant Biochemical Networks and Potential Drug Vulnerabilities Induced by Tumor Suppressor Loss in Malignant Pleural Mesothelioma. Cancers 2020, 12, 2310. https://doi.org/10.3390/cancers12082310
Yang H, Xu D, Yang Z, Yao F, Zhao H, Schmid RA, Peng R-W. Systematic Analysis of Aberrant Biochemical Networks and Potential Drug Vulnerabilities Induced by Tumor Suppressor Loss in Malignant Pleural Mesothelioma. Cancers. 2020; 12(8):2310. https://doi.org/10.3390/cancers12082310
Chicago/Turabian StyleYang, Haitang, Duo Xu, Zhang Yang, Feng Yao, Heng Zhao, Ralph A. Schmid, and Ren-Wang Peng. 2020. "Systematic Analysis of Aberrant Biochemical Networks and Potential Drug Vulnerabilities Induced by Tumor Suppressor Loss in Malignant Pleural Mesothelioma" Cancers 12, no. 8: 2310. https://doi.org/10.3390/cancers12082310
APA StyleYang, H., Xu, D., Yang, Z., Yao, F., Zhao, H., Schmid, R. A., & Peng, R. -W. (2020). Systematic Analysis of Aberrant Biochemical Networks and Potential Drug Vulnerabilities Induced by Tumor Suppressor Loss in Malignant Pleural Mesothelioma. Cancers, 12(8), 2310. https://doi.org/10.3390/cancers12082310