Deciphering the Role of Innate Immune NF-ĸB Pathway in Pancreatic Cancer


Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
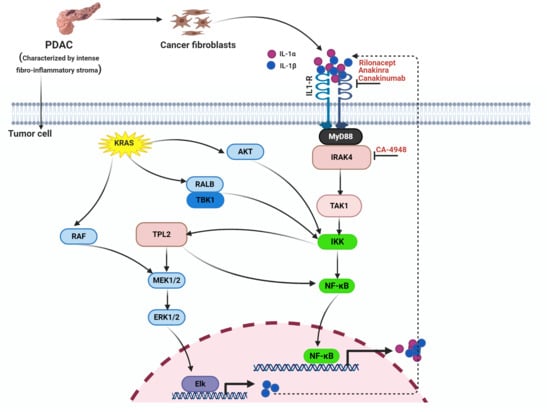
1. Introduction
2. Chronic Inflammation Drives Desmoplasia and Neoplastic Progression in PDAC
3. NF-κB Pathway: A Major Driver of Inflammation in PDAC
3.1. The Role of NF-κB in PDAC Cells
3.2. The Role of NF-κB in CAFs
3.3. The Role of NF-κB in Immune Cells
4. Mechanisms that Activate the NF-κB Pathway in PDAC
4.1. Toll-Like Receptors
4.1.1. TLR4
4.1.2. TLR7/8
4.1.3. TLR9
4.2. IL-1α/β and IL-1R
4.3. TNF-α and TNFR
4.4. IRAK4
4.5. TAK1
4.6. TPL2
5. Intricate Crosstalk between the KRAS and NF-κB Pathways
6. Therapeutic Targeting of the NF-ĸB Pathway in PDAC
6.1. IL-1R Blockade
6.2. IRAK4
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivaldi, C.; Fornaro, L.; Vasile, E. FOLFIRINOX Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2019, 380, 1187–1188. [Google Scholar] [CrossRef] [PubMed]
- Neoptolemos, J.P.; Palmer, D.H.; Ghaneh, P.; Psarelli, E.E.; Valle, J.W.; Halloran, C.M.; Faluyi, O.; O’Reilly, D.A.; Cunningham, D.; Wadsley, J.; et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): A multicentre, open-label, randomised, phase 3 trial. Lancet 2017, 389, 1011–1024. [Google Scholar] [CrossRef]
- Oettle, H.; Neuhaus, P.; Hochhaus, A.; Hartmann, J.T.; Gellert, K.; Ridwelski, K.; Niedergethmann, M.; Zulke, C.; Fahlke, J.; Arning, M.B.; et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: The CONKO-001 randomized trial. JAMA 2013, 310, 1473–1481. [Google Scholar] [CrossRef] [Green Version]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Makohon-Moore, A.; Iacobuzio-Donahue, C.A. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat. Rev. Cancer 2016, 16, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Neesse, A.; Algul, H.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: A changing paradigm. Gut 2015, 64, 1476–1484. [Google Scholar] [CrossRef] [Green Version]
- Goedegebuure, P.; Mitchem, J.B.; Porembka, M.R.; Tan, M.C.; Belt, B.A.; Wang-Gillam, A.; Gillanders, W.E.; Hawkins, W.G.; Linehan, D.C. Myeloid-derived suppressor cells: General characteristics and relevance to clinical management of pancreatic cancer. Curr. Cancer Drug Targets 2011, 11, 734–751. [Google Scholar] [CrossRef] [Green Version]
- Porembka, M.R.; Mitchem, J.B.; Belt, B.A.; Hsieh, C.S.; Lee, H.M.; Herndon, J.; Gillanders, W.E.; Linehan, D.C.; Goedegebuure, P. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol. Immunother. 2012, 61, 1373–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [Green Version]
- Ramanathan, R.K.; McDonough, S.L.; Philip, P.A.; Hingorani, S.R.; Lacy, J.; Kortmansky, J.S.; Thumar, J.; Chiorean, E.G.; Shields, A.F.; Behl, D.; et al. Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase Versus FOLFIRINOX Alone in Patients With Metastatic Pancreatic Adenocarcinoma: SWOG S1313. J. Clin. Oncol. 2019, 37, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Tempero, M.A.; Sigal, D.; Oh, D.Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa With Nab-Paclitaxel Plus Gemcitabine for Patients With Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2020, 38, 3185–3194. [Google Scholar] [CrossRef]
- De Jesus-Acosta, A.; Sugar, E.A.; O’Dwyer, P.J.; Ramanathan, R.K.; Von Hoff, D.D.; Rasheed, Z.; Zheng, L.; Begum, A.; Anders, R.; Maitra, A.; et al. Phase 2 study of vismodegib, a hedgehog inhibitor, combined with gemcitabine and nab-paclitaxel in patients with untreated metastatic pancreatic adenocarcinoma. Br. J. Cancer 2020, 122, 498–505. [Google Scholar] [CrossRef]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Lowenfels, A.B.; Maisonneuve, P.; Cavallini, G.; Ammann, R.W.; Lankisch, P.G.; Andersen, J.R.; Dimagno, E.P.; Andren-Sandberg, A.; Domellof, L. Pancreatitis and the Risk of Pancreatic Cancer. N. Engl. J. Med. 1993, 328, 1433–1437. [Google Scholar] [CrossRef]
- Lee, K.E.; Bar-Sagi, D. Oncogenic KRas suppresses inflammation-associated senescence of pancreatic ductal cells. Cancer Cell 2010, 18, 448–458. [Google Scholar] [CrossRef] [Green Version]
- Guerra, C.; Barbacid, M. Genetically engineered mouse models of pancreatic adenocarcinoma. Mol. Oncol. 2013, 7, 232–247. [Google Scholar] [CrossRef]
- Herreros-Villanueva, M.; Hijona, E.; Cosme, A.; Bujanda, L. Mouse models of pancreatic cancer. World J. Gastroenterol. 2012, 18, 1286–1294. [Google Scholar] [CrossRef] [PubMed]
- Guerra, C.; Schuhmacher, A.J.; Cañamero, M.; Grippo, P.J.; Verdaguer, L.; Pérez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic Pancreatitis Is Essential for Induction of Pancreatic Ductal Adenocarcinoma by K-Ras Oncogenes in Adult Mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carriere, C.; Young, A.L.; Gunn, J.R.; Longnecker, D.S.; Korc, M. Acute pancreatitis accelerates initiation and progression to pancreatic cancer in mice expressing oncogenic Kras in the nestin cell lineage. PLoS ONE 2011, 6, e27725. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Torres, M.P.; Kaur, S.; Rachagani, S.; Joshi, S.; Johansson, S.L.; Momi, N.; Baine, M.J.; Gilling, C.E.; Smith, L.M.; et al. Smoking accelerates pancreatic cancer progression by promoting differentiation of MDSCs and inducing HB-EGF expression in macrophages. Oncogene 2015, 34, 2052–2060. [Google Scholar] [CrossRef] [Green Version]
- Incio, J.; Liu, H.; Suboj, P.; Chin, S.M.; Chen, I.X.; Pinter, M.; Ng, M.R.; Nia, H.T.; Grahovac, J.; Kao, S.; et al. Obesity-Induced Inflammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discov. 2016, 6, 852–869. [Google Scholar] [CrossRef] [Green Version]
- Malka, D.; Hammel, P.; Maire, F.; Rufat, P.; Madeira, I.; Pessione, F.; Lévy, P.; Ruszniewski, P. Risk of pancreatic adenocarcinoma in chronic pancreatitis. Gut 2002, 51, 849–852. [Google Scholar] [CrossRef]
- Yadav, D.; Lowenfels, A.B. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 2013, 144, 1252–1261. [Google Scholar] [CrossRef] [Green Version]
- Bartsch, D.K.; Gress, T.M.; Langer, P. Familial pancreatic cancer--current knowledge. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 445–453. [Google Scholar] [CrossRef]
- Tan, C.R.; Yaffee, P.M.; Jamil, L.H.; Lo, S.K.; Nissen, N.; Pandol, S.J.; Tuli, R.; Hendifar, A.E. Pancreatic cancer cachexia: A review of mechanisms and therapeutics. Front. Physiol. 2014, 5, 88. [Google Scholar] [CrossRef] [Green Version]
- Mulero, M.C.; Huxford, T.; Ghosh, G. NF-kappaB, IkappaB, and IKK: Integral Components of Immune System Signaling. Adv. Exp. Med. Biol. 2019, 1172, 207–226. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-kappa B and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-kappaB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Wang, W.; Abbruzzese, J.L.; Evans, D.B.; Larry, L.; Cleary, K.R.; Chiao, P.J. The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin. Cancer Res. 1999, 5, 119–127. [Google Scholar]
- Zhang, D.; Li, L.; Jiang, H.; Knolhoff, B.L.; Lockhart, A.C.; Wang-Gillam, A.; DeNardo, D.G.; Ruzinova, M.B.; Lim, K.H. Constitutive IRAK4 Activation Underlies Poor Prognosis and Chemoresistance in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2017, 23, 1748–1759. [Google Scholar] [CrossRef] [Green Version]
- Karin, M.; Greten, F.R. NF-kappaB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-kappaB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Zhang, X.; Ren, D.; Wu, X.; Lin, X.; Ye, L.; Lin, C.; Wu, S.; Zhu, J.; Peng, X.; Song, L. miR-1266 Contributes to Pancreatic Cancer Progression and Chemoresistance by the STAT3 and NF-κB Signaling Pathways. Mol. Ther. Nucleic Acids 2018, 11, 142–158. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Muñoz, A.R.; Pingali, S.; Payton-Stewart, F.; Chan, D.E.; Freeman, J.W.; Ghosh, R.; Kumar, A.P. Downregulation of STAT3/NF-κB potentiates gemcitabine activity in pancreatic cancer cells. Mol. Carcinog. 2017, 56, 402–411. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Arumugam, T.; Yamamoto, T.; Levin, P.A.; Ramachandran, V.; Ji, B.; Lopez-Berestein, G.; Vivas-Mejia, P.E.; Sood, A.K.; McConkey, D.J.; et al. Nuclear factor-κB p65/relA silencing induces apoptosis and increases gemcitabine effectiveness in a subset of pancreatic cancer cells. Clin. Cancer Res. 2008, 14, 8143–8151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; Chen, S.; Guo, Y.; Sun, C. Oncogenic TRIM31 confers gemcitabine resistance in pancreatic cancer via activating the NF-κB signaling pathway. Theranostics 2018, 8, 3224–3236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Duan, Q.; Zhao, H.; Liu, T.; Wu, H.; Shen, Q.; Wang, C.; Yin, T. Gemcitabine treatment promotes pancreatic cancer stemness through the Nox/ROS/NF-kappaB/STAT3 signaling cascade. Cancer Lett. 2016, 382, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Godwin, P.; Baird, A.M.; Heavey, S.; Barr, M.P.; O’Byrne, K.J.; Gately, K. Targeting nuclear factor-kappa B to overcome resistance to chemotherapy. Front. Oncol. 2013, 3, 120. [Google Scholar] [CrossRef] [Green Version]
- Norton, J.; Foster, D.; Chinta, M.; Titan, A.; Longaker, M. Pancreatic Cancer Associated Fibroblasts (CAF): Under-Explored Target for Pancreatic Cancer Treatment. Cancers 2020, 12, 1347. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Zhang, D.; Li, L.; Jiang, H.; Li, Q.; Wang-Gillam, A.; Yu, J.; Head, R.; Liu, J.; Ruzinova, M.B.; Lim, K.H. Tumor-Stroma IL1beta-IRAK4 Feedforward Circuitry Drives Tumor Fibrosis, Chemoresistance, and Poor Prognosis in Pancreatic Cancer. Cancer Res. 2018, 78, 1700–1712. [Google Scholar] [CrossRef] [Green Version]
- Garg, B.; Giri, B.; Modi, S.; Sethi, V.; Castro, I.; Umland, O.; Ban, Y.; Lavania, S.; Dawra, R.; Banerjee, S.; et al. NFkappaB in Pancreatic Stellate Cells Reduces Infiltration of Tumors by Cytotoxic T Cells and Killing of Cancer Cells, via Up-regulation of CXCL12. Gastroenterology 2018, 155, 880–891. [Google Scholar] [CrossRef]
- Thyagarajan, A.; Alshehri, M.S.A.; Miller, K.L.R.; Sherwin, C.M.; Travers, J.B.; Sahu, R.P. Myeloid-Derived Suppressor Cells and Pancreatic Cancer: Implications in Novel Therapeutic Approaches. Cancers 2019, 11, 1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Bosch, N.; Vinaixa, J.; Navarro, P. Immune Evasion in Pancreatic Cancer: From Mechanisms to Therapy. Cancers 2018, 10, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, S.J.; Baldwin, A.S. Deletion of the NF-kappaB subunit p65/RelA in the hematopoietic compartment leads to defects in hematopoietic stem cell function. Blood 2013, 121, 5015–5024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Xiu, Y.; Ashton, J.; Xing, L.; Morita, Y.; Jordan, C.T.; Boyce, B.F. Noncanonical NF-kappaB signaling regulates hematopoietic stem cell self-renewal and microenvironment interactions. Stem Cells 2012, 30, 709–718. [Google Scholar] [CrossRef] [Green Version]
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.C.; Goode, J.; Miething, C.; Goktuna, S.I.; Neuenhahn, M.; Fierer, J.; Paxian, S.; et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 2007, 130, 918–931. [Google Scholar] [CrossRef] [Green Version]
- Senftleben, U.; Li, Z.W.; Baud, V.; Karin, M. IKKbeta is essential for protecting T cells from TNFalpha-induced apoptosis. Immunity 2001, 14, 217–230. [Google Scholar] [CrossRef] [Green Version]
- Jimi, E.; Strickland, I.; Voll, R.E.; Long, M.; Ghosh, S. Differential role of the transcription factor NF-kappaB in selection and survival of CD4+ and CD8+ thymocytes. Immunity 2008, 29, 523–537. [Google Scholar] [CrossRef] [Green Version]
- Messina, N.; Fulford, T.; O’Reilly, L.; Loh, W.X.; Motyer, J.M.; Ellis, D.; McLean, C.; Naeem, H.; Lin, A.; Gugasyan, R.; et al. The NF-kappaB transcription factor RelA is required for the tolerogenic function of Foxp3(+) regulatory T cells. J. Autoimmun. 2016, 70, 52–62. [Google Scholar] [CrossRef]
- Mancino, A.; Lawrence, T. Nuclear factor-kappaB and tumor-associated macrophages. Clin. Cancer Res. 2010, 16, 784–789. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.W.; Omori, S.A.; Labuda, T.; Karin, M.; Rickert, R.C. IKK beta is required for peripheral B cell survival and proliferation. J. Immunol. 2003, 170, 4630–4637. [Google Scholar] [CrossRef] [Green Version]
- Ren, H.; Schmalstieg, A.; Yuan, D.; Gaynor, R.B. I-kappa B kinase beta is critical for B cell proliferation and antibody response. J. Immunol. 2002, 168, 577–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasparakis, M.; Schmidt-Supprian, M.; Rajewsky, K. IkappaB kinase signaling is essential for maintenance of mature B cells. J. Exp. Med. 2002, 196, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Agnellini, P.; Wolint, P.; Rehr, M.; Cahenzli, J.; Karrer, U.; Oxenius, A. Impaired NFAT nuclear translocation results in split exhaustion of virus-specific CD8+ T cell functions during chronic viral infection. Proc. Natl. Acad. Sci. USA 2007, 104, 4565–4570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, G.J.; Pereira, R.M.; Aijo, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity 2015, 42, 265–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, K.; Gabriel, S.S.; Liao, Y.; Gloury, R.; Preston, S.; Henstridge, D.C.; Pellegrini, M.; Zehn, D.; Berberich-Siebelt, F.; Febbraio, M.A.; et al. Transcription Factor IRF4 Promotes CD8(+) T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity 2017, 47, 1129–1141. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Villar, M.; Gautron, A.S.; de Marcken, M.; Keller, M.J.; Hafler, D.A. TLR7 induces anergy in human CD4(+) T cells. Nat. Immunol. 2015, 16, 118–128. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef]
- Lim, K.H.; Staudt, L.M. Toll-like receptor signaling. Cold Spring Harb. Perspect. Biol. 2013, 5, a011247. [Google Scholar] [CrossRef] [Green Version]
- Blonska, M.; Shambharkar, P.B.; Kobayashi, M.; Zhang, D.; Sakurai, H.; Su, B.; Lin, X. TAK1 is recruited to the tumor necrosis factor-alpha (TNF-alpha) receptor 1 complex in a receptor-interacting protein (RIP)-dependent manner and cooperates with MEKK3 leading to NF-kappaB activation. J. Biol. Chem. 2005, 280, 43056–43063. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Kobayashi, M.; Blonska, M.; You, Y.; Lin, X. Ubiquitination of RIP is required for tumor necrosis factor alpha-induced NF-kappaB activation. J. Biol. Chem. 2006, 281, 13636–13643. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.J. Glycolytic reprogramming by TLRs in dendritic cells. Nat. Immunol. 2014, 15, 314–315. [Google Scholar] [CrossRef] [PubMed]
- Santoni, M.; Andrikou, K.; Sotte, V.; Bittoni, A.; Lanese, A.; Pellei, C.; Piva, F.; Conti, A.; Nabissi, M.; Santoni, G.; et al. Toll like receptors and pancreatic diseases: From a pathogenetic mechanism to a therapeutic target. Cancer Treat. Rev. 2015, 41, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Ochi, A.; Nguyen, A.H.; Bedrosian, A.S.; Mushlin, H.M.; Zarbakhsh, S.; Barilla, R.; Zambirinis, C.P.; Fallon, N.C.; Rehman, A.; Pylayeva-Gupta, Y.; et al. MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. J. Exp. Med. 2012, 209, 1671–1687. [Google Scholar] [CrossRef] [PubMed]
- Ikebe, M.; Kitaura, Y.; Nakamura, M.; Tanaka, H.; Yamasaki, A.; Nagai, S.; Wada, J.; Yanai, K.; Koga, K.; Sato, N.; et al. Lipopolysaccharide (LPS) increases the invasive ability of pancreatic cancer cells through the TLR4/MyD88 signaling pathway. J. Surg. Oncol. 2009, 100, 725–731. [Google Scholar] [CrossRef]
- Ochi, A.; Graffeo, C.S.; Zambirinis, C.P.; Rehman, A.; Hackman, M.; Fallon, N.; Barilla, R.M.; Henning, J.R.; Jamal, M.; Rao, R.; et al. Toll-like receptor 7 regulates pancreatic carcinogenesis in mice and humans. J. Clin. Investig. 2012, 122, 4118–4129. [Google Scholar] [CrossRef]
- Grimmig, T.; Matthes, N.; Hoeland, K.; Tripathi, S.; Chandraker, A.; Grimm, M.; Moench, R.; Moll, E.M.; Friess, H.; Tsaur, I.; et al. TLR7 and TLR8 expression increases tumor cell proliferation and promotes chemoresistance in human pancreatic cancer. Int. J. Oncol. 2015, 47, 857–866. [Google Scholar] [CrossRef] [Green Version]
- Schölch, S.; Rauber, C.; Tietz, A.; Rahbari, N.N.; Bork, U.; Schmidt, T.; Kahlert, C.; Haberkorn, U.; Tomai, M.A.; Lipson, K.E.; et al. Radiotherapy combined with TLR7/8 activation induces strong immune responses against gastrointestinal tumors. Oncotarget 2015, 6, 4663–4676. [Google Scholar] [CrossRef] [Green Version]
- Shankara Narayanan, J.S.; Ray, P.; Hayashi, T.; Whisenant, T.C.; Vicente, D.; Carson, D.A.; Miller, A.M.; Schoenberger, S.P.; White, R.R. Irreversible electroporation combined with checkpoint blockade and TLR7 stimulation induces antitumor immunity in a murine pancreatic cancer model. Cancer Immunol. Res. 2019, 7, 1714–1726. [Google Scholar] [CrossRef]
- Michaelis, K.A.; Norgard, M.A.; Zhu, X.; Levasseur, P.R.; Sivagnanam, S.; Liudahl, S.M.; Burfeind, K.G.; Olson, B.; Pelz, K.R.; Angeles Ramos, D.M.; et al. The TLR7/8 agonist R848 remodels tumor and host responses to promote survival in pancreatic cancer. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Zambirinis, C.P.; Levie, E.; Nguy, S.; Avanzi, A.; Barilla, R.; Xu, Y.; Seifert, L.; Daley, D.; Greco, S.H.; Deutsch, M.; et al. TLR9 ligation in pancreatic stellate cells promotes tumorigenesis. J. Exp. Med. 2015, 212, 2077–2094. [Google Scholar] [CrossRef]
- Dodhiawala, P.B.; Khurana, N.; Zhang, D.; Cheng, Y.; Li, L.; Wei, Q.; Seehra, K.; Jiang, H.; Grierson, P.M.; Wang-Gillam, A.; et al. TPL2 enforces RAS-induced inflammatory signaling and is activated by point mutations. J. Clin. Investig. 2020, 130, 4771–4790. [Google Scholar] [CrossRef] [PubMed]
- Grimmig, T.; Moench, R.; Kreckel, J.; Haack, S.; Rueckert, F.; Rehder, R.; Tripathi, S.; Ribas, C.; Chandraker, A.; Germer, C.T.; et al. Toll Like Receptor 2, 4, and 9 Signaling Promotes Autoregulative Tumor Cell Growth and VEGF/PDGF Expression in Human Pancreatic Cancer. Int. J. Mol. Sci. 2016, 17, 2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, R.; Melisi, D.; Damiano, V.; Bianco, R.; Garofalo, S.; Gelardi, T.; Agrawal, S.; Di Nicolantonio, F.; Scarpa, A.; Bardelli, A.; et al. Toll-like receptor 9 agonist IMO cooperates with cetuximab in K-ras mutant colorectal and pancreatic cancers. Clin. Cancer Res. 2011, 17, 6531–6541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krieg, A.M. Therapeutic potential of Toll-like receptor 9 activation. Nat. Rev. Drug Discov. 2006, 5, 471–484. [Google Scholar] [CrossRef]
- Pratesi, G.; Petrangolini, G.; Tortoreto, M.; Addis, A.; Belluco, S.; Rossini, A.; Selleri, S.; Rumio, C.; Menard, S.; Balsari, A. Therapeutic synergism of gemcitabine and CpG-oligodeoxynucleotides in an orthotopic human pancreatic carcinoma xenograft. Cancer Res. 2005, 65, 6388–6393. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, C.; Duewell, P.; Heckelsmiller, K.; Wei, J.; Bauernfeind, F.; Ellermeier, J.; Kisser, U.; Bauer, C.A.; Dauer, M.; Eigler, A.; et al. An ISCOM vaccine combined with a TLR9 agonist breaks immune evasion mediated by regulatory T cells in an orthotopic model of pancreatic carcinoma. Int. J. Cancer 2011, 128, 897–907. [Google Scholar] [CrossRef]
- Ling, J.; Kang, Y.; Zhao, R.; Xia, Q.; Lee, D.F.; Chang, Z.; Li, J.; Peng, B.; Fleming, J.B.; Wang, H.; et al. KrasG12D-induced IKK2/β/NF-κB activation by IL-1α and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 105–120. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Shapiro, B.; Vucic, E.A.; Vogt, S.; Bar-Sagi, D. Tumor Cell-Derived IL1beta Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer. Cancer Res. 2020, 80, 1088–1101. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.Y.; Doppler, H.; Necela, B.; Krishna, M.; Crawford, H.C.; Raimondo, M.; Storz, P. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-kappaB and MMPs. J. Cell Biol. 2013, 202, 563–577. [Google Scholar] [CrossRef]
- Egberts, J.H.; Cloosters, V.; Noack, A.; Schniewind, B.; Thon, L.; Klose, S.; Kettler, B.; von Forstner, C.; Kneitz, C.; Tepel, J.; et al. Anti-tumor necrosis factor therapy inhibits pancreatic tumor growth and metastasis. Cancer Res. 2008, 68, 1443–1450. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Fan, W.; Xu, Z.; Chen, H.; He, Y.; Yang, G.; Yang, G.; Hu, H.; Tang, S.; Wang, P.; et al. Inhibiting tumor necrosis factor-alpha diminishes desmoplasia and inflammation to overcome chemoresistance in pancreatic ductal adenocarcinoma. Oncotarget 2016, 7, 81110–81122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Fernandez, S.A.; Criswell, T.; Chidiac, T.A.; Guttridge, D.; Villalona-Calero, M.; Bekaii-Saab, T.S. Disrupting cytokine signaling in pancreatic cancer: A phase I/II study of etanercept in combination with gemcitabine in patients with advanced disease. Pancreas 2013, 42, 813–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowie, A.G. Insights from vaccinia virus into Toll-like receptor signalling proteins and their regulation by ubiquitin: Role of IRAK-2. Biochem Soc Trans 2008, 36, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Suzuki, S.; Yeh, W.C. IRAK-4 as the central TIR signaling mediator in innate immunity. Trends Immunol. 2002, 23, 503–506. [Google Scholar] [CrossRef]
- Mukhopadhyay, H.; Lee, N.Y. Multifaceted roles of TAK1 signaling in cancer. Oncogene 2020, 39, 1402–1413. [Google Scholar] [CrossRef] [PubMed]
- Pandol, S.; Edderkaoui, M.; Gukovsky, I.; Lugea, A.; Gukovskaya, A. Desmoplasia of Pancreatic Ductal Adenocarcinoma. Clin. Gastroenterol. Hepatol. 2009, 7, S44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, N.; Suzuki, S.; Duncan, G.S.; Millar, D.G.; Wada, T.; Mirtsos, C.; Takada, H.; Wakeham, A.; Itie, A.; Li, S.; et al. Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature 2002, 416, 750–756. [Google Scholar] [CrossRef] [PubMed]
- von Bernuth, H.; Picard, C.; Puel, A.; Casanova, J.L. Experimental and natural infections in MyD88- and IRAK-4-deficient mice and humans. Eur. J. Immunol. 2012, 42, 3126–3135. [Google Scholar] [CrossRef]
- Picard, C.; von Bernuth, H.; Ghandil, P.; Chrabieh, M.; Levy, O.; Arkwright, P.D.; McDonald, D.; Geha, R.S.; Takada, H.; Krause, J.C.; et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine 2010, 89, 403–425. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, N.; Suzuki, S.; Millar, D.G.; Unno, M.; Hara, H.; Calzascia, T.; Yamasaki, S.; Yokosuka, T.; Chen, N.J.; Elford, A.R.; et al. A critical role for the innate immune signaling molecule IRAK-4 in T cell activation. Science 2006, 311, 1927–1932. [Google Scholar] [CrossRef]
- Kawagoe, T.; Sato, S.; Jung, A.; Yamamoto, M.; Matsui, K.; Kato, H.; Uematsu, S.; Takeuchi, O.; Akira, S. Essential role of IRAK-4 protein and its kinase activity in Toll-like receptor-mediated immune responses but not in TCR signaling. J. Exp. Med. 2007, 204, 1013–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferris, R.L.; Lu, B.; Kane, L.P. Too much of a good thing? Tim-3 and TCR signaling in T cell exhaustion. J. Immunol. 2014, 193, 1525–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, R.; Carbone, C.; Piro, G.; Chiao, P.J.; Melisi, D. TAK-ing aim at chemoresistance: The emerging role of MAP3K7 as a target for cancer therapy. Drug Resist. Updates 2017, 33–35, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H. Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharmacol. Sci. 2012, 33, 522–530. [Google Scholar] [CrossRef]
- Ajibade, A.A.; Wang, H.Y.; Wang, R.F. Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol. 2013, 34, 307–316. [Google Scholar] [CrossRef]
- Fan, Y.; Yu, Y.; Mao, R.; Zhang, H.; Yang, J. TAK1 Lys-158 but not Lys-209 is required for IL-1beta-induced Lys63-linked TAK1 polyubiquitination and IKK/NF-kappaB activation. Cell Signal. 2011, 23, 660–665. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Yu, Y.; Shi, Y.; Sun, W.; Xie, M.; Ge, N.; Mao, R.; Chang, A.; Xu, G.; Schneider, M.D.; et al. Lysine 63-linked polyubiquitination of TAK1 at lysine 158 is required for tumor necrosis factor alpha- and interleukin-1beta-induced IKK/NF-kappaB and JNK/AP-1 activation. J. Biol. Chem. 2010, 285, 5347–5360. [Google Scholar] [CrossRef] [Green Version]
- Singhirunnusorn, P.; Suzuki, S.; Kawasaki, N.; Saiki, I.; Sakurai, H. Critical roles of threonine 187 phosphorylation in cellular stress-induced rapid and transient activation of transforming growth factor-beta-activated kinase 1 (TAK1) in a signaling complex containing TAK1-binding protein TAB1 and TAB2. J. Biol. Chem. 2005, 280, 7359–7368. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Ge, N.; Xie, M.; Sun, W.; Burlingame, S.; Pass, A.K.; Nuchtern, J.G.; Zhang, D.; Fu, S.; Schneider, M.D.; et al. Phosphorylation of Thr-178 and Thr-184 in the TAK1 T-loop is required for interleukin (IL)-1-mediated optimal NFkappaB and AP-1 activation as well as IL-6 gene expression. J. Biol. Chem. 2008, 283, 24497–24505. [Google Scholar] [CrossRef] [Green Version]
- Scholz, R.; Sidler, C.L.; Thali, R.F.; Winssinger, N.; Cheung, P.C.; Neumann, D. Autoactivation of transforming growth factor beta-activated kinase 1 is a sequential bimolecular process. J. Biol. Chem. 2010, 285, 25753–25766. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.H.; Yu, Y.; Mao, R.F.; Tan, X.J.; Xu, G.F.; Zhang, H.; Lu, X.B.; Fu, S.B.; Yang, J. USP4 targets TAK1 to downregulate TNFalpha-induced NF-kappaB activation. Cell Death Differ. 2011, 18, 1547–1560. [Google Scholar] [CrossRef] [PubMed]
- Reiley, W.W.; Jin, W.; Lee, A.J.; Wright, A.; Wu, X.; Tewalt, E.F.; Leonard, T.O.; Norbury, C.C.; Fitzpatrick, L.; Zhang, M.; et al. Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J. Exp. Med. 2007, 204, 1475–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N.; Zeng, M.; Sinha, I.; Polin, L.; Wei, W.Z.; Rathinam, C.; Flavell, R.; Massoumi, R.; Venuprasad, K. The E3 ligase Itch and deubiquitinase Cyld act together to regulate Tak1 and inflammation. Nat. Immunol. 2011, 12, 1176–1183. [Google Scholar] [CrossRef] [Green Version]
- Kajino, T.; Ren, H.; Iemura, S.; Natsume, T.; Stefansson, B.; Brautigan, D.L.; Matsumoto, K.; Ninomiya-Tsuji, J. Protein phosphatase 6 down-regulates TAK1 kinase activation in the IL-1 signaling pathway. J. Biol. Chem. 2006, 281, 39891–39896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.G.; Katsura, K.; Nomiyama, H.; Komaki, K.; Ninomiya-Tsuji, J.; Matsumoto, K.; Kobayashi, T.; Tamura, S. Regulation of the interleukin-1-induced signaling pathways by a novel member of the protein phosphatase 2C family (PP2Cepsilon). J. Biol. Chem. 2003, 278, 12013–12021. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Li, Q.; Chen, R.; Zhang, J.; Ran, Y.; He, X.; Li, S.; Shu, H.B. The dual-specificity phosphatase DUSP14 negatively regulates tumor necrosis factor- and interleukin-1-induced nuclear factor-kappaB activation by dephosphorylating the protein kinase TAK1. J. Biol. Chem. 2013, 288, 819–825. [Google Scholar] [CrossRef] [Green Version]
- Jadrich, J.L.; O’Connor, M.B.; Coucouvanis, E. The TGF beta activated kinase TAK1 regulates vascular development in vivo. Development 2006, 133, 1529–1541. [Google Scholar] [CrossRef] [Green Version]
- Shim, J.H.; Xiao, C.; Paschal, A.E.; Bailey, S.T.; Rao, P.; Hayden, M.S.; Lee, K.Y.; Bussey, C.; Steckel, M.; Tanaka, N.; et al. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 2005, 19, 2668–2681. [Google Scholar] [CrossRef] [Green Version]
- Kajino-Sakamoto, R.; Inagaki, M.; Lippert, E.; Akira, S.; Robine, S.; Matsumoto, K.; Jobin, C.; Ninomiya-Tsuji, J. Enterocyte-derived TAK1 signaling prevents epithelium apoptosis and the development of ileitis and colitis. J. Immunol. 2008, 181, 1143–1152. [Google Scholar] [CrossRef]
- Singh, A.; Sweeney, M.F.; Yu, M.; Burger, A.; Greninger, P.; Benes, C.; Haber, D.A.; Settleman, J. TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell 2012, 148, 639–650. [Google Scholar] [CrossRef] [Green Version]
- Hinz, M.; Stilmann, M.; Arslan, S.Ç.; Khanna, K.K.; Dittmar, G.; Scheidereit, C. A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-κB activation. Mol. Cell 2010, 40, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.H.; Wong, E.T.; Shi, Y.; Niu, J.; Chen, Z.; Miyamoto, S.; Tergaonkar, V. ATM- and NEMO-dependent ELKS ubiquitination coordinates TAK1-Mediated IKK activation in response to genotoxic stress. Mol. Cell 2010, 40, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Jagadeeshan, S.; Subramanian, A.; Tentu, S.; Beesetti, S.; Singhal, M.; Raghavan, S.; Surabhi, R.P.; Mavuluri, J.; Bhoopalan, H.; Biswal, J.; et al. P21-activated kinase 1 (Pak1) signaling influences therapeutic outcome in pancreatic cancer. Ann. Oncol. 2016, 27, 1546–1556. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Xia, Q.; Paradiso, G.; Ling, J.; Moccia, T.; Carbone, C.; Budillon, A.; Abbruzzese, J.L.; Chiao, P.J. Modulation of pancreatic cancer chemoresistance by inhibition of TAK1. J. Natl. Cancer Inst. 2011, 103, 1190–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, M.; Wei, X.; Guo, Y.; Breslin, P.; Zhang, S.; Zhang, S.; Wei, W.; Xia, Z.; Diaz, M.; Akira, S.; et al. TAK1 is required for the survival of hematopoietic cells and hepatocytes in mice. J. Exp. Med. 2008, 205, 1611–1619. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.H.; Xie, M.; Schneider, M.D.; Chen, Z.J. Essential role of TAK1 in thymocyte development and activation. Proc. Natl. Acad. Sci. USA 2006, 103, 11677–11682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.Y.; Chi, H.; Xie, M.; Schneider, M.D.; Flavell, R.A. The kinase TAK1 integrates antigen and cytokine receptor signaling for T cell development, survival and function. Nat. Immunol. 2006, 7, 851–858. [Google Scholar] [CrossRef]
- Sato, S.; Sanjo, H.; Takeda, K.; Ninomiya-Tsuji, J.; Yamamoto, M.; Kawai, T.; Matsumoto, K.; Takeuchi, O.; Akira, S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 2005, 6, 1087–1095. [Google Scholar] [CrossRef]
- Schuman, J.; Chen, Y.; Podd, A.; Yu, M.; Liu, H.H.; Wen, R.; Chen, Z.J.; Wang, D. A critical role of TAK1 in B-cell receptor-mediated nuclear factor kappaB activation. Blood 2009, 113, 4566–4574. [Google Scholar] [CrossRef] [Green Version]
- Ajibade, A.A.; Wang, Q.; Cui, J.; Zou, J.; Xia, X.; Wang, M.; Tong, Y.; Hui, W.; Liu, D.; Su, B.; et al. TAK1 negatively regulates NF-kappaB and p38 MAP kinase activation in Gr-1+CD11b+ neutrophils. Immunity 2012, 36, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Totzke, J.; Gurbani, D.; Raphemot, R.; Hughes, P.F.; Bodoor, K.; Carlson, D.A.; Loiselle, D.R.; Bera, A.K.; Eibschutz, L.S.; Perkins, M.M.; et al. Takinib, a Selective TAK1 Inhibitor, Broadens the Therapeutic Efficacy of TNF-alpha Inhibition for Cancer and Autoimmune Disease. Cell Chem. Biol. 2017, 24, 1029–1039.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mielke, L.A.; Elkins, K.L.; Wei, L.; Starr, R.; Tsichlis, P.N.; O’Shea, J.J.; Watford, W.T. Tumor progression locus 2 (Map3k8) is critical for host defense against Listeria monocytogenes and IL-1 beta production. J. Immunol. 2009, 183, 7984–7993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattison, M.J.; Mitchell, O.; Flynn, H.R.; Chen, C.S.; Yang, H.T.; Ben-Addi, H.; Boeing, S.; Snijders, A.P.; Ley, S.C. TLR and TNF-R1 activation of the MKK3/MKK6-p38alpha axis in macrophages is mediated by TPL-2 kinase. Biochem. J. 2016, 473, 2845–2861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beinke, S.; Deka, J.; Lang, V.; Belich, M.P.; Walker, P.A.; Howell, S.; Smerdon, S.J.; Gamblin, S.J.; Ley, S.C. NF-kappaB1 p105 negatively regulates TPL-2 MEK kinase activity. Mol. Cell. Biol. 2003, 23, 4739–4752. [Google Scholar] [CrossRef] [Green Version]
- Beinke, S.; Robinson, M.J.; Hugunin, M.; Ley, S.C. Lipopolysaccharide activation of the TPL-2/MEK/extracellular signal-regulated kinase mitogen-activated protein kinase cascade is regulated by IkappaB kinase-induced proteolysis of NF-kappaB1 p105. Mol. Cell. Biol. 2004, 24, 9658–9667. [Google Scholar] [CrossRef] [Green Version]
- Ben-Addi, A.; Mambole-Dema, A.; Brender, C.; Martin, S.R.; Janzen, J.; Kjaer, S.; Smerdon, S.J.; Ley, S.C. IkappaB kinase-induced interaction of TPL-2 kinase with 14-3-3 is essential for Toll-like receptor activation of ERK-1 and -2 MAP kinases. Proc. Natl. Acad. Sci. USA 2014, 111, E2394–E2403. [Google Scholar] [CrossRef] [Green Version]
- Roget, K.; Ben-Addi, A.; Mambole-Dema, A.; Gantke, T.; Yang, H.T.; Janzen, J.; Morrice, N.; Abbott, D.; Ley, S.C. IkappaB kinase 2 regulates TPL-2 activation of extracellular signal-regulated kinases 1 and 2 by direct phosphorylation of TPL-2 serine 400. Mol. Cell. Biol. 2012, 32, 4684–4690. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.; Tsichlis, P.N. Phosphorylation at Thr-290 regulates Tpl2 binding to NF-kappaB1/p105 and Tpl2 activation and degradation by lipopolysaccharide. Proc. Natl. Acad. Sci. USA 2005, 102, 2350–2355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belich, M.P.; Salmeron, A.; Johnston, L.H.; Ley, S.C. TPL-2 kinase regulates the proteolysis of the NF-kappaB-inhibitory protein NF-kappaB1 p105. Nature 1999, 397, 363–368. [Google Scholar] [CrossRef]
- Salmeron, A.; Ahmad, T.B.; Carlile, G.W.; Pappin, D.; Narsimhan, R.P.; Ley, S.C. Activation of MEK-1 and SEK-1 by Tpl-2 proto-oncoprotein, a novel MAP kinase kinase kinase. EMBO J. 1996, 15, 817–826. [Google Scholar] [CrossRef]
- Senger, K.; Pham, V.C.; Varfolomeev, E.; Hackney, J.A.; Corzo, C.A.; Collier, J.; Lau, V.W.C.; Huang, Z.; Hamidzhadeh, K.; Caplazi, P.; et al. The kinase TPL2 activates ERK and p38 signaling to promote neutrophilic inflammation. Sci. Signal. 2017, 10, eaah4273. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Cho, J.; Lambertz, I.; Kelliher, M.A.; Eliopoulos, A.G.; Du, K.; Tsichlis, P.N. Tpl2/cot signals activate ERK, JNK, and NF-kappaB in a cell-type and stimulus-specific manner. J. Biol. Chem. 2005, 280, 23748–23757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef] [PubMed]
- Madrid, L.V.; Mayo, M.W.; Reuther, J.Y.; Baldwin, A.S., Jr. Akt stimulates the transactivation potential of the RelA/p65 Subunit of NF-kappa B through utilization of the Ikappa B kinase and activation of the mitogen-activated protein kinase p38. J. Biol. Chem. 2001, 276, 18934–18940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madrid, L.V.; Wang, C.Y.; Guttridge, D.C.; Schottelius, A.J.; Baldwin, A.S., Jr.; Mayo, M.W. Akt suppresses apoptosis by stimulating the transactivation potential of the RelA/p65 subunit of NF-kappaB. Mol. Cell. Biol. 2000, 20, 1626–1638. [Google Scholar] [CrossRef] [Green Version]
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.; Baldwin, A.S. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008, 22, 1490–1500. [Google Scholar] [CrossRef] [Green Version]
- Chien, Y.; Kim, S.; Bumeister, R.; Loo, Y.M.; Kwon, S.W.; Johnson, C.L.; Balakireva, M.G.; Romeo, Y.; Kopelovich, L.; Gale, M., Jr.; et al. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell 2006, 127, 157–170. [Google Scholar] [CrossRef] [Green Version]
- Bang, D.; Wilson, W.; Ryan, M.; Yeh, J.J.; Baldwin, A.S. GSK-3α promotes oncogenic KRAS function in pancreatic cancer via TAK1-TAB stabilization and regulation of noncanonical NF-κB. Cancer Discov. 2013, 3, 690–703. [Google Scholar] [CrossRef] [Green Version]
- Daniluk, J.; Liu, Y.; Deng, D.; Chu, J.; Huang, H.; Gaiser, S.; Cruz-Monserrate, Z.; Wang, H.; Ji, B.; Logsdon, C.D. An NF-kappaB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J. Clin. Investig. 2012, 122, 1519–1528. [Google Scholar] [CrossRef] [Green Version]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef] [Green Version]
- Takada, Y.; Singh, S.; Aggarwal, B.B. Identification of a p65 peptide that selectively inhibits NF-kappa B activation induced by various inflammatory stimuli and its role in down-regulation of NF-kappaB-mediated gene expression and up-regulation of apoptosis. J. Biol. Chem. 2004, 279, 15096–15104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.P.; Hori, M.; Sanda, T.; Okamoto, T. Identification of a novel inhibitor of nuclear factor-kappaB, RelA-associated inhibitor. J. Biol. Chem. 1999, 274, 15662–15670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moles, A.; Sanchez, A.M.; Banks, P.S.; Murphy, L.B.; Luli, S.; Borthwick, L.; Fisher, A.; O’Reilly, S.; van Laar, J.M.; White, S.A.; et al. Inhibition of RelA-Ser536 phosphorylation by a competing peptide reduces mouse liver fibrosis without blocking the innate immune response. Hepatology 2013, 57, 817–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Rayes, B.F.; Ali, S.; Sarkar, F.H.; Philip, P.A. Cyclooxygenase-2-dependent and -independent effects of celecoxib in pancreatic cancer cell lines. Mol. Cancer 2004, 3, 1421–1426. [Google Scholar]
- Dragovich, T.; Burris, H., 3rd; Loehrer, P.; Von Hoff, D.D.; Chow, S.; Stratton, S.; Green, S.; Obregon, Y.; Alvarez, I.; Gordon, M. Gemcitabine plus celecoxib in patients with advanced or metastatic pancreatic adenocarcinoma: Results of a phase II trial. Am. J. Clin. Oncol. 2008, 31, 157–162. [Google Scholar] [CrossRef]
- Kanai, M.; Yoshimura, K.; Asada, M.; Imaizumi, A.; Suzuki, C.; Matsumoto, S.; Nishimura, T.; Mori, Y.; Masui, T.; Kawaguchi, Y.; et al. A phase I/II study of gemcitabine-based chemotherapy plus curcumin for patients with gemcitabine-resistant pancreatic cancer. Cancer Chemother. Pharm. 2011, 68, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, N.; Aggarwal, B.B.; Newman, R.A.; Wolff, R.A.; Kunnumakkara, A.B.; Abbruzzese, J.L.; Ng, C.S.; Badmaev, V.; Kurzrock, R. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin. Cancer Res. 2008, 14, 4491–4499. [Google Scholar] [CrossRef] [Green Version]
- Alberts, S.R.; Foster, N.R.; Morton, R.F.; Kugler, J.; Schaefer, P.; Wiesenfeld, M.; Fitch, T.R.; Steen, P.; Kim, G.P.; Gill, S. PS-341 and gemcitabine in patients with metastatic pancreatic adenocarcinoma: A North Central Cancer Treatment Group (NCCTG) randomized phase II study. Ann. Oncol. 2005, 16, 1654–1661. [Google Scholar] [CrossRef]
- Lamberti, M.J.; Nigro, A.; Mentucci, F.M.; Rumie Vittar, N.B.; Casolaro, V.; Dal Col, J. Dendritic Cells and Immunogenic Cancer Cell Death: A Combination for Improving Antitumor Immunity. Pharmaceutics 2020, 12, 256. [Google Scholar] [CrossRef] [Green Version]
- Konduri, V.; Li, D.; Halpert, M.M.; Liang, D.; Liang, Z.; Chen, Y.; Fisher, W.E.; Paust, S.; Levitt, J.M.; Yao, Q.C.; et al. Chemo-immunotherapy mediates durable cure of orthotopic Kras(G12D)/p53(−/−) pancreatic ductal adenocarcinoma. Oncoimmunology 2016, 5, e1213933. [Google Scholar] [CrossRef] [Green Version]
- Younes, A.; Nowakowski, G.; Rosenthal, A.C.; Leslie, L.A.; Tun, H.W.; Lunning, M.A.; Isufi, I.; Martell, R.; Patel, K. Phase 1 Dose-Finding Study Investigating CA-4948, an IRAK4 Kinase Inhibitor, in Patients with R/R NHL: Report of Initial Efficacy and Updated Safety Information. Blood 2019, 134, 5327. [Google Scholar] [CrossRef]
- Li, Q.; Chen, Y.; Zhang, D.; Grossman, J.; Li, L.; Khurana, N.; Jiang, H.; Grierson, P.M.; Herndon, J.; DeNardo, D.G.; et al. IRAK4 mediates colitis-induced tumorigenesis and chemoresistance in colorectal cancer. JCI Insight 2019, 4, e130867. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khurana, N.; Dodhiawala, P.B.; Bulle, A.; Lim, K.-H. Deciphering the Role of Innate Immune NF-ĸB Pathway in Pancreatic Cancer. Cancers 2020, 12, 2675. https://doi.org/10.3390/cancers12092675
Khurana N, Dodhiawala PB, Bulle A, Lim K-H. Deciphering the Role of Innate Immune NF-ĸB Pathway in Pancreatic Cancer. Cancers. 2020; 12(9):2675. https://doi.org/10.3390/cancers12092675
Chicago/Turabian StyleKhurana, Namrata, Paarth B. Dodhiawala, Ashenafi Bulle, and Kian-Huat Lim. 2020. "Deciphering the Role of Innate Immune NF-ĸB Pathway in Pancreatic Cancer" Cancers 12, no. 9: 2675. https://doi.org/10.3390/cancers12092675
APA StyleKhurana, N., Dodhiawala, P. B., Bulle, A., & Lim, K. -H. (2020). Deciphering the Role of Innate Immune NF-ĸB Pathway in Pancreatic Cancer. Cancers, 12(9), 2675. https://doi.org/10.3390/cancers12092675