Landscape of Genome-Wide DNA Methylation of Colorectal Cancer Metastasis

 ,
,  , , ,
, , ,  , ,
, ,
Abstract
:Simple Summary
Abstract
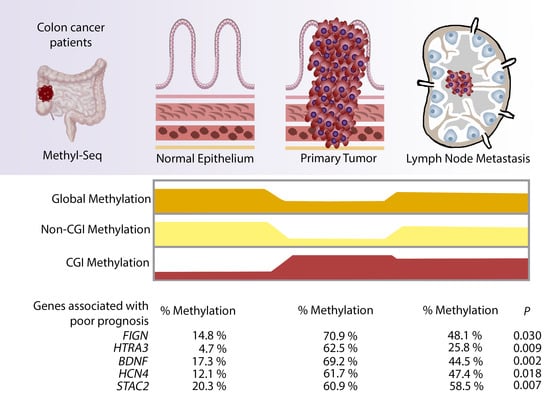
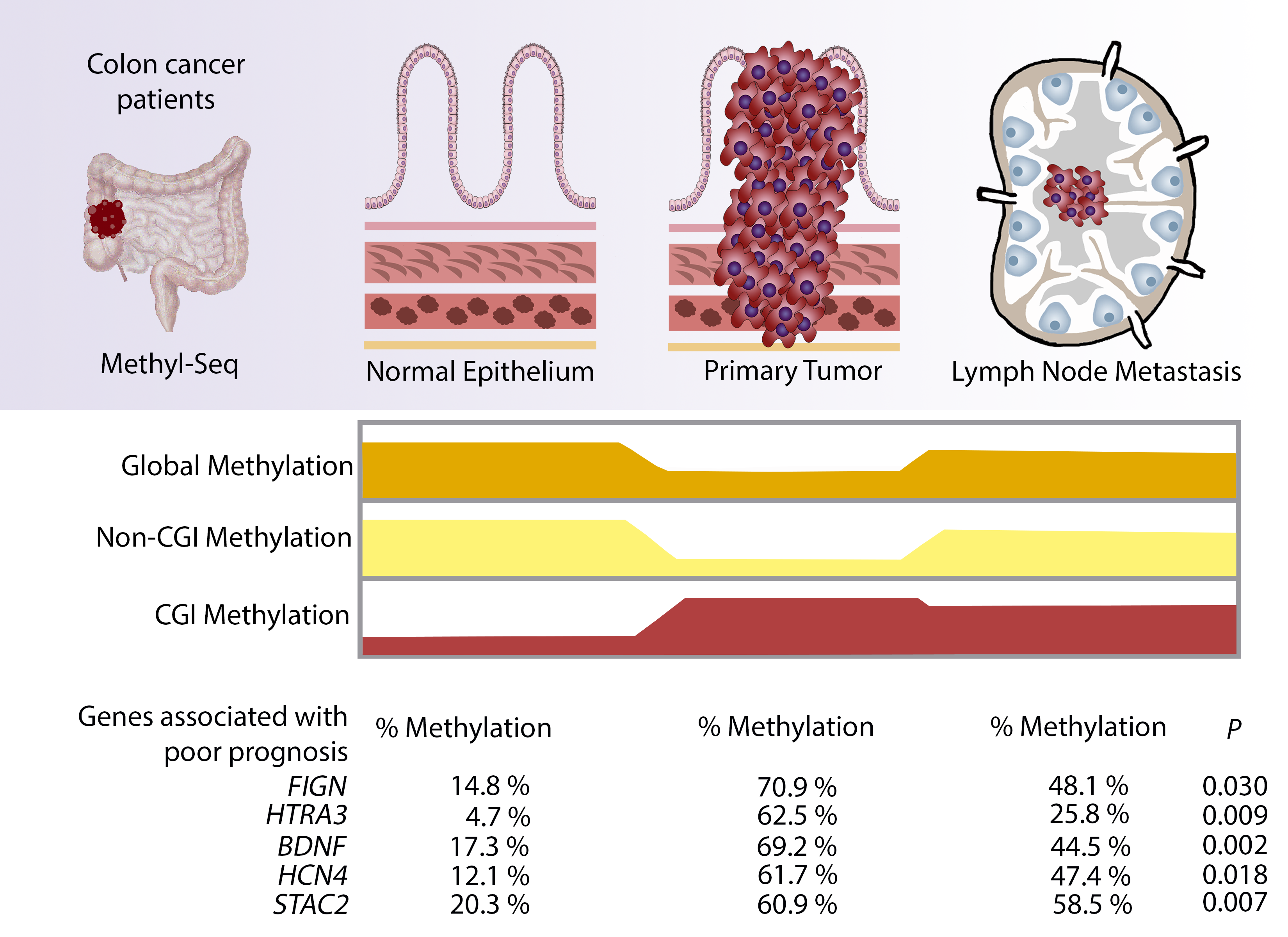
1. Introduction
2. Results
2.1. Clinical and Pathological Characteristics of Patient Cohort
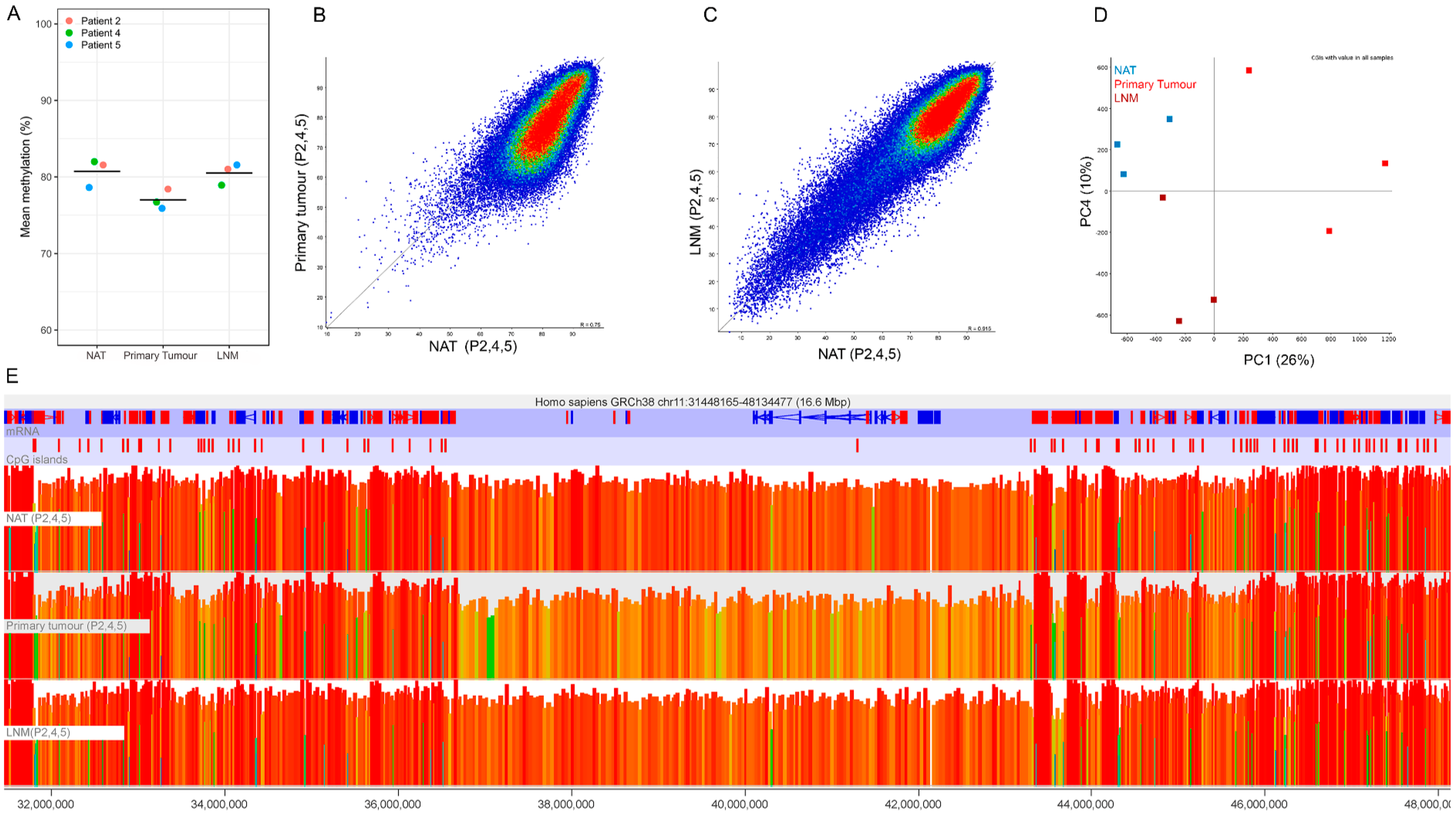
2.2. DNA Methylation QC Analysis
2.3. Global Hypomethylation of Non-CGI Regions in Primary Tumour but Not Metastasis Samples
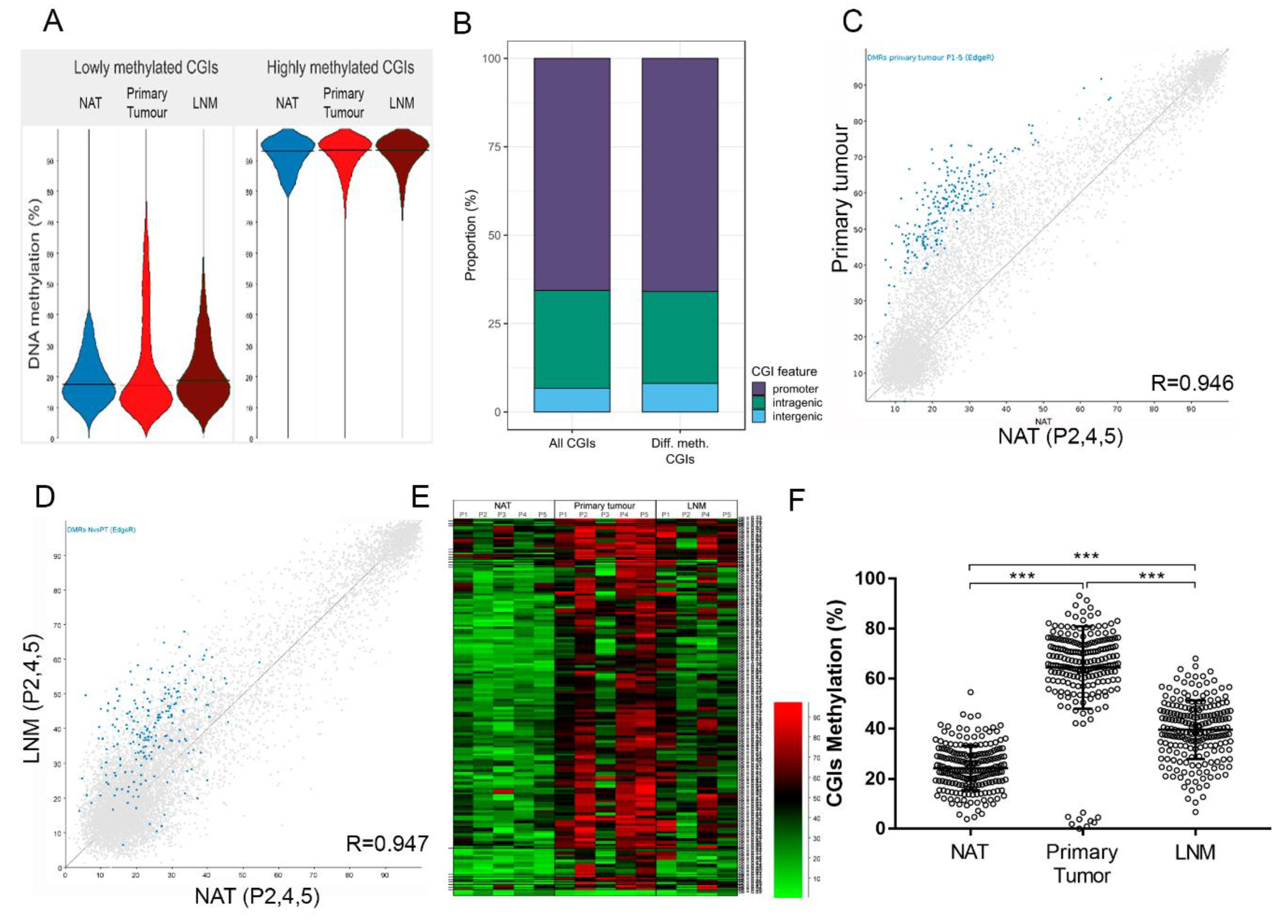
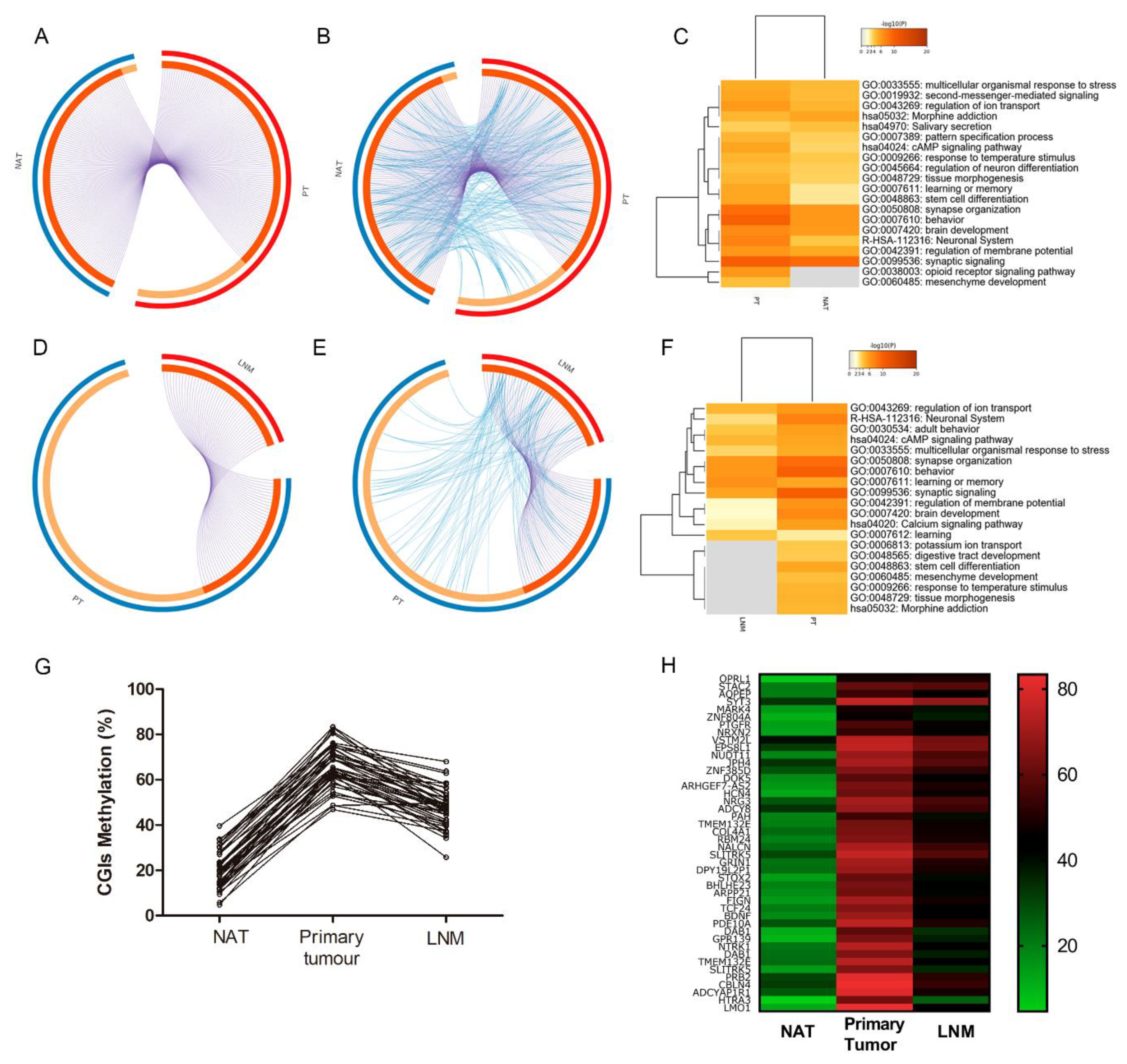
2.4. Identification of Hypermethylated CGIs in Primary Tumour and Lymph Node Metastasis Samples
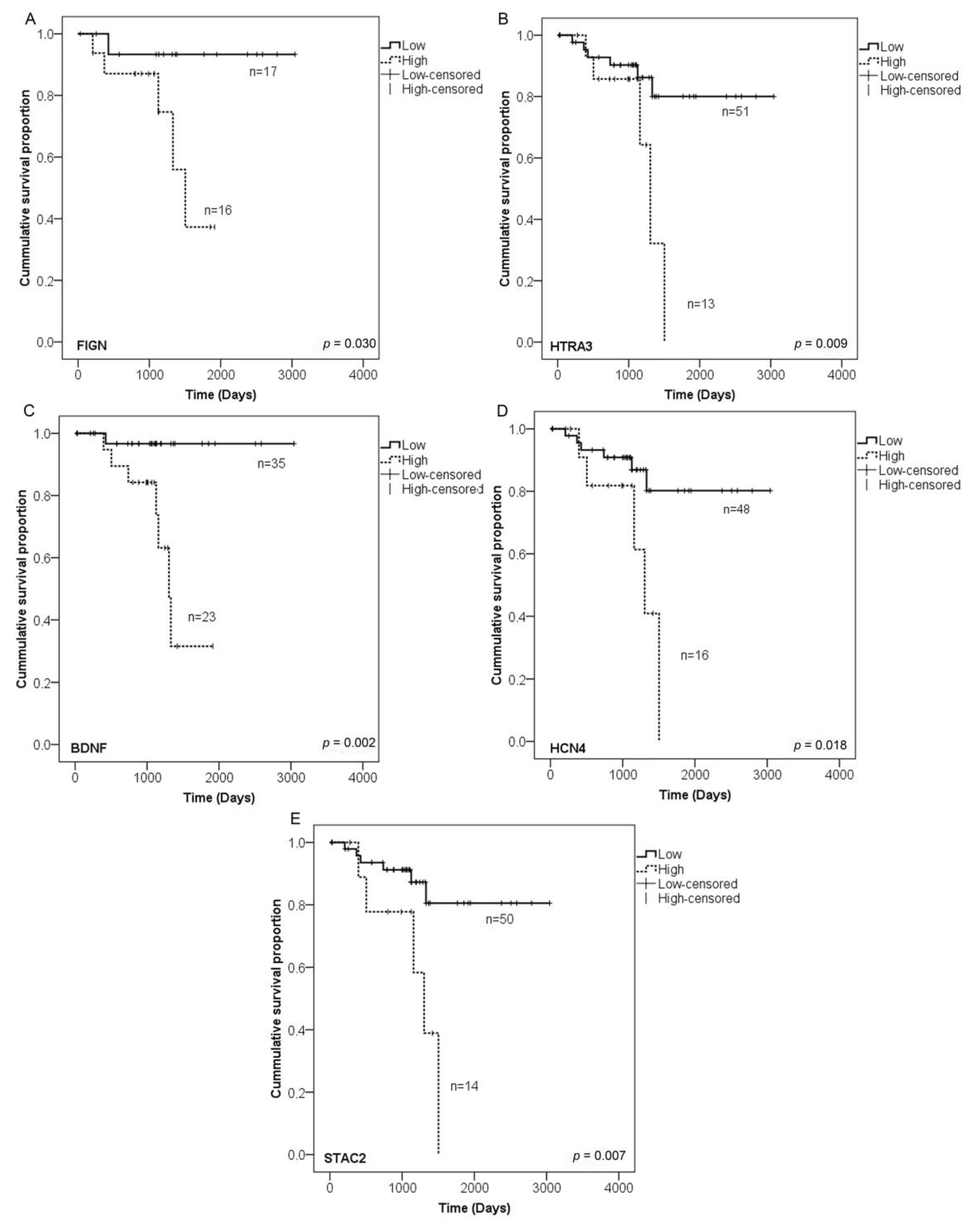
2.5. Validation of Hypermethylated Metastasis CGIs in a Larger Cohort
3. Discussion
4. Materials and Methods
4.1. Discovery Cohort
4.2. DNA Extraction
4.3. DNA Quality Control Prior Sequencing
4.4. Genome Wide Methyl-Seq Bisulphite Sequencing
4.5. Library Mapping and Trimming
4.6. DNA Methylation Sequencing Analysis
4.7. Gene Ontology Analysis
4.8. TCGA Methylation Array Data
4.9. Candidates Genes Selection and Survival Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jemal, A.; Center, M.M.; DeSantis, C.; Ward, E.M. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1893–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzig, D.O.; Tsikitis, V.L. Molecular markers for colon diagnosis, prognosis and targeted therapy. J. Surg. Oncol. 2015, 111, 96–102. [Google Scholar] [CrossRef]
- Binefa, G.; Rodríguez-Moranta, F.; Teule, À.; Medina-Hayas, M. Colorectal cancer: From prevention to personalized medicine. World J. Gastroenterol. 2014, 20, 6786–6808. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; Van De Velde, C.J.H.; Watanabe, T. Colorectal cancer. Nat. Rev. Dis. Prim. 2015, 1, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Lao, V.V.; Grady, W.M. Epigenetics and colorectal cancer. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 686–700. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar]
- Zheng, R.; Gao, D.; He, T.; Zhang, M.; Zhang, X.; Linghu, E.; Wei, L.; Guo, M. Methylation of DIRAS1 promotes colorectal cancer progression and may serve as a marker for poor prognosis. Clin. Epigenetics 2017, 9, 1–9. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2010, 1, 239–259. [Google Scholar] [CrossRef] [Green Version]
- Deng, G.; Nguyen, A.; Tanaka, H.; Matsuzaki, K.; Bell, I.; Mehta, K.R.; Terdiman, J.P.; Waldman, F.M.; Kakar, S.; Gum, J.; et al. Regional hypermethylation and global hypomethylation are associated with altered chromatin conformation and histone acetylation in colorectal cancer. Int. J. Cancer 2006, 118, 2999–3005. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Lee, J.; Sidransky, D. DNA methylation markers in colorectal cancer. Cancer Metastasis Rev. 2010, 29, 181–206. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, M.R.; Hussong, M.; Röhr, C.; Lehrach, H. Genomics and epigenomics of colorectal cancer. Wiley Interdiscip. Rev. Syst. Biol. Med. 2013, 5, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Walther, A.; Johnstone, E.; Swanton, C.; Midgley, R.; Tomlinson, I.; Kerr, D. Genetic prognostic and predictive markers in colorectal cancer. Nat. Rev. Cancer 2009, 9, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Scheel, C.; Weinberg, R.A. Cancer stem cells and epithelial-mesenchymal transition: Concepts and molecular links. Semin. Cancer Biol. 2012, 22, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Miranda, E.; Destro, A.; Malesci, A.; Balladore, E.; Bianchi, P.; Baryshnikova, E.; Franchi, G.; Morenghi, E.; Laghi, L.; Gennari, L.; et al. Genetic and epigenetic changes in primary metastatic and nonmetastatic colorectal cancer. Br. J. Cancer 2006, 95, 1101–1107. [Google Scholar] [CrossRef] [Green Version]
- Hohla, F.; Winder, T.; Greil, R.; Rick, F.G.; Block, N.L.; Schally, A.V. Targeted therapy in advanced metastatic colorectal cancer: Current concepts and perspectives. World J. Gastroenterol. 2014, 20, 6102–6112. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Liang, X.; Jiang, W.; Li, J.; Xu, J.; Cai, X. Cyclin B1 suppresses colorectal cancer invasion and metastasis by regulating E-cadherin. PLoS ONE 2015. [Google Scholar] [CrossRef]
- Ju, H.X.; An, B.; Okamoto, Y.; Shinjo, K.; Kanemitsu, Y.; Komori, K.; Hirai, T.; Shimizu, Y.; Sano, T.; Sawaki, A.; et al. Distinct profiles of epigenetic evolution between colorectal cancers with and without metastasis. Am. J. Pathol. 2011, 178, 1835–1846. [Google Scholar] [CrossRef]
- Yan, W.; Wu, K.; Herman, J.G.; Brock, M.V.; Fuks, F.; Yang, L.; Zhu, H.; Li, Y.; Yang, Y.; Guo, M. Epigenetic regulation of DACH1, a novel Wnt signaling component in colorectal cancer. Epigenetics 2013, 8, 1373–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef] [Green Version]
- Hoque, M.O.; Kim, M.S.; Ostrow, K.L.; Liu, J.; Wisman, G.B.A.; Park, H.L.; Poeta, M.L.; Jeronimo, C.; Henrique, R.; Lendvai, Á.; et al. Genome-wide promoter analysis uncovers portions of the cancer methylome. Cancer Res. 2008. [Google Scholar] [CrossRef] [Green Version]
- Kass, S.U.; Pruss, D.; Wolffe, A.P. How does DNA methylation repress transcription? Trends Genet. 1997, 13, 444–449. [Google Scholar] [CrossRef]
- Hawkins, N.; Norrie, M.; Cheong, K.; Mokany, E.; Ku, S.L.; Meagher, A.; O’Connor, T.; Ward, R. CpG island methylation in sporadic colorectal cancers and its relationship to microsatellite instability. Gastroenterology 2002. [Google Scholar] [CrossRef] [PubMed]
- Barault, L.; Charon-Barra, C.; Jooste, V.; De La Vega, M.F.; Martin, L.; Roignot, P.; Rat, P.; Bouvier, A.M.; Laurent-Puig, P.; Faivre, J.; et al. Hypermethylator phenotype in sporadic colon cancer: Study on a population-based series of 582 cases. Cancer Res. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattesen, T.B.; Rasmussen, M.H.; Sandoval, J.; Ongen, H.; Árnadóttir, S.S.; Gladov, J.; Martinez-Cardus, A.; Castro de Moura, M.; Madsen, A.H.; Laurberg, S.; et al. MethCORR modelling of methylomes from formalin-fixed paraffin-embedded tissue enables characterization and prognostication of colorectal cancer. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hanley, M.P.; Hahn, M.A.; Li, A.X.; Wu, X.; Lin, J.; Wang, J.; Choi, A.H.; Ouyang, Z.; Fong, Y.; Pfeifer, G.P.; et al. Genome-wide DNA methylation profiling reveals cancer-associated changes within early colonic neoplasia. Oncogene 2017, 36, 5035–5044. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Mei, Y.; Xiang, P.; Zhai, G.; Zhao, N.; Xu, C.; Liu, M.; Pan, Z.; Tang, K.; Jia, D. DNA methylation affects metastasis of renal cancer and is associated with TGF-β/RUNX3 inhibition. Cancer Cell Int. 2018, 18, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Yin, A.; Etcheverry, A.; He, Y.; Aubry, M.; Barnholtz-Sloan, J.; Zhang, L.; Mao, X.; Chen, W.; Liu, B.; Zhang, W.; et al. Integrative analysis of novel hypomethylation and gene expression signatures in glioblastomas. Oncotarget 2017, 8, 89607–89619. [Google Scholar] [CrossRef] [PubMed]
- Salas, L.A.; Johnson, K.C.; Koestler, D.C.; O’Sullivan, D.E.; Christensen, B.C. Integrative epigenetic and genetic pan-cancer somatic alteration portraits. Epigenetics 2017, 12, 561–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Y.; Lennartsson, A.; Gaidzik, V.I.; Deneberg, S.; Karimi, M.; Bengtzén, S.; Höglund, M.; Bullinger, L.; Döhner, K.; Lehmann, S. Differential methylation in CN-AML preferentially targets non-CGI regions and is dictated by DNMT3A mutational status and associated with predominant hypomethylation of HOX genes. Epigenetics 2014, 9, 1108–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Chow, J.; Wang, Z.; Fan, G. Abnormal CpG island methylation occurs during in vitro differentiation of human embryonic stem cells. Hum. Mol. Genet. 2006, 15, 2623–2635. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zöller, M. Exosomes, metastases, and the miracle of cancer stem cell markers. Cancer Metastasis Rev. 2019, 38, 259–295. [Google Scholar] [CrossRef]
- Ferrer, A.I.; Trinidad, J.R.; Sandiford, O.; Etchegaray, J.P.; Rameshwar, P. Epigenetic dynamics in cancer stem cell dormancy. Cancer Metastasis Rev. 2020. [Google Scholar] [CrossRef]
- Fernández, E.A.; Mahmoud, Y.D.; Veigas, F.; Rocha, D.; Balzarini, M.; Lujan, H.D.; Rabinovich, G.A.; Girotti, M.R. MIXTURE: An improved algorithm for immune tumor microenvironment estimation based on gene expression data. bioRxiv 2019, 726562. [Google Scholar] [CrossRef]
- Gómez, S.; Castellano, G.; Mayol, G.; Suñol, M.; Queiros, A.; Bibikova, M.; Nazor, K.L.; Loring, J.F.; Lemos, I.; Rodríguez, E.; et al. DNA methylation fingerprint of neuroblastoma reveals new biological and clinical insights. Epigenomics 2015, 7, 1137–1153. [Google Scholar] [CrossRef] [Green Version]
- Vidal, E.; Sayols, S.; Moran, S.; Guillaumet-Adkins, A.; Schroeder, M.P.; Royo, R.; Orozco, M.; Gut, M.; Gut, I.; Lopez-Bigas, N.; et al. A DNA methylation map of human cancer at single base-pair resolution. Oncogene 2017, 36, 5648–5657. [Google Scholar] [CrossRef]
- Pistore, C.; Giannoni, E.; Colangelo, T.; Rizzo, F.; Magnani, E.; Muccillo, L.; Giurato, G.; Mancini, M.; Rizzo, S.; Riccardi, M.; et al. DNA methylation variations are required for epithelial-to-mesenchymal transition induced by cancer-associated fibroblasts in prostate cancer cells. Oncogene 2017, 36, 5551–5566. [Google Scholar] [CrossRef]
- Luo, Y.; Wong, C.J.; Kaz, A.M.; Dzieciatkowski, S.; Carter, K.T.; Morris, S.M.; Wang, J.; Willis, J.E.; Makar, K.W.; Ulrich, C.M.; et al. Differences in DNA methylation signatures reveal multiple pathways of progression from adenoma to colorectal cancer. Gastroenterology 2014, 147, 418–429.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, W.; Wu, X.; Wang, F.; Han, P.; Cui, B. Genome-wide methylation analysis identifies novel prognostic methylation markers in colon adenocarcinoma. Biomed. Pharmacother. 2018, 108, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Schulten, H.J.; Hussein, D. Array expression meta-analysis of cancer stem cell genes identifies upregulation of PODXL especially in DCC low expression meningiomas. PLoS ONE 2019, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Yao, H.; Li, A.; Wang, M. CSCdb: A Cancer stem cells portal for markers, related genes and functional information. Database 2016, 2016, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Xing, T.; Yang, Z.; Dudek, R.; Lu, Q.; Chen, Y.-H. Epithelial Mesenchymal Transition in Embryonic Development, Tissue Repair and Cancer: A Comprehensive Overview. J. Clin. Med. 2017, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- Puigdevall, P.; Piccari, L.; Blanco, I.; Barberà, J.A.; Geiger, D.; Badenas, C.; Milà, M.; Castelo, R.; Madrigal, I. Genetic linkage analysis of a large family identifies FIGN as a candidate modulator of reduced penetrance in heritable pulmonary arterial hypertension. J. Med. Genet. 2019, 56, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Riordan, J.D.; Feddersen, C.R.; Tschida, B.R.; Beckmann, P.J.; Keng, V.W.; Linden, M.A.; Amin, K.; Stipp, C.S.; Largaespada, D.A.; Dupuy, A.J. Chronic liver injury alters driver mutation profiles in hepatocellular carcinoma in mice. Hepatology 2018. [Google Scholar] [CrossRef] [Green Version]
- Wolter, S.; Kloth, C.; Golombek, M.; Dittmar, F.; Försterling, L.; Seifert, R. CCMP causes caspase-dependent apoptosis in mouse lymphoma cell lines. Biochem. Pharmacol. 2015, 98, 119–131. [Google Scholar] [CrossRef]
- Forse, C.L.; Rahimi, M.; Diamandis, E.P.; Assarzadegan, N.; Dawson, H.; Grin, A.; Kennedy, E.; O’Connor, B.; Messenger, D.E.; Riddell, R.H.; et al. HtrA3 stromal expression is correlated with tumor budding in stage II colorectal cancer. Exp. Mol. Pathol. 2017, 103, 94–100. [Google Scholar] [CrossRef]
- Zhao, J.; Feng, M.; Liu, D.; Liu, H.; Shi, M.; Zhang, J.; Qu, J. Antagonism between HTRA3 and TGFb1 contributes to metastasis in non–small cell lung cancer. Cancer Res. 2019, 79, 2853–2864. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Ding, J.X.; Nie, G.Y.; Wei, J.; Li, Y.; Yin, X.Y.; Chen, Q. HTRA3 is reduced in ovarian cancers regardless of stage. Cancer Investig. 2014, 32, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Wu, M.; Nie, G.; Wang, K.; Wei, J.; Zhao, M.; Chen, Q. HtrA3 is negatively correlated with lymph node metastasis in invasive ductal breast cancer. Tumor Biol. 2013, 34, 3611–3617. [Google Scholar] [CrossRef] [PubMed]
- Monteggia, L.M.; Björkholm, C. BDNF—A key transducer of antidepressant effects. Neuropharmacology 2017, 102, 72–79. [Google Scholar] [CrossRef]
- Contreras-Zárate, M.J.; Day, N.L.; Ormond, D.R.; Borges, V.F.; Tobet, S.; Gril, B.; Steeg, P.S.; Cittelly, D.M. Estradiol induces BDNF/TrkB signaling in triple-negative breast cancer to promote brain metastases. Oncogene 2019, 38, 4685–4699. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Ye, H.Q.; Ren, Q.C. Upregulation of the BDNF/TrKB pathway promotes epithelial-mesenchymal transition, as well as the migration and invasion of cervical cancer. Int. J. Oncol. 2018, 52, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, A.; Kemp, K.; Ginty, M.; Hares, K.; Mallam, E.; Scolding, N. Human bone marrow-derived mesenchymal stem cells secrete brain-derived neurotrophic factor which promotes neuronal survival in vitro. Stem Cell Res. 2009, 3, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Möller, M.; Silbernagel, N.; Wrobel, E.; Stallmayer, B.; Amedonu, E.; Rinné, S.; Peischard, S.; Meuth, S.G.; Wünsch, B.; Strutz-Seebohm, N.; et al. In vitro analyses of novel HCN4 gene mutations. Cell. Physiol. Biochem. 2018, 49, 1238–1248. [Google Scholar] [CrossRef]
- Jeong, E.; Choi, H.K.; Park, J.H.; Lee, S.Y. STAC2 negatively regulates osteoclast formation by targeting the RANK signaling complex. Cell Death Differ. 2018, 25, 1364–1374. [Google Scholar] [CrossRef] [Green Version]
- Polster, A.; Dittmer, P.J.; Perni, S.; Bichraoui, H.; Sather, W.A.; Beam, K.G. Stac proteins suppress Ca2+-dependent inactivation of neuronal L-type Ca2+ channels. J. Neurosci. 2018, 38, 9215–9227. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Zhou, L.; Chen, J.; Su, J.; Shen, W.; Liu, B.; Zhou, J.; Yu, S.; Qian, J. A four-gene signature for prognosis in breast cancer patients with hypermethylated IL15RA. Oncol. Lett. 2019, 17, 4245–4254. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Wang, L.; Shi, L.; Yun, F.; Liu, X.; Chen, Y.; Chen, C.; Ren, Y.; Jia, Y. Transcriptome profiling revealed multiple genes and ECM-receptor interaction pathways that may be associated with breast cancer. Cell. Mol. Biol. Lett. 2019, 24, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.B.; Greene, F.L.; Edge, S.B.; Compton, C.C.; Gershenwald, J.E.; Brookland, R.K.; Meyer, L.; Gress, D.M.; Byrd, D.R.; Winchester, D.P. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J. Clin. 2017. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nearest Gene | Description | Chr | Start | End | Feature Strand | CGI Orientation in Relation to Nearest Gene | Proposed Function |
|---|---|---|---|---|---|---|---|
| FIGN | Fidgetin | chr2 | 163,736,135 | 163,737,001 | − | downstream | ATP-dependent microtubule severing protein |
| HTRA3 | HtrA serine peptidase 3 | chr4 | 8,269,488 | 8,270,364 | + | overlapping | Serine protease that cleaves beta-casein/CSN2 as well as several extracellular matrix (ECM) proteoglycans. |
| BDNF | Brain-derived neurotrophic factor | chr11 | 27,721,926 | 27,723,017 | − | overlapping | Important signalling molecule that activates signalling cascades downstream of NTRK2 |
| HCN4 | Hyperpolarisation activated cyclic nucleotide-gated potassium channel 4 | chr15 | 73,367,520 | 73,369,674 | − | overlapping | Activated by cAMP. cAMP binding causes a conformation change that leads to the assembly of an active tetramer and channel opening. |
| STAC2 | SH3 and cysteine rich domain 2 | chr17 | 39,224,701 | 39,226,110 | − | overlapping | Plays a redundant role in promoting the expression of calcium channel CACNA1S at the cell membrane, and thereby contributes to increased channel activity. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ili, C.; Buchegger, K.; Demond, H.; Castillo-Fernandez, J.; Kelsey, G.; Zanella, L.; Abanto, M.; Riquelme, I.; López, J.; Viscarra, T.; et al. Landscape of Genome-Wide DNA Methylation of Colorectal Cancer Metastasis. Cancers 2020, 12, 2710. https://doi.org/10.3390/cancers12092710
Ili C, Buchegger K, Demond H, Castillo-Fernandez J, Kelsey G, Zanella L, Abanto M, Riquelme I, López J, Viscarra T, et al. Landscape of Genome-Wide DNA Methylation of Colorectal Cancer Metastasis. Cancers. 2020; 12(9):2710. https://doi.org/10.3390/cancers12092710
Chicago/Turabian StyleIli, Carmen, Kurt Buchegger, Hannah Demond, Juan Castillo-Fernandez, Gavin Kelsey, Louise Zanella, Michel Abanto, Ismael Riquelme, Jaime López, Tamara Viscarra, and et al. 2020. "Landscape of Genome-Wide DNA Methylation of Colorectal Cancer Metastasis" Cancers 12, no. 9: 2710. https://doi.org/10.3390/cancers12092710
APA StyleIli, C., Buchegger, K., Demond, H., Castillo-Fernandez, J., Kelsey, G., Zanella, L., Abanto, M., Riquelme, I., López, J., Viscarra, T., García, P., Bellolio, E., Saavedra, D., & Brebi, P. (2020). Landscape of Genome-Wide DNA Methylation of Colorectal Cancer Metastasis. Cancers, 12(9), 2710. https://doi.org/10.3390/cancers12092710