Kv10.1 Regulates Microtubule Dynamics during Mitosis



Abstract
:1. Introduction
2. Results
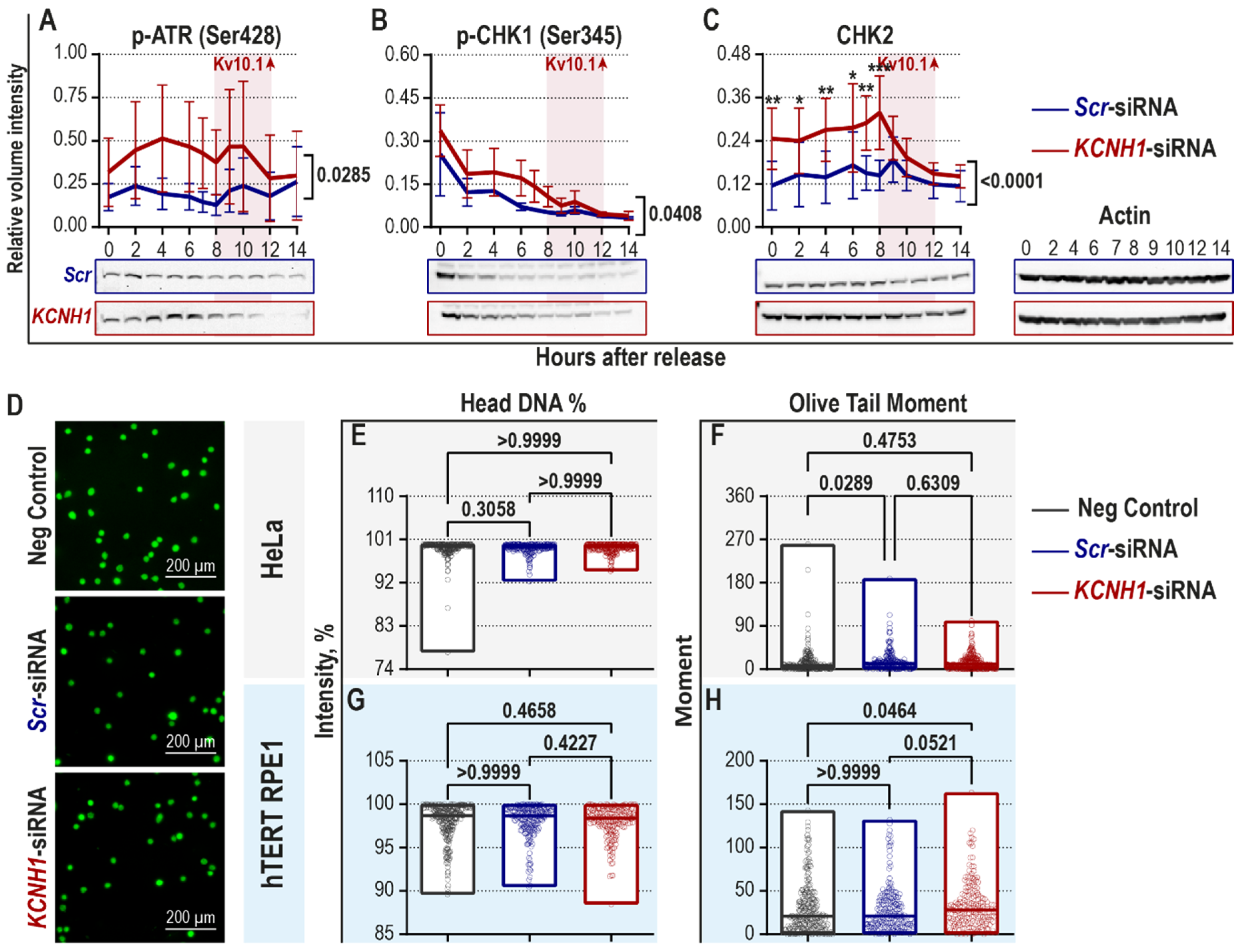
2.1. Downregulation of Kv10.1 Partially Activates the DNA Damage-Induced Checkpoint in the Absence of DNA Damage
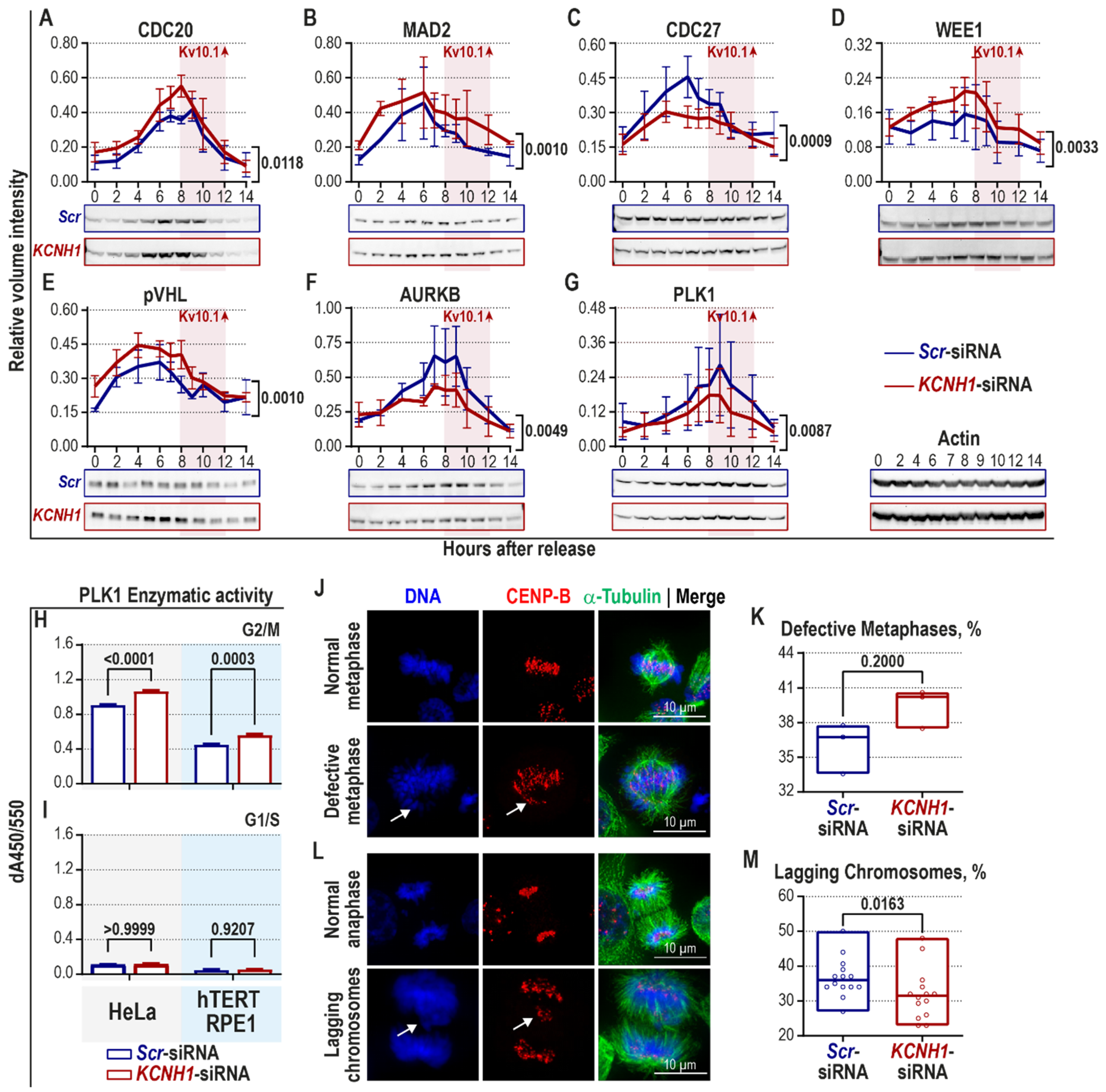
2.2. Kv10.1 Downregulation Leads to Activation of Spindle Assembly Checkpoint and Decreased Occurrence of Lagging Chromosomes in HeLa Cells
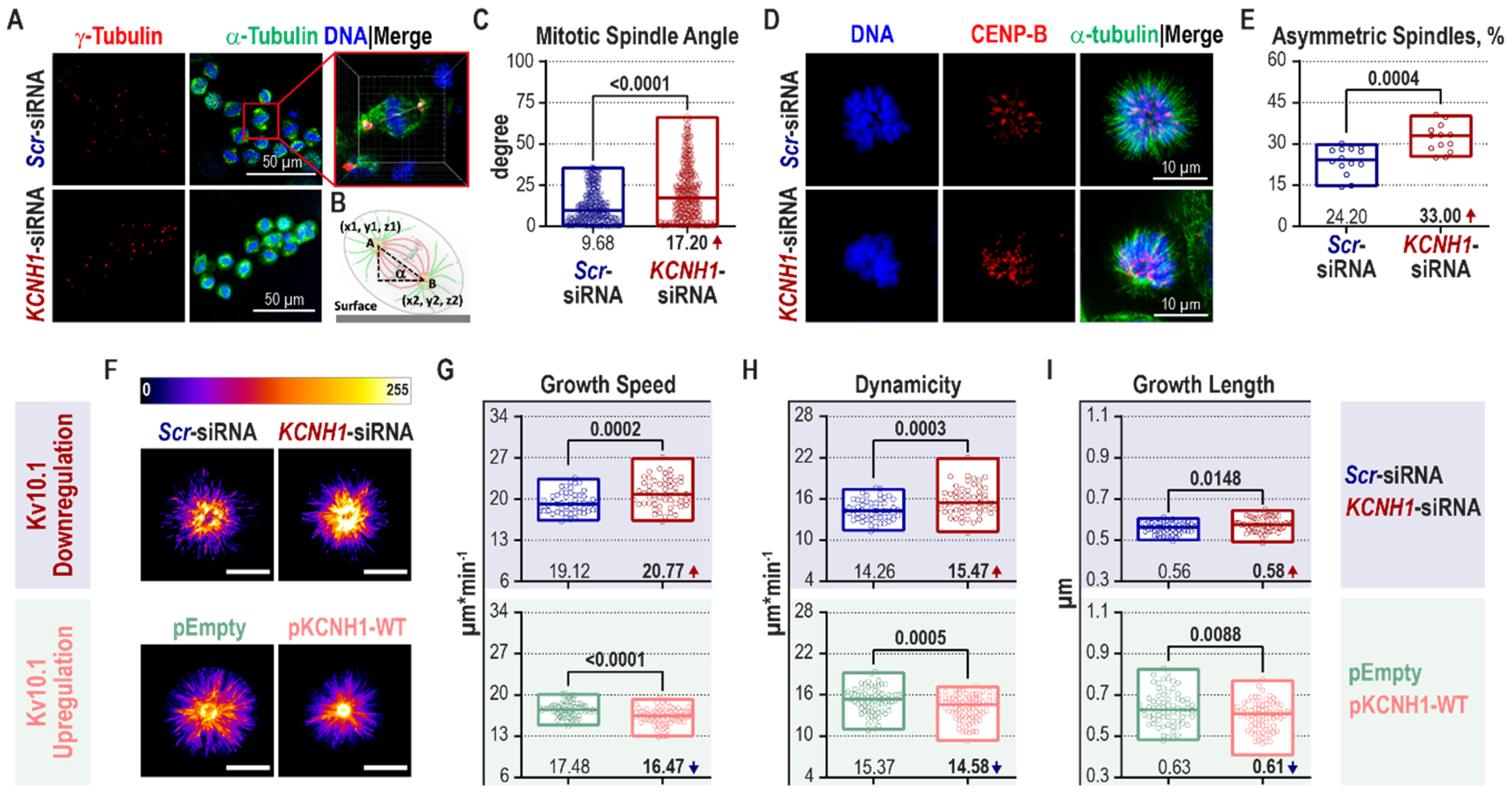
2.3. Kv10.1 Knockdown Leads to Mitotic Spindle Tilting and Increased Microtubule Plus-End Growth
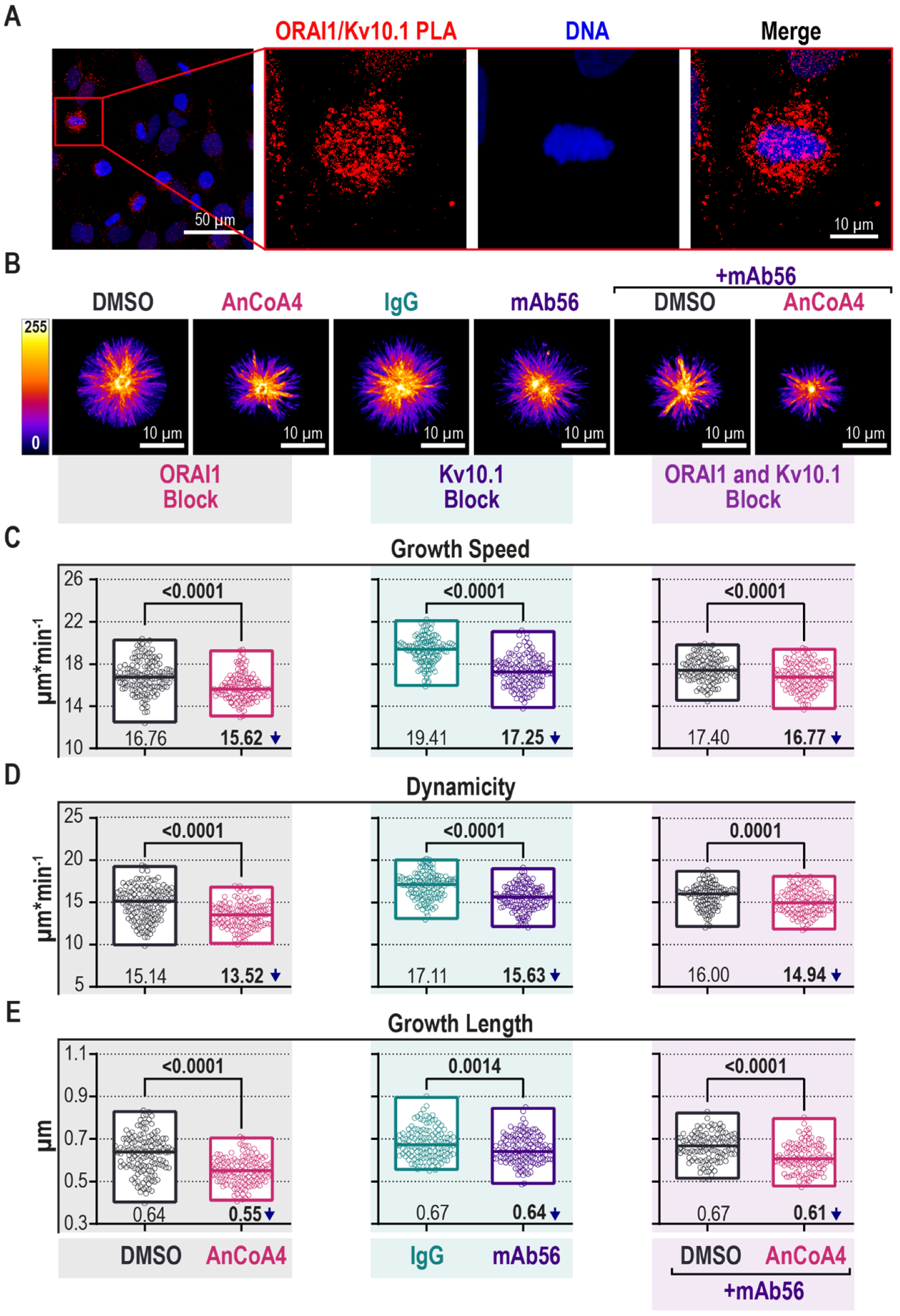
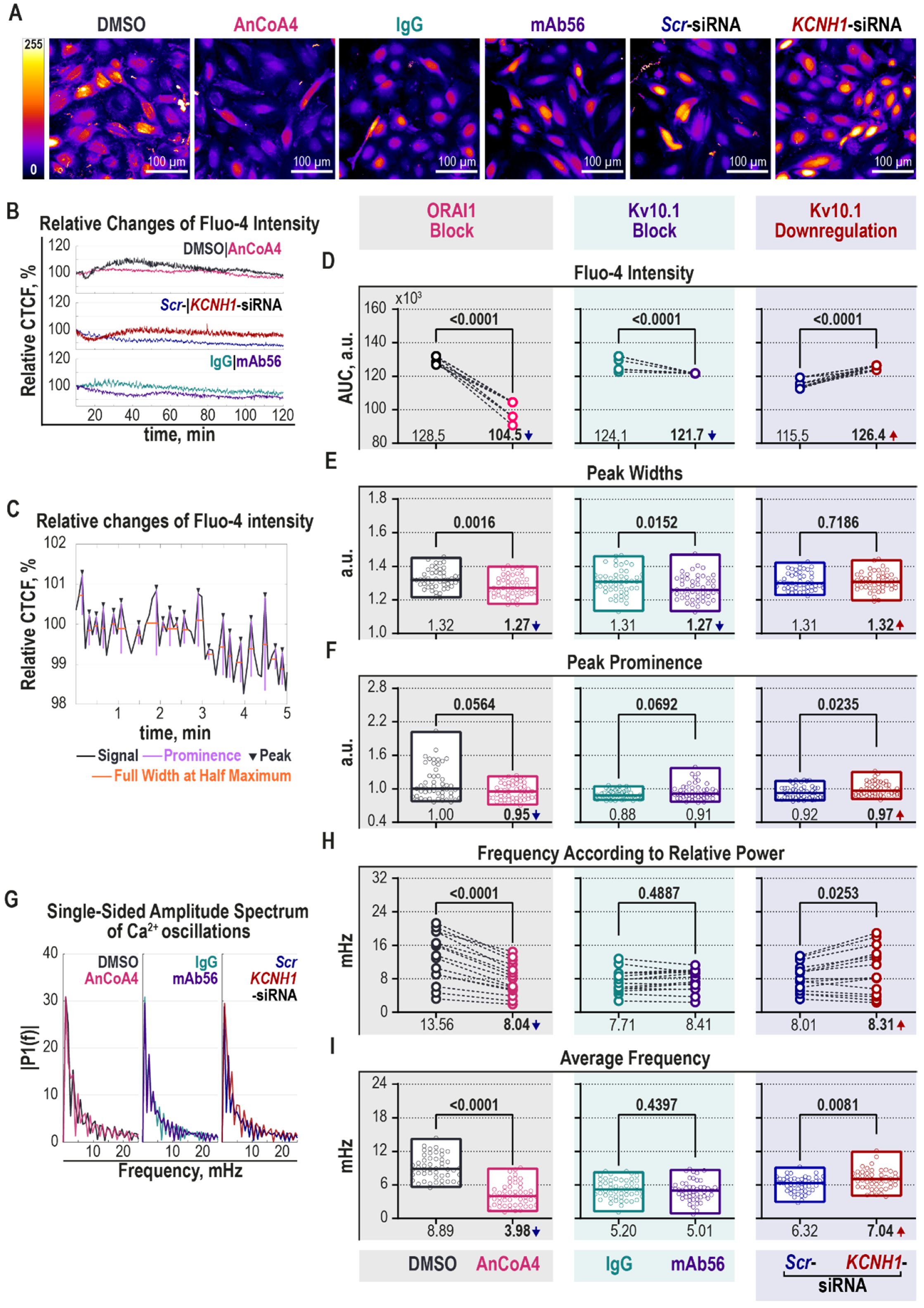
2.4. Block of Kv10.1 or ORAI1 Currents Reduces Microtubule Dynamics
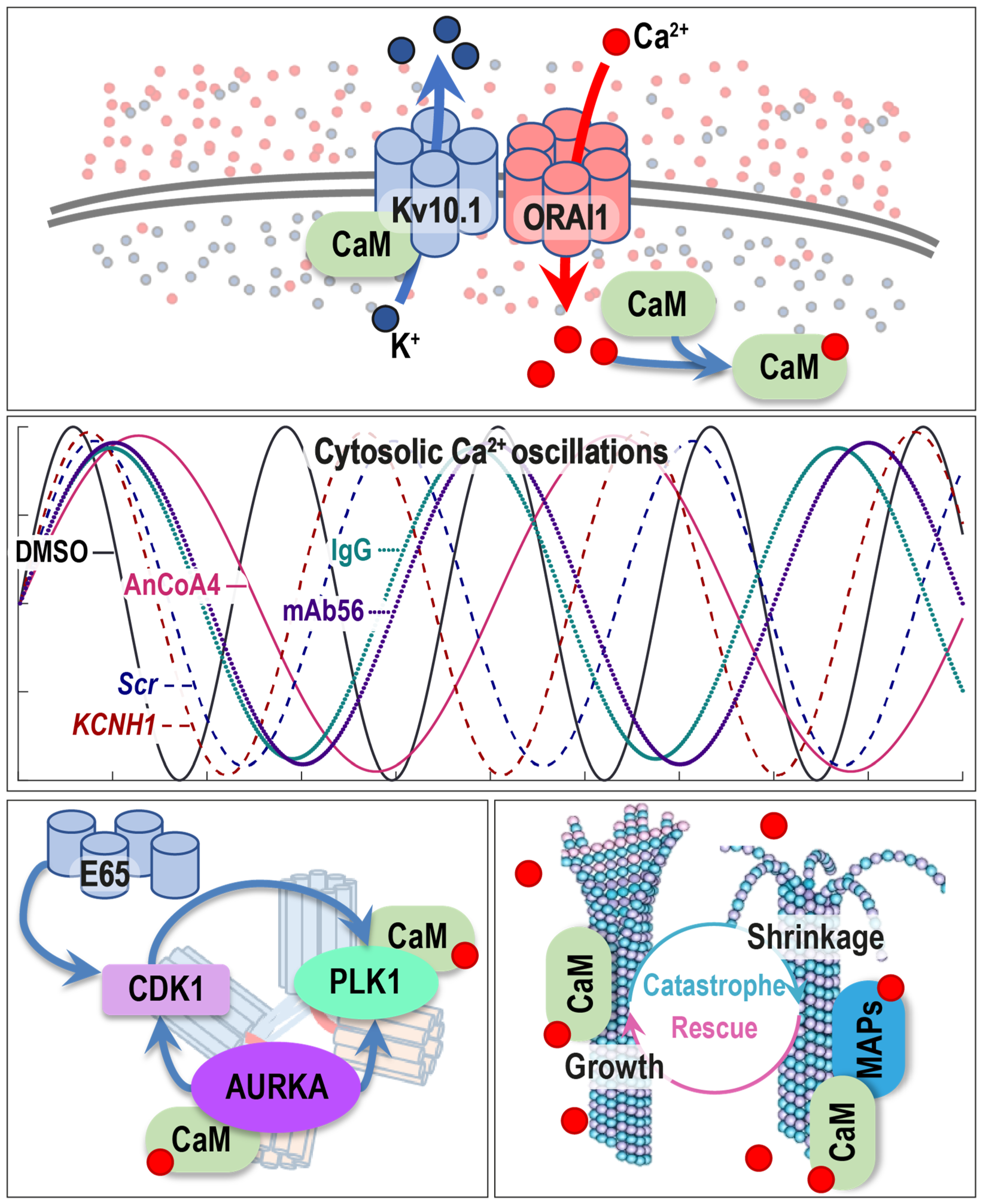
2.5. Kv10.1 Regulates Intracellular [Ca2+] Oscillations
3. Materials and Methods
3.1. Cell Lines
3.2. Transfections
3.3. Cell Cycle Synchronization
3.4. Protein Extracts
3.5. SDS-PAGE and Immunoblotting
3.6. Immunoprecipitation
3.7. Single-Cell Gel Electrophoresis
3.8. Immunofluorescence
3.9. Determination of Fan-Shaped Mitotic Spindles
3.10. Determination of Lagging Chromosomes and Defective Metaphases
3.11. Determination of Mitotic Spindle Angle
3.12. Determination of the Polo-Like Kinase 1 Enzymatic Activity
3.13. Proximity Ligation Assay
3.14. Live Cell Imaging
3.15. Assessment of Microtubule Dynamics
3.16. Intracellular Calcium Imaging in HeLa Cells
3.17. Statistical Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gutman, G.A.; Chandy, K.G.; Grissmer, S.; Lazdunski, M.; McKinnon, D.; Pardo, L.A.; Robertson, G.A.; Rudy, B.; Sanguinetti, M.C.; Stuhmer, W.; et al. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol. Rev. 2005, 57, 473–508. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Lino de Oliveira, C.; Mello de Queiroz, F.; Pardo, L.A.; Stuhmer, W.; Del Bel, E. Eag1 potassium channel immunohistochemistry in the CNS of adult rat and selected regions of human brain. Neuroscience 2008, 155, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, L.S.; Schmidt, H.; Farsi, Z.; Barrantes-Freer, A.; Rubio, M.E.; Ufartes, R.; Eilers, J.; Sakaba, T.; Stuhmer, W.; Pardo, L.A. KV 10.1 opposes activity-dependent increase in Ca2+ influx into the presynaptic terminal of the parallel fibre-Purkinje cell synapse. J. Physiol. 2015, 593, 181–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Martinson, A.S.; Layden, M.J.; Diatta, F.H.; Sberna, A.P.; Simmons, D.K.; Martindale, M.Q.; Jegla, T.J. Ether-a-go-go family voltage-gated K+ channels evolved in an ancestral metazoan and functionally diversified in a cnidarian-bilaterian ancestor. J. Exp. Biol. 2015, 218, 526–536. [Google Scholar] [CrossRef] [Green Version]
- Pardo, L.A.; del Camino, D.; Sanchez, A.; Alves, F.; Bruggemann, A.; Beckh, S.; Stuhmer, W. Oncogenic potential of EAG K(+) channels. EMBO J. 1999, 18, 5540–5547. [Google Scholar] [CrossRef]
- Agarwal, J.R.; Griesinger, F.; Stuhmer, W.; Pardo, L.A. The potassium channel Ether a go-go is a novel prognostic factor with functional relevance in acute myeloid leukemia. Mol. Cancer 2010, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Asher, V.; Khan, R.; Warren, A.; Shaw, R.; Schalkwyk, G.V.; Bali, A.; Sowter, H.M. The Eag potassium channel as a new prognostic marker in ovarian cancer. Diagn. Pathol. 2010, 5, 78. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.W.; Luo, H.S.; Jin, X.; Yan, J.J.; Ai, Y.W. Aberrant expression of Eag1 potassium channels in gastric cancer patients and cell lines. Med. Oncol. 2007, 24, 345–350. [Google Scholar] [CrossRef]
- Ding, X.W.; Wang, X.G.; Luo, H.S.; Tan, S.Y.; Gao, S.; Luo, B.; Jiang, H. Expression and prognostic roles of Eag1 in resected esophageal squamous cell carcinomas. Dig. Dis. Sci. 2008, 53, 2039–2044. [Google Scholar] [CrossRef]
- Hemmerlein, B.; Weseloh, R.M.; Mello de Queiroz, F.; Knotgen, H.; Sanchez, A.; Rubio, M.E.; Martin, S.; Schliephacke, T.; Jenke, M.; Heinz Joachim, R.; et al. Overexpression of Eag1 potassium channels in clinical tumours. Mol. Cancer 2006, 5, 41. [Google Scholar] [CrossRef] [Green Version]
- Martinez, R.; Stuhmer, W.; Martin, S.; Schell, J.; Reichmann, A.; Rohde, V.; Pardo, L. Analysis of the expression of Kv10.1 potassium channel in patients with brain metastases and glioblastoma multiforme: Impact on survival. BMC Cancer 2015, 15, 839. [Google Scholar] [CrossRef] [Green Version]
- Mello de Queiroz, F.; Suarez-Kurtz, G.; Stuhmer, W.; Pardo, L.A. Ether a go-go potassium channel expression in soft tissue sarcoma patients. Mol. Cancer 2006, 5, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ousingsawat, J.; Spitzner, M.; Puntheeranurak, S.; Terracciano, L.; Tornillo, L.; Bubendorf, L.; Kunzelmann, K.; Schreiber, R. Expression of voltage-gated potassium channels in human and mouse colonic carcinoma. Clin. Cancer Res. 2007, 13, 824–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadhwa, S.; Wadhwa, P.; Dinda, A.K.; Gupta, N.P. Differential expression of potassium ion channels in human renal cell carcinoma. Int. Urol. Nephrol. 2009, 41, 251–257. [Google Scholar] [CrossRef]
- Gomez-Varela, D.; Zwick-Wallasch, E.; Knotgen, H.; Sanchez, A.; Hettmann, T.; Ossipov, D.; Weseloh, R.; Contreras-Jurado, C.; Rothe, M.; Stuhmer, W.; et al. Monoclonal antibody blockade of the human Eag1 potassium channel function exerts antitumor activity. Cancer Res. 2007, 67, 7343–7349. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wu, X.; Zhong, D.; Zhai, W.; Ding, Z.; Zhou, Y. Short Hairpin RNA (shRNA) Ether a go-go 1 (Eag1) inhibition of human osteosarcoma angiogenesis via VEGF/PI3K/AKT signaling. Int. J. Mol. Sci. 2012, 13, 12573–12583. [Google Scholar] [CrossRef]
- Wu, X.; Zhong, D.; Gao, Q.; Zhai, W.; Ding, Z.; Wu, J. MicroRNA-34a inhibits human osteosarcoma proliferation by downregulating ether a go-go 1 expression. Int. J. Med. Sci. 2013, 10, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Zhong, D.; Fu, X.; Liu, Q.; Kang, L.; Ding, Z. Silencing of Ether a go-go 1 by shRNA inhibits osteosarcoma growth and cell cycle progression. Int. J. Mol. Sci. 2014, 15, 5570–5581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spitzner, M.; Ousingsawat, J.; Scheidt, K.; Kunzelmann, K.; Schreiber, R. Voltage-gated K+ channels support proliferation of colonic carcinoma cells. FASEB J. 2007, 21, 35–44. [Google Scholar] [CrossRef]
- Garcia-Quiroz, J.; Garcia-Becerra, R.; Santos-Martinez, N.; Barrera, D.; Ordaz-Rosado, D.; Avila, E.; Halhali, A.; Villanueva, O.; Ibarra-Sanchez, M.J.; Esparza-Lopez, J.; et al. In vivo dual targeting of the oncogenic Ether-a-go-go-1 potassium channel by calcitriol and astemizole results in enhanced antineoplastic effects in breast tumors. BMC Cancer 2014, 14, 745. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, A.; Urrego, D.; Pardo, L.A. Cyclic expression of the voltage-gated potassium channel KV10.1 promotes disassembly of the primary cilium. EMBO Rep. 2016, 17, 708–723. [Google Scholar] [CrossRef] [PubMed]
- Urrego, D.; Movsisyan, N.; Ufartes, R.; Pardo, L.A. Periodic expression of Kv10.1 driven by pRb/E2F1 contributes to G2/M progression of cancer and non-transformed cells. Cell Cycle 2016, 15, 799–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heim, A.; Rymarczyk, B.; Mayer, T.U. Regulation of Cell Division. Adv. Exp. Med. Biol 2017, 953, 83–116. [Google Scholar] [CrossRef]
- Shaltiel, I.A.; Krenning, L.; Bruinsma, W.; Medema, R.H. The same, only different—DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci. 2015, 128, 607–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musacchio, A.; Salmon, E.D. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 2007, 8, 379–393. [Google Scholar] [CrossRef]
- Saurin, A.T. Kinase and Phosphatase Cross-Talk at the Kinetochore. Front. Cell Dev. Biol. 2018, 6, 62. [Google Scholar] [CrossRef] [Green Version]
- Barghouth, P.G.; Thiruvalluvan, M.; Oviedo, N.J. Bioelectrical regulation of cell cycle and the planarian model system. Biochim. Biophys. Acta 2015, 1848, 2629–2637. [Google Scholar] [CrossRef] [Green Version]
- Blackiston, D.J.; McLaughlin, K.A.; Levin, M. Bioelectric controls of cell proliferation: Ion channels, membrane voltage and the cell cycle. Cell Cycle 2009, 8, 3527–3536. [Google Scholar] [CrossRef] [Green Version]
- Kahl, C.R.; Means, A.R. Regulation of cell cycle progression by calcium/calmodulin-dependent pathways. Endocr. Rev. 2003, 24, 719–736. [Google Scholar] [CrossRef] [Green Version]
- Roderick, H.L.; Cook, S.J. Ca2+ signalling checkpoints in cancer: Remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef] [Green Version]
- Humeau, J.; Bravo-San Pedro, J.M.; Vitale, I.; Nunez, L.; Villalobos, C.; Kroemer, G.; Senovilla, L. Calcium signaling and cell cycle: Progression or death. Cell Calcium 2018, 70, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Kao, J.P.; Alderton, J.M.; Tsien, R.Y.; Steinhardt, R.A. Active involvement of Ca2+ in mitotic progression of Swiss 3T3 fibroblasts. J. Cell Biol. 1990, 111, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Poenie, M.; Alderton, J.; Steinhardt, R.; Tsien, R. Calcium rises abruptly and briefly throughout the cell at the onset of anaphase. Science 1986, 233, 886–889. [Google Scholar] [CrossRef] [PubMed]
- Ratan, R.R.; Maxfield, F.R.; Shelanski, M.L. Long-lasting and rapid calcium changes during mitosis. J. Cell Biol. 1988, 107, 993–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tombes, R.M.; Borisy, G.G. Intracellular free calcium and mitosis in mammalian cells: Anaphase onset is calcium modulated, but is not triggered by a brief transient. J. Cell Biol. 1989, 109, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, M.; Larman, M.G. Calcium and mitosis. Semin. Cell Dev. Biol. 2001, 12, 53–58. [Google Scholar] [CrossRef]
- Nishida, E.; Kumagai, H.; Ohtsuki, I.; Sakai, H. The Interactions between Calcium-Dependent Regulator Protein of Cyclic Nucleotide Phosphodiesterase and Microtubule Proteins. I. Effect of Calcium-Dependent Regulator Protein on the Calcium Sensitivity of Microtubule Assembly. J. Biochem. 1979, 85, 1257–1266. [Google Scholar]
- Sandoval, I.V.; Weber, K. Calcium-induced inactivation of microtubule formation in brain extracts. Presence of a calcium-dependent protease acting on polymerization-stimulating microtubule-associated proteins. Eur. J. Biochem. 1978, 92, 463–470. [Google Scholar] [CrossRef]
- Xu, N.; Luo, K.Q.; Chang, D.C. Ca2+ signal blockers can inhibit M/A transition in mammalian cells by interfering with the spindle checkpoint. Biochem. Biophys. Res. Commun. 2003, 306, 737–745. [Google Scholar] [CrossRef]
- Buljan, V.; Ivanova, E.P.; Cullen, K.M. How calcium controls microtubule anisotropic phase formation in the presence of microtubule-associated proteins in vitro. Biochem. Biophys. Res. Commun. 2009, 381, 224–228. [Google Scholar] [CrossRef]
- Badaoui, M.; Mimsy-Julienne, C.; Saby, C.; Van Gulick, L.; Peretti, M.; Jeannesson, P.; Morjani, H.; Ouadid-Ahidouch, H. Collagen type 1 promotes survival of human breast cancer cells by overexpressing Kv10.1 potassium and Orai1 calcium channels through DDR1-dependent pathway. Oncotarget 2018, 9, 24653–24671. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [CrossRef] [PubMed]
- Vauzour, D.; Vafeiadou, K.; Rice-Evans, C.; Cadenas, E.; Spencer, J.P. Inhibition of cellular proliferation by the genistein metabolite 5,7,3′,4′-tetrahydroxyisoflavone is mediated by DNA damage and activation of the ATR signalling pathway. Arch. Biochem. Biophys. 2007, 468, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Pereira, E.; Maxfield, M.; Russell, B.; Goudelock, D.M.; Sanchez, Y. Regulation of Chk1 includes chromatin association and 14-3-3 binding following phosphorylation on Ser-345. J. Biol. Chem. 2003, 278, 25207–25217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell Biol. 2001, 21, 4129–4139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.Y.; Li, K.; Stewart, G.S.; Elledge, S.J. Human Claspin works with BRCA1 to both positively and negatively regulate cell proliferation. Proc. Natl. Acad. Sci. USA 2004, 101, 6484–6489. [Google Scholar] [CrossRef] [Green Version]
- Speit, G.; Rothfuss, A. The comet assay: A sensitive genotoxicity test for the detection of DNA damage and repair. Methods Mol. Biol. 2012, 920, 79–90. [Google Scholar] [CrossRef]
- Baumann, K. Nuclear envelope: ATR senses mechanical stress. Nat. Rev. Mol. Cell Biol. 2014, 15, 559. [Google Scholar] [CrossRef]
- Burgess, R.C.; Burman, B.; Kruhlak, M.J.; Misteli, T. Activation of DNA damage response signaling by condensed chromatin. Cell Rep. 2014, 9, 1703–1717. [Google Scholar] [CrossRef] [Green Version]
- Eliezer, Y.; Argaman, L.; Kornowski, M.; Roniger, M.; Goldberg, M. Interplay between the DNA damage proteins MDC1 and ATM in the regulation of the spindle assembly checkpoint. J. Biol. Chem. 2014, 289, 8182–8193. [Google Scholar] [CrossRef] [Green Version]
- Kidiyoor, G.R.; Kumar, A.; Foiani, M. ATR-mediated regulation of nuclear and cellular plasticity. DNA Repair 2016, 44, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, K.S.; Chau, T.; Engebrecht, J. DNA damage response and spindle assembly checkpoint function throughout the cell cycle to ensure genomic integrity. PLoS Genet. 2015, 11, e1005150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Hemmerich, P.; Grosse, F. Centrosomal localization of DNA damage checkpoint proteins. J. Cell Biochem. 2007, 101, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Chabalier-Taste, C.; Racca, C.; Dozier, C.; Larminat, F. BRCA1 is regulated by Chk2 in response to spindle damage. Biochim. Biophys. Acta 2008, 1783, 2223–2233. [Google Scholar] [CrossRef]
- Golan, A.; Pick, E.; Tsvetkov, L.; Nadler, Y.; Kluger, H.; Stern, D.F. Centrosomal Chk2 in DNA damage responses and cell cycle progression. Cell Cycle 2010, 9, 2647–2656. [Google Scholar] [CrossRef] [Green Version]
- Ertych, N.; Stolz, A.; Valerius, O.; Braus, G.H.; Bastians, H. CHK2-BRCA1 tumor-suppressor axis restrains oncogenic Aurora-A kinase to ensure proper mitotic microtubule assembly. Proc. Natl. Acad. Sci. USA 2016, 113, 1817–1822. [Google Scholar] [CrossRef] [Green Version]
- Shang, Z.; Yu, L.; Lin, Y.F.; Matsunaga, S.; Shen, C.Y.; Chen, B.P. DNA-PKcs activates the Chk2-Brca1 pathway during mitosis to ensure chromosomal stability. Oncogenesis 2014, 3, e85. [Google Scholar] [CrossRef] [Green Version]
- Stolz, A.; Ertych, N.; Kienitz, A.; Vogel, C.; Schneider, V.; Fritz, B.; Jacob, R.; Dittmar, G.; Weichert, W.; Petersen, I.; et al. The CHK2-BRCA1 tumour suppressor pathway ensures chromosomal stability in human somatic cells. Nat. Cell Biol. 2010, 12, 492–499. [Google Scholar] [CrossRef]
- Yeh, C.W.; Yu, Z.C.; Chen, P.H.; Cheng, Y.C.; Shieh, S.Y. Phosphorylation at threonine 288 by cell cycle checkpoint kinase 2 (CHK2) controls human monopolar spindle 1 (Mps1) kinetochore localization. J. Biol. Chem. 2014, 289, 15319–15327. [Google Scholar] [CrossRef] [Green Version]
- King, R.W.; Peters, J.-M.; Tugendreich, S.; Rolfe, M.; Hieter, P.; Kirschner, M.W. A 20s complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell 1995, 81, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Rohe, A.; Platzer, C.; Najjar, A.; Erdmann, F.; Sippl, W. Regulation of G2/M Transition by Inhibition of WEE1 and PKMYT1 Kinases. Molecules 2017, 22, 2045. [Google Scholar] [CrossRef] [Green Version]
- Chow, J.P.; Poon, R.Y.; Ma, H.T. Inhibitory phosphorylation of cyclin-dependent kinase 1 as a compensatory mechanism for mitosis exit. Mol. Cell Biol. 2011, 31, 1478–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Fiore, B.; Pines, J. How cyclin A destruction escapes the spindle assembly checkpoint. J. Cell Biol. 2010, 190, 501–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collin, P.; Nashchekina, O.; Walker, R.; Pines, J. The spindle assembly checkpoint works like a rheostat rather than a toggle switch. Nat. Cell Biol. 2013, 15, 1378–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoma, C.R.; Toso, A.; Gutbrodt, K.L.; Reggi, S.P.; Frew, I.J.; Schraml, P.; Hergovich, A.; Moch, H.; Meraldi, P.; Krek, W. VHL loss causes spindle misorientation and chromosome instability. Nat. Cell Biol. 2009, 11, 994–1001. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Huang, Y.; Zhang, L.; Yuan, K.; Chu, Y.; Dou, Z.; Jin, C.; Garcia-Barrio, M.; Liu, X.; Yao, X. Spatiotemporal dynamics of Aurora B-PLK1-MCAK signaling axis orchestrates kinetochore bi-orientation and faithful chromosome segregation. Sci. Rep. 2015, 5, 12204. [Google Scholar] [CrossRef]
- Krenn, V.; Musacchio, A. The Aurora B Kinase in Chromosome Bi-Orientation and Spindle Checkpoint Signaling. Front. Oncol. 2015, 5, 225. [Google Scholar] [CrossRef] [Green Version]
- Shandilya, J.; Medler, K.F.; Roberts, S.G. Regulation of AURORA B function by mitotic checkpoint protein MAD2. Cell Cycle 2016, 15, 2196–2201. [Google Scholar] [CrossRef] [Green Version]
- Zachos, G.; Black, E.J.; Walker, M.; Scott, M.T.; Vagnarelli, P.; Earnshaw, W.C.; Gillespie, D.A.F. Chk1 Is Required for Spindle Checkpoint Function. Dev. Cell 2007, 12, 247–260. [Google Scholar] [CrossRef] [Green Version]
- Peddibhotla, S.; Lam, M.H.; Gonzalez-Rimbau, M.; Rosen, J.M. The DNA-damage effector checkpoint kinase 1 is essential for chromosome segregation and cytokinesis. Proc. Natl. Acad. Sci. USA 2009, 106, 5159–5164. [Google Scholar] [CrossRef] [Green Version]
- Enders, G.H. Expanded roles for Chk1 in genome maintenance. J. Biol. Chem. 2008, 283, 17749–17752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petsalaki, E.; Zachos, G. Chk2 prevents mitotic exit when the majority of kinetochores are unattached. J. Cell Biol. 2014, 205, 339–356. [Google Scholar] [CrossRef] [PubMed]
- Lera, R.F.; Burkard, M.E. High mitotic activity of Polo-like kinase 1 is required for chromosome segregation and genomic integrity in human epithelial cells. J. Biol. Chem. 2012, 287, 42812–42825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charnley, M.; Anderegg, F.; Holtackers, R.; Textor, M.; Meraldi, P. Effect of Cell Shape and Dimensionality on Spindle Orientation and Mitotic Timing. PLoS ONE 2013, 8, e66918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hergovich, A.; Lisztwan, J.; Barry, R.; Ballschmieter, P.; Krek, W. Regulation of microtubule stability by the von Hippel-Lindau tumour suppressor protein pVHL. Nat. Cell Biol. 2003, 5, 64–70. [Google Scholar] [CrossRef]
- Lolkema, M.P.; Mans, D.A.; Snijckers, C.M.; van Noort, M.; van Beest, M.; Voest, E.E.; Giles, R.H. The von Hippel-Lindau tumour suppressor interacts with microtubules through kinesin-2. FEBS Lett. 2007, 581, 4571–4576. [Google Scholar] [CrossRef] [Green Version]
- Lolkema, M.P.; Mehra, N.; Jorna, A.S.; van Beest, M.; Giles, R.H.; Voest, E.E. The von Hippel-Lindau tumor suppressor protein influences microtubule dynamics at the cell periphery. Exp. Cell Res. 2004, 301, 139–146. [Google Scholar] [CrossRef]
- Hanafusa, H.; Kedashiro, S.; Tezuka, M.; Funatsu, M.; Usami, S.; Toyoshima, F.; Matsumoto, K. PLK1-dependent activation of LRRK1 regulates spindle orientation by phosphorylating CDK5RAP2. Nat. Cell Biol. 2015, 17, 1024–1035. [Google Scholar] [CrossRef]
- Miyamoto, T.; Akutsu, S.N.; Fukumitsu, A.; Morino, H.; Masatsuna, Y.; Hosoba, K.; Kawakami, H.; Yamamoto, T.; Shimizu, K.; Ohashi, H.; et al. PLK1-mediated phosphorylation of WDR62/MCPH2 ensures proper mitotic spindle orientation. Hum. Mol. Genet. 2017, 26, 4429–4440. [Google Scholar] [CrossRef] [Green Version]
- Kiyomitsu, T.; Cheeseman, I.M. Chromosome- and spindle-pole-derived signals generate an intrinsic code for spindle position and orientation. Nat. Cell Biol. 2012, 14, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Sana, S.; Keshri, R.; Rajeevan, A.; Kapoor, S.; Kotak, S. Plk1 regulates spindle orientation by phosphorylating NuMA in human cells. Life Sci. Alliance 2018, 1, e201800223. [Google Scholar] [CrossRef] [PubMed]
- Kotak, S. Mechanisms of Spindle Positioning: Lessons from Worms and Mammalian Cells. Biomolecules 2019, 9, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.Y.; Nazockdast, E.; Shelley, M.J.; Needleman, D.J. Forces positioning the mitotic spindle: Theories, and now experiments. Bioessays 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Ertych, N.; Stolz, A.; Stenzinger, A.; Weichert, W.; Kaulfuss, S.; Burfeind, P.; Aigner, A.; Wordeman, L.; Bastians, H. Increased microtubule assembly rates influence chromosomal instability in colorectal cancer cells. Nat. Cell Biol. 2014, 16, 779–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolz, A.; Ertych, N.; Bastians, H. A phenotypic screen identifies microtubule plus end assembly regulators that can function in mitotic spindle orientation. Cell Cycle 2015, 14, 827–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaubke, E.; Bastians, H. A Cell-Based Assay for Mitotic Spindle Orientation. Methods Mol. Biol. 2018, 1787, 67–75. [Google Scholar] [CrossRef]
- Ouadid-Ahidouch, H.; Ahidouch, A. K(+) channels and cell cycle progression in tumor cells. Front. Physiol. 2013, 4, 220. [Google Scholar] [CrossRef] [Green Version]
- Hepler, P.K. The Cytoskeleton and Its Regulation by Calcium and Protons. Plant Physiol. 2016, 170, 3–22. [Google Scholar] [CrossRef] [Green Version]
- Erneux, C.; Passareiro, H.; Nunez, J. Interaction between calmodulin and microtubule-associated proteins prepared at different stages of brain development. FEBS Lett. 1984, 172, 315–320. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.C.; Wolff, J. Two opposing effects of calmodulin on microtubule assembly depend on the presence of microtubule-associated proteins. J. Biol. Chem. 1982, 257, 6306–6310. [Google Scholar]
- Berkowitz, S.A.; Wolff, J. Intrinsic calcium sensitivity of tubulin polymerization. The contributions of temperature, tubulin concentration, and associated proteins. J. Biol. Chem. 1981, 256, 11216–11223. [Google Scholar] [PubMed]
- Peretti, M.; Badaoui, M.; Girault, A.; Van Gulick, L.; Mabille, M.P.; Tebbakha, R.; Sevestre, H.; Morjani, H.; Ouadid-Ahidouch, H. Original association of ion transporters mediates the ECM-induced breast cancer cell survival: Kv10.1-Orai1-SPCA2 partnership. Sci. Rep. 2019, 9, 1175. [Google Scholar] [CrossRef]
- Sadaghiani, A.M.; Lee, S.M.; Odegaard, J.I.; Leveson-Gower, D.B.; McPherson, O.M.; Novick, P.; Kim, M.R.; Koehler, A.N.; Negrin, R.; Dolmetsch, R.E.; et al. Identification of Orai1 channel inhibitors by using minimal functional domains to screen small molecule microarrays. Chem. Biol. 2014, 21, 1278–1292. [Google Scholar] [CrossRef] [PubMed]
- Samanta, K.; Parekh, A.B. Spatial Ca2+ profiling: Decrypting the universal cytosolic Ca2+ oscillation. J. Physiol. 2017, 595, 3053–3062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, W.A.; Taylor, S.S.; Shokat, K.M. Recognizing and exploiting differences between RNAi and small-molecule inhibitors. Nat. Chem. Biol. 2007, 3, 739–744. [Google Scholar] [CrossRef]
- Downie, B.R.; Sanchez, A.; Knotgen, H.; Contreras-Jurado, C.; Gymnopoulos, M.; Weber, C.; Stuhmer, W.; Pardo, L.A. Eag1 expression interferes with hypoxia homeostasis and induces angiogenesis in tumors. J. Biol. Chem. 2008, 283, 36234–36240. [Google Scholar] [CrossRef] [Green Version]
- Ramos Gomes, F.; Romaniello, V.; Sanchez, A.; Weber, C.; Narayanan, P.; Psol, M.; Pardo, L.A. Alternatively Spliced Isoforms of KV10.1 Potassium Channels Modulate Channel Properties and Can Activate Cyclin-dependent Kinase in Xenopus Oocytes. J. Biol. Chem. 2015, 290, 30351–30365. [Google Scholar] [CrossRef] [Green Version]
- Smyth, J.T.; Putney, J.W. Regulation of store-operated calcium entry during cell division. Biochem. Soc. Trans. 2012, 40, 119–123. [Google Scholar] [CrossRef]
- Smaardijk, S.; Chen, J.; Wuytack, F.; Vangheluwe, P. SPCA2 couples Ca2+ influx via Orai1 to Ca2+ uptake into the Golgi/secretory pathway. Tissue Cell 2017, 49, 141–149. [Google Scholar] [CrossRef]
- Cross, B.M.; Hack, A.; Reinhardt, T.A.; Rao, R. SPCA2 regulates Orai1 trafficking and store independent Ca2+ entry in a model of lactation. PLoS ONE 2013, 8, e67348. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Grice, D.M.; Faddy, H.M.; Nguyen, N.; Leitch, S.; Wang, Y.; Muend, S.; Kenny, P.A.; Sukumar, S.; Roberts-Thomson, S.J.; et al. Store-independent activation of Orai1 by SPCA2 in mammary tumors. Cell 2010, 143, 84–98. [Google Scholar] [CrossRef] [Green Version]
- Stansfeld, C.E.; Roper, J.; Ludwig, J.; Weseloh, R.M.; Marsh, S.J.; Brown, D.A.; Pongs, O. Elevation of intracellular calcium by muscarinic receptor activation induces a block of voltage-activated rat ether-a-go-go channels in a stably transfected cell line. Proc. Natl. Acad. Sci. USA 1996, 93, 9910–9914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonherr, R.; Lober, K.; Heinemann, S.H. Inhibition of human ether a go-go potassium channels by Ca(2+)/calmodulin. EMBO J. 2000, 19, 3263–3271. [Google Scholar] [CrossRef] [PubMed]
- Merriam, E.B.; Millette, M.; Lumbard, D.C.; Saengsawang, W.; Fothergill, T.; Hu, X.; Ferhat, L.; Dent, E.W. Synaptic regulation of microtubule dynamics in dendritic spines by calcium, F-actin, and drebrin. J. Neurosci. 2013, 33, 16471–16482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azqueta, A.; Collins, A.R. The Comet Assay: High Throughput Use of FPG. In Genotoxicity and DNA Repair; Humana Press: New York, NY, USA, 2014; pp. 199–217. [Google Scholar]
- Końca, K.; Lankoff, A.; Banasik, A.; Lisowska, H.; Kuszewski, T.; Góźdź, S.; Koza, Z.; Wojcik, A. A cross-platform public domain PC image-analysis program for the comet assay. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2003, 534, 15–20. [Google Scholar] [CrossRef]
- Collins, T.J. ImageJ for microscopy. Biotechniques 2007, 43, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Applegate, K.T.; Besson, S.; Matov, A.; Bagonis, M.H.; Jaqaman, K.; Danuser, G. plusTipTracker: Quantitative image analysis software for the measurement of microtubule dynamics. J. Struct. Biol. 2011, 176, 168–184. [Google Scholar] [CrossRef] [Green Version]
- Jaqaman, K.; Loerke, D.; Mettlen, M.; Kuwata, H.; Grinstein, S.; Schmid, S.L.; Danuser, G. Robust single-particle tracking in live-cell time-lapse sequences. Nat. Methods 2008, 5, 695–702. [Google Scholar] [CrossRef] [Green Version]
- Matov, A.; Applegate, K.; Kumar, P.; Thoma, C.; Krek, W.; Danuser, G.; Wittmann, T. Analysis of microtubule dynamic instability using a plus-end growth marker. Nat. Methods 2010, 7, 761–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gee, K.R.; Brown, K.A.; Chen, W.N.; Bishop-Stewart, J.; Gray, D.; Johnson, I. Chemical and physiological characterization of fluo-4 Ca2+-indicator dyes. Cell Calcium 2000, 27, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Schatten, H.; Sun, Q.Y. Functions and dysfunctions of the mammalian centrosome in health, disorders, disease, and aging. Histochem. Cell Biol. 2018, 150, 303–325. [Google Scholar] [CrossRef]
- Dai, G.; Qian, Y.; Chen, J.; Meng, F.L.; Pan, F.Y.; Shen, W.G.; Zhang, S.Z.; Xue, B.; Li, C.J. Calmodulin activation of polo-like kinase 1 is required during mitotic entry. Biochem. Cell Biol. 2013, 91, 287–294. [Google Scholar] [CrossRef]
- Plotnikova, O.V.; Pugacheva, E.N.; Dunbrack, R.L.; Golemis, E.A. Rapid calcium-dependent activation of Aurora-A kinase. Nat. Commun. 2010, 1, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macůrek, L.; Lindqvist, A.; Lim, D.; Lampson, M.A.; Klompmaker, R.; Freire, R.; Clouin, C.; Taylor, S.S.; Yaffe, M.B.; Medema, R.H. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 2008, 455, 119–123. [Google Scholar] [CrossRef]
- Parrilla, A.; Cirillo, L.; Thomas, Y.; Gotta, M.; Pintard, L.; Santamaria, A. Mitotic entry: The interplay between Cdk1, Plk1 and Bora. Cell Cycle 2016, 15, 3177–3182. [Google Scholar] [CrossRef] [Green Version]
- Thomas, Y.; Cirillo, L.; Panbianco, C.; Martino, L.; Tavernier, N.; Schwager, F.; Van Hove, L.; Joly, N.; Santamaria, A.; Pintard, L.; et al. Cdk1 Phosphorylates SPAT-1/Bora to Promote Plk1 Activation in C. elegans and Human Cells. Cell Rep. 2016, 15, 510–518. [Google Scholar] [CrossRef] [Green Version]
- Van Horn, R.D.; Chu, S.; Fan, L.; Yin, T.; Du, J.; Beckmann, R.; Mader, M.; Zhu, G.; Toth, J.; Blanchard, K.; et al. Cdk1 activity is required for mitotic activation of aurora A during G2/M transition of human cells. J. Biol. Chem. 2010, 285, 21849–21857. [Google Scholar] [CrossRef] [Green Version]
- Vigneron, S.; Sundermann, L.; Labbe, J.C.; Pintard, L.; Radulescu, O.; Castro, A.; Lorca, T. Cyclin A-cdk1-Dependent Phosphorylation of Bora Is the Triggering Factor Promoting Mitotic Entry. Dev. Cell 2018, 45, 637–650.e7. [Google Scholar] [CrossRef]
- Colicino, E.G.; Hehnly, H. Regulating a key mitotic regulator, polo-like kinase 1 (PLK1). Cytoskeleton 2018, 75, 481–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmena, M.; Ruchaud, S.; Earnshaw, W.C. Making the Auroras glow: Regulation of Aurora A and B kinase function by interacting proteins. Curr. Opin. Cell Biol. 2009, 21, 796–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serpico, A.F.; Grieco, D. Recent advances in understanding the role of Cdk1 in the Spindle Assembly Checkpoint. F1000Research 2020, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Checkpoint | Effect |
|---|---|---|
| p-ATR (Ser428) | DDR | Increased |
| p-CHK1 (Ser345) | DDR | Increased |
| CHK2 | DDR | Increased |
| pVHL | SAC | Increased |
| AURKB | SAC | Decreased |
| PLK1 | SAC | Decreased |
| MAD2 | SAC | Increased |
| CDC20 | SAC (APC/C) | Increased |
| WEE1 | SAC (APC/C) | Increased |
| CDC27 | APC/C | Decreased |
| Antibody | Source | Dilution | Cat. Number | Manufacturer |
|---|---|---|---|---|
| p-ATR (Ser428) | Rb/pcl | 1:1000 | 2853 | Cell Signaling |
| p-BRCA1 (Ser1524) | Rb/pcl | 1:1000 | 9009 | Cell Signaling |
| p-CHK1 (Ser345) (133D3) | Rb/mcl | 1:1000 | 2348 | Cell Signaling |
| CHK1 (FL-476) | Rb/pcl | 1:1000 | sc-7898 | Santa Cruz Biotechnology |
| p-CHK2 (Thr68)(C13C1) | Rb/mcl | 1:1000 | 2197 | Cell Signaling |
| CHK2 (DCS-270) | Ms/mcl | 1:1000 | sc-56296 | Santa Cruz Biotechnology |
| p21 Waf1/Cip1 | Ms/mcl | 1:1000 | 05-345 | Merck Millipore |
| p-histone H2AX (Ser139) | Rb/mcl | 1:1000 | sc-101696 | Cell Signaling |
| CDC27 (AF3.1) | Ms/mcl | 1:1000 | sc-9972 | Santa Cruz Biotechnology |
| CDC20 | Rb/pcl | 1:1000 | ab26483 | Abcam |
| AURKB/AIM1 | Rb/pcl | 1:1000 | 3094 | Cell Signaling |
| PLK1 | Ms/mcl | 1:500 | sc-17783 | Santa Cruz Biotechnology |
| WEE1 | MS/mcl | 1:500 | sc-5285 | Santa Cruz Biotechnology |
| BUB3 | Ms/mcl | 1:1000 | sc-376506 | Santa Cruz Biotechnology |
| MAD2L1 (D8A7) | Rb/mcl | 1:1000 | 4636 | Cell Signaling |
| pVHL | Ms/mcl | 1:200 | 556347 | BD Pharmingen |
| Actin (C-11) | G/pcl | 1:1000 | sc-1615 | Santa Cruz Biotechnology |
| Kv10.1 | Rb/pcl | 1:1500 | 9391 | |
| Normal Mouse IgG | Ms | Isotype control | sc-2025 | Santa Cruz Biotechnology |
| Normal Rabbit IgG | Rb | Isotype control | sc-3888 | Santa Cruz Biotechnology |
| ECL Anti-Rabbit IgG, HRP Linked | D/pcl | 1:10,000 | NA934 | GE Healthcare |
| ECL Anti-Mouse IgG, HRP Linked | Sh/pcl | 1:10,000 | NA931 | GE Healthcare |
| Anti-Goat IgG (H + L)-HRP Conjugate | Rb/pcl | 1:10,000 | 172-1034 | BIO-RAD |
| Antibody | Source | Dilution | Cat. Number | Manufacturer |
|---|---|---|---|---|
| α-Tubulin | G/pcl | 1:350 | sc-7396 | Santa Cruz Biotechnology |
| γ-Tubulin | Ms/mcl | 1:700 | sc-23948 | Santa Cruz Biotechnology |
| CENP-B (H-65) | Rb/pcl | 1:350 | sc-22788 | Santa Cruz Biotechnology |
| Alexa Fluor 488 anti-mouse IgG (H + L) | D/pcl | 1:1000 | A-21202 | Invitrogen |
| Alexa Fluor 546 anti-rabbit IgG (H + L) | D/pcl | 1:1000 | A-11056 | Invitrogen |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Movsisyan, N.; Pardo, L.A. Kv10.1 Regulates Microtubule Dynamics during Mitosis. Cancers 2020, 12, 2409. https://doi.org/10.3390/cancers12092409
Movsisyan N, Pardo LA. Kv10.1 Regulates Microtubule Dynamics during Mitosis. Cancers. 2020; 12(9):2409. https://doi.org/10.3390/cancers12092409
Chicago/Turabian StyleMovsisyan, Naira, and Luis A. Pardo. 2020. "Kv10.1 Regulates Microtubule Dynamics during Mitosis" Cancers 12, no. 9: 2409. https://doi.org/10.3390/cancers12092409
APA StyleMovsisyan, N., & Pardo, L. A. (2020). Kv10.1 Regulates Microtubule Dynamics during Mitosis. Cancers, 12(9), 2409. https://doi.org/10.3390/cancers12092409