Novel Epigenetic Eight-Gene Signature Predictive of Poor Prognosis and MSI-Like Phenotype in Human Metastatic Colorectal Carcinomas

 , , ,
, , ,  and
and
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
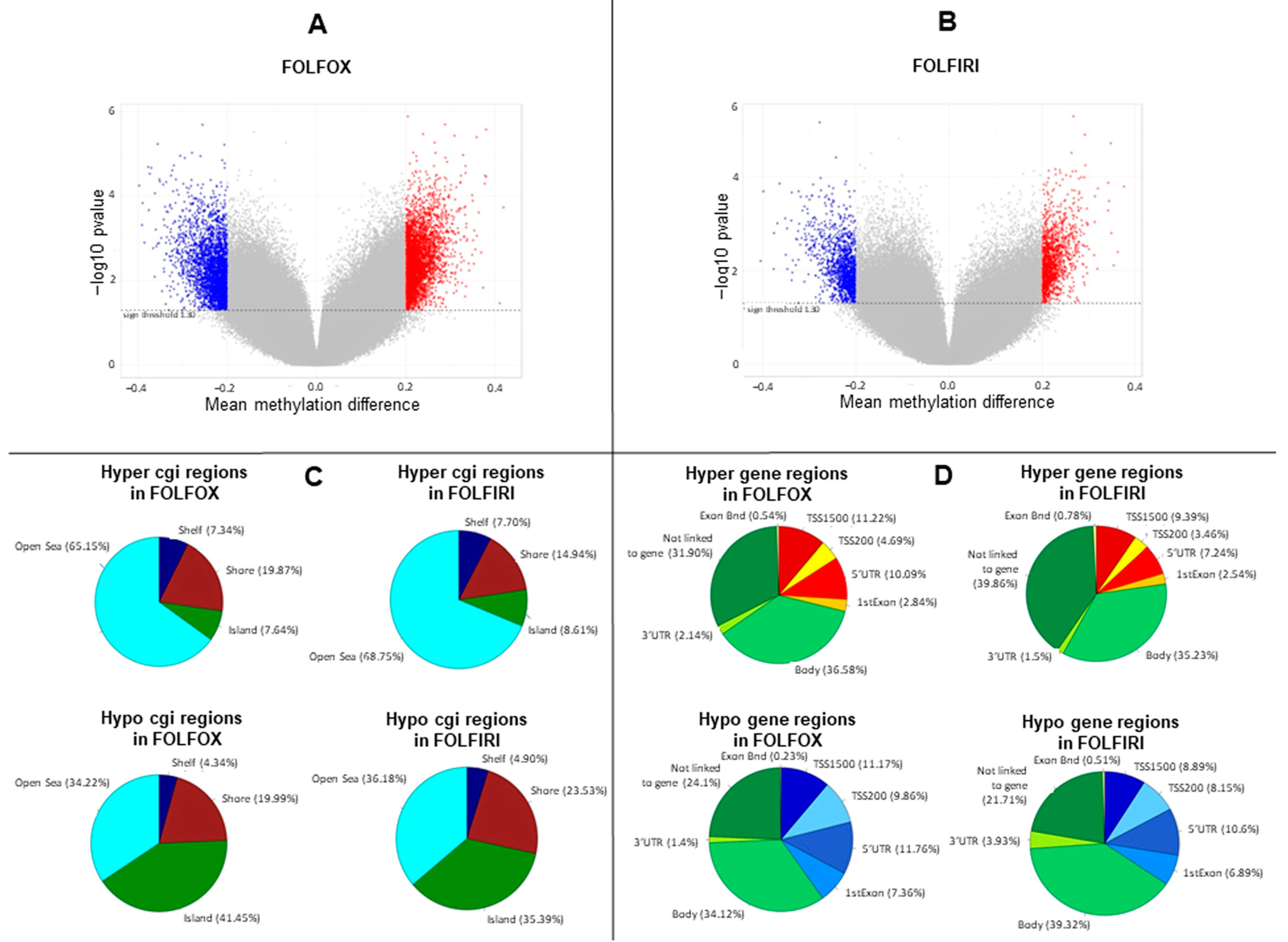
2.1. DNA Methylation Profile Is Remodeled in Primary-Resistant mCRCs
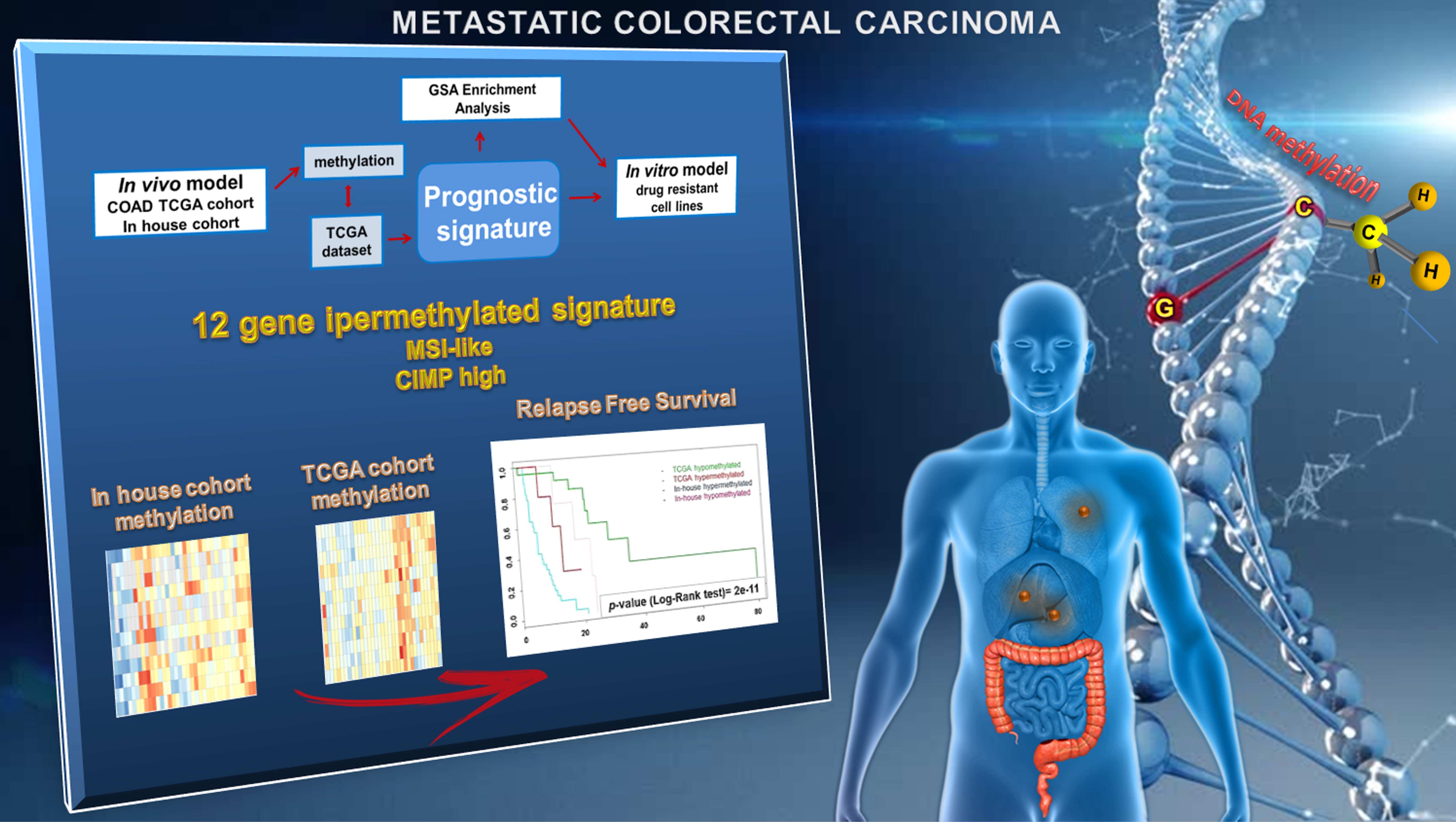
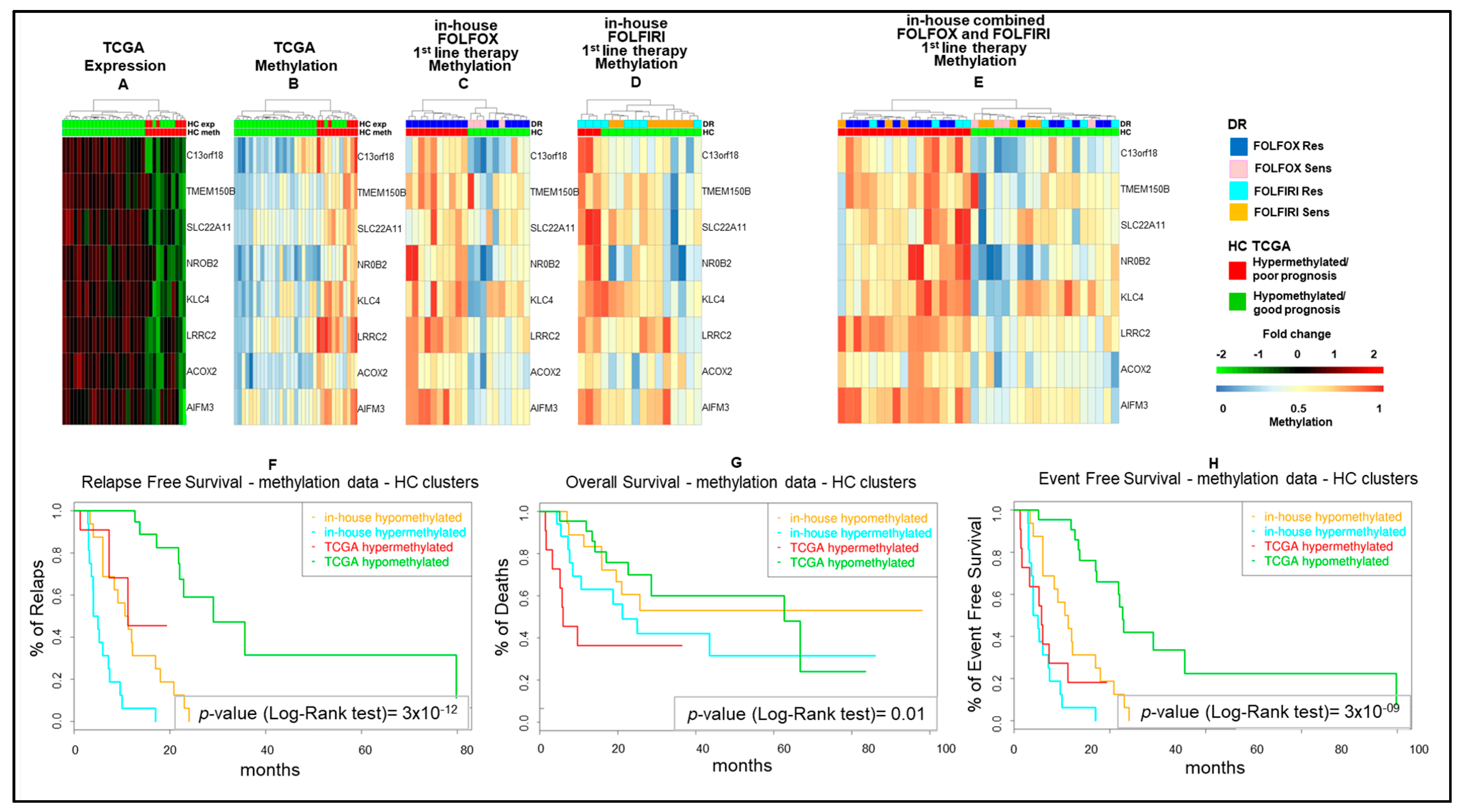
2.2. Epigenetic Alterations Predict Prognosis in Human mCRCs
2.3. Differential Epigenetic Alterations Obtained According to the Eight-Gene Signature Are Similar in in-House, TCGA, and GSE48684 Datasets
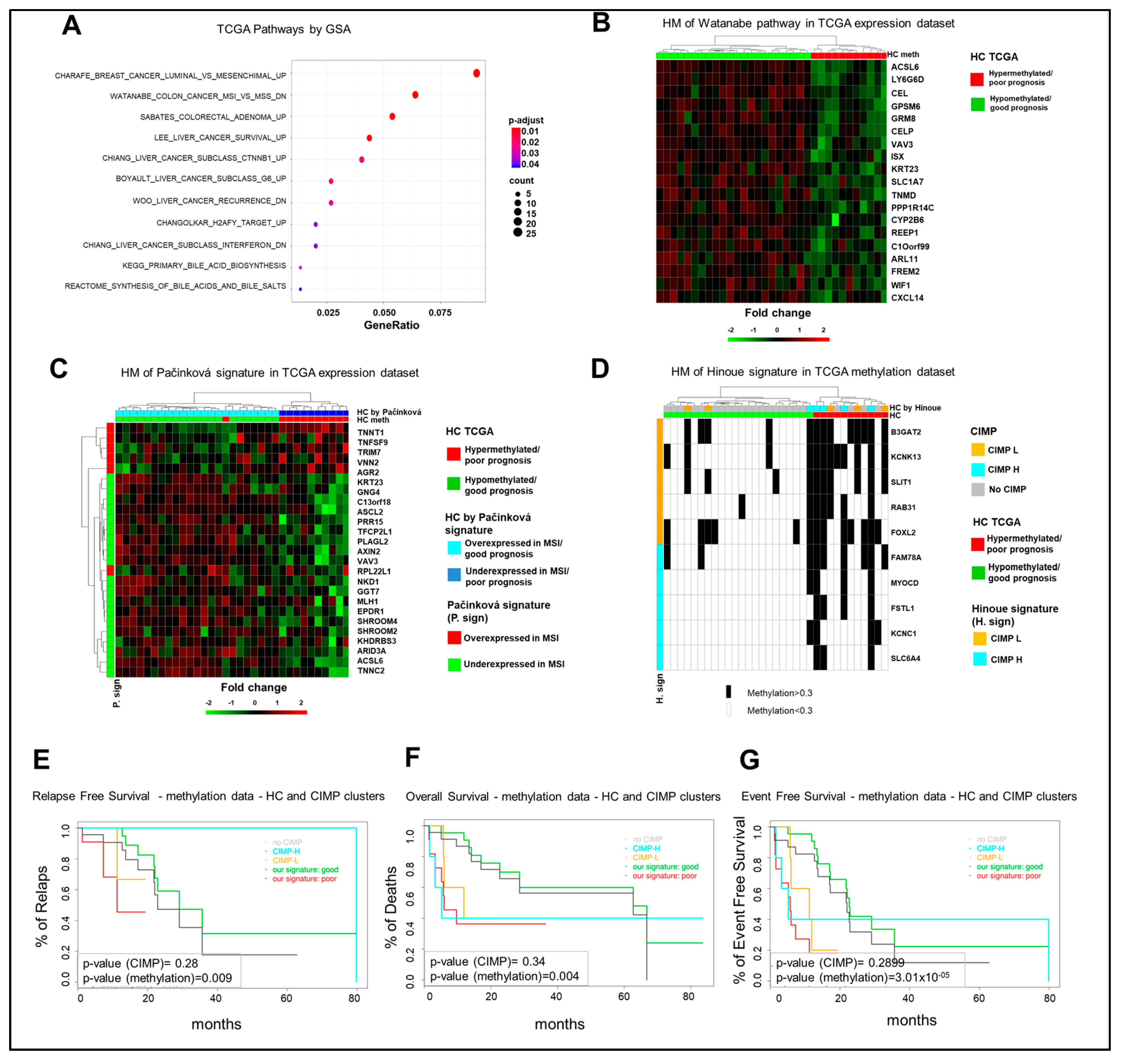
2.4. The Poor Prognosis Hypermethylated Cluster Is Characterized by an MSI-Like Phenotype and Is Enriched of CIMP-High Tumors
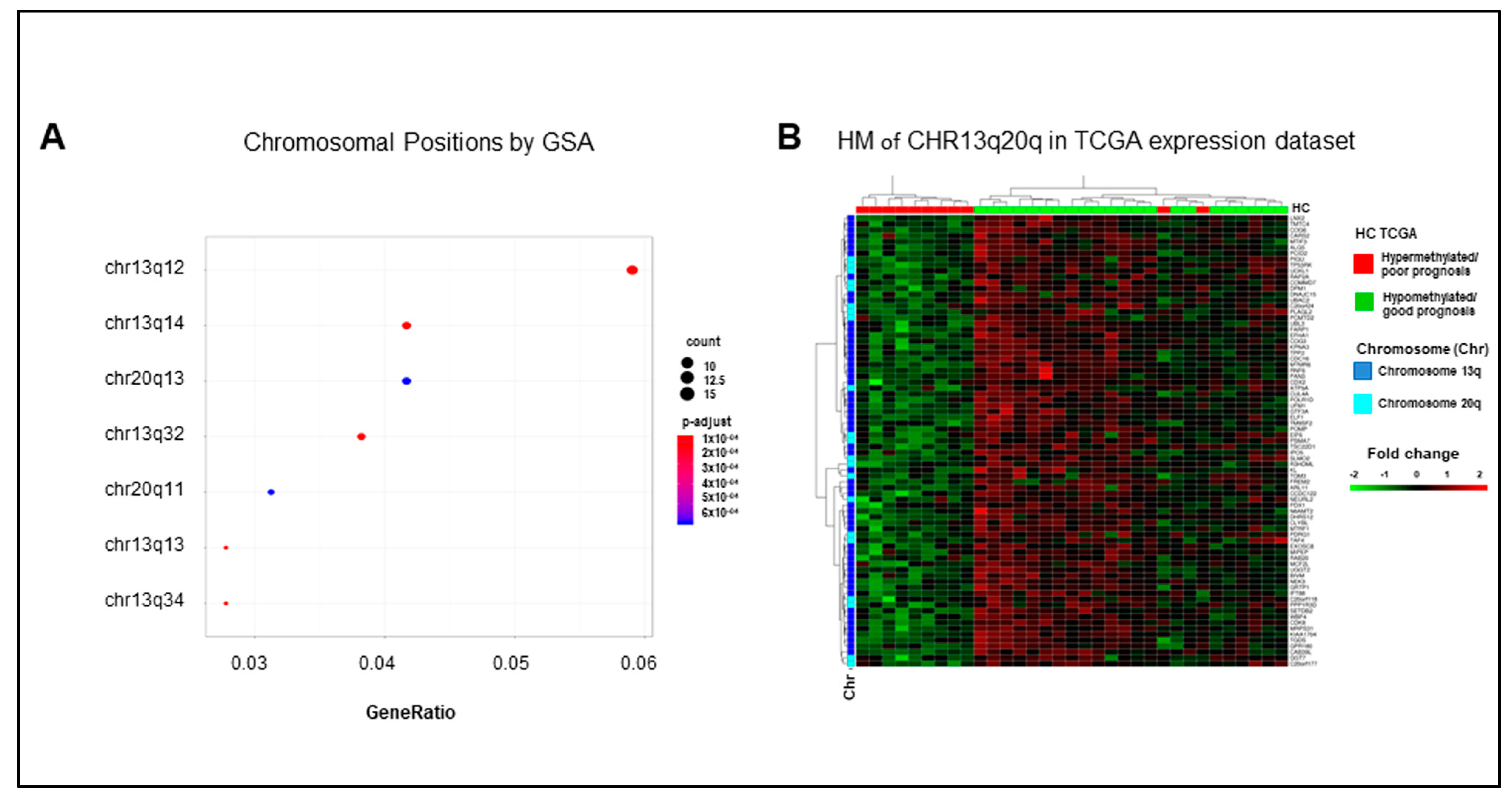
2.5. Hypermethylated Genes Are Enriched on Arms q of Chromosomes 13 and 20
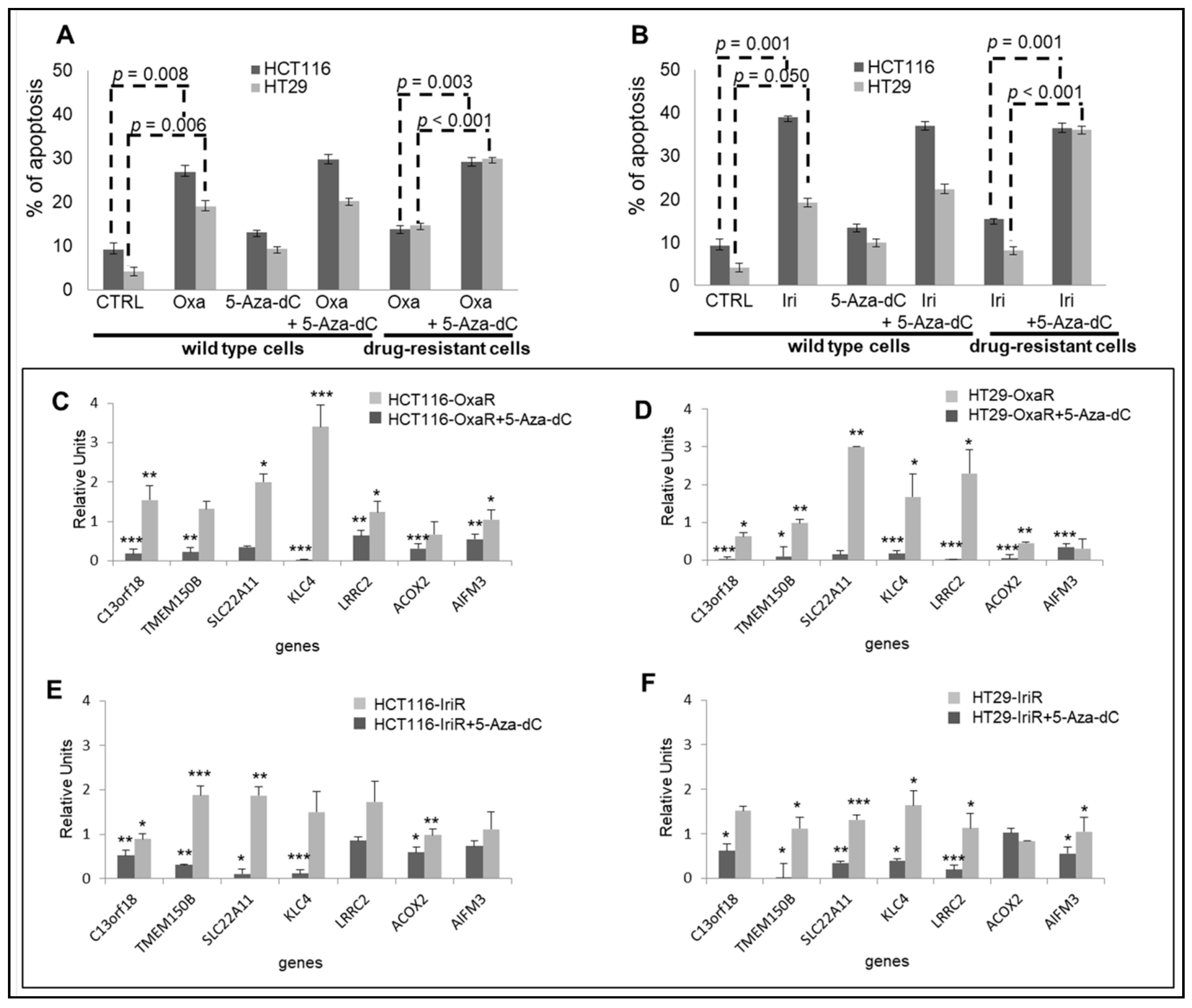
2.6. Epigenetic Modifications Are Reproduced in Drug-Resistant Cell Models
3. Discussion
4. Materials and Methods
4.1. Patients and Samples Collection
4.2. Cell Lines and In Vitro Drug-Resistant Models
4.3. Array-Based DNA Methylation Profiling
4.4. Bioinformatics Analysis
4.5. RNA Extraction and Real-Time RT-PCR Analysis
4.6. Apoptosis Assay
4.7. Data Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Màrmol, L.; Sánchez-de-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M.J. Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef] [Green Version]
- Chakedis, J.; Schmidt, C.R. Surgical Treatment of Metastatic Colorectal Cancer. Surg. Oncol. Clin. N. Am. 2018, 27, 377–399. [Google Scholar] [CrossRef]
- Elez, E.; Argilés, G.; Tabernero, J. First-Line Treatment of Metastatic Colorectal Cancer: Interpreting FIRE-3, PEAK, and CALGB/SWOG 80405. Curr. Treat Options Oncol. 2015, 16, 52. [Google Scholar] [CrossRef] [PubMed]
- Loupakis, F.; Depetris, I.; Biason, P.; Intini, R.; Prete, A.A.; Leone, F.; Lombardi, P.; Filippi, R.; Spallanzani, A.; Casciu, S.; et al. Prediction of Benefit from Checkpoint Inhibitors in Mismatch Repair Deficient Metastatic Colorectal Cancer: Role of Tumor Infiltrating Lymphocytes. Oncologist 2020, 25, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Park, J.W.; Han, J.W. Targeting epigenetics for cancer therapy. Arch. Pharm. Res. 2019, 42, 159–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mummaneni, P.; Shord, S.S. Epigenetics and oncology. Pharmacotherapy 2014, 34, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Vacante, M.; Borzì, A.M.; Basile, F.; Biondi, A. Biomarkers in colorectal cancer: Current clinical utility and future perspectives. World J. Clin. Cases 2018, 6, 869–881. [Google Scholar] [CrossRef]
- Jones, P.A.; Issa, J.P.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet 2016, 17, 630–641. [Google Scholar] [CrossRef]
- Condelli, V.; Crispo, F.; Pietrafes, M.; Lettini, G.; Matassa, D.S.; Esposito, F.; Landriscina, M.; Maddalena, F. HSP90 Molecular Chaperones, Metabolic Rewiring, and Epigenetics: Impact on Tumor Progression and Perspective for Anticancer Therapy. Cells 2019, 8, 532. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Lay, F.; Han, H.; Jones, P.A. Targeting DNA methylation for epigenetic therapy. Trends Pharmacol. Sci. 2010, 31, 536–546. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A. At the tipping point for epigenetic therapies in cancer. J. Clin. Investig. 2014, 124, 14–16. [Google Scholar] [CrossRef] [Green Version]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyn, H.; Esteller, M. DNA methylation profiling in the clinic: Applications and challenges. Nat. Rev. Genet. 2012, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Glasspool, R.M.; Teodoridis, J.M.; Brown, R. Epigenetics as a mechanism driving polygenic clinical drug resistance. Br. J. Cancer 2006, 94, 1087–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasan, H.S.; Gibbs, P.; Sharma, N.K.; Taieb, J.; Heinemann, V.; Ricke, J.; Peeters, M.; Findlay, M.; Weaver, A.; Mills, J.; et al. First-line selective internal radiotherapy plus chemotherapy versus chemotherapy alone in patients with liver metastases from colorectal cancer (FOXFIRE, SIRFLOX, and FOXFIRE-Global): A combined analysis of three multicentre, randomised, phase 3 trials. Lancet Oncol. 2017, 18, 1159–1171. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Haikova, P.; Ecker, J.T. Dynamic DNA methylation: In the right place at the right time. Science 2018, 361, 1336–1340. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Kobunai, T.; Toda, E.; Yamamoto, Y.; Kanazawa, T.; Kazama, Y.; Tanaka, J.; Tanaka, T.; Konishi, T.; Sugimoto, Y.; et al. Distal colorectal cancers with microsatellite instability (MSI) display distinct gene expression profiles that are different from proximal MSI cancers. Cancer Res. 2006, 66, 9804–9808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pačínková, A.; Popovici, V. Cross-platform Data Analysis Reveals a Generic Gene Expression Signature for Microsatellite Instability in Colorectal Cancer. BioMed Res. Int. 2019, 2019, 6763596. [Google Scholar] [CrossRef] [Green Version]
- Advani, S.; Advani, P.; DeSantis, S.M.; Brown, D.; VonVille, H.M.; Lam, M.; Loree, J.M.; Sarshekeh, A.M.; Bressler, J.; Lopez, D.S.; et al. Clinical, Pathological, and Molecular Characteristics of CpG Island Methylator Phenotype in Colorectal Cancer: A Systematic Review and Meta-analysis. Transl. Oncol. 2018, 11, 1188–1201. [Google Scholar] [CrossRef]
- Hinoue, T.; Weisenberger, D.J.; Lange, C.P.; Shen, H.; Byun, H.-M.; Berg, D.V.D.; Malik, S.; Pan, F.; Noushmehr, H.; Van Dijk, C.M.; et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012, 22, 271–282. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nat. Cell Biol. 2008, 455, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Nikolsky, Y.; Sviridov, E.; Yao, J.; Dosymbekov, D.; Ustyansky, V.; Kaznacheev, V.; Dezso, Z.; Mulvey, L.; MacConaill, L.E.; Winckler, W.; et al. Genome-Wide Functional Synergy between Amplified and Mutated Genes in Human Breast Cancer. Cancer Res. 2008, 68, 9532–9540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leichsenring, J.; Koppelle, A.; Reinacher-Schick, A. Colorectal Cancer: Personalized Therapy. Gastrointest Tumors 2014, 1, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Zhang, W.; Lenz, H.-J. Molecular Landscape and Treatment Options for Patients with Metastatic Colorectal Cancer. Indian J. Surg. Oncol. 2017, 8, 580–590. [Google Scholar] [CrossRef]
- Ciombor, K.K.; Bekaii-Saab, T. A Comprehensive Review of Sequencing and Combination Strategies of Targeted Agents in Metastatic Colorectal Cancer. Oncologist 2018, 23, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Fang, F.; Cardenas, H.; Huang, H.; Jiang, G.; Perkins, S.M.; Zhang, C.; Keer, H.N.; Liu, Y.; Nephew, K.P.; Matei, D. Genomic and Epigenomic Signatures in Ovarian Cancer Associated with Resensitization to Platinum Drugs. Cancer Res. 2018, 78, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Miranda Furtado, C.L.; Dos Santos Luciano, M.C.; Silva Santos, R.D.; Furtado, G.P.; Moraes, M.O.; Pessoa, C. Epidrugs: Targeting epigenetic marks in cancer treatment. Epigenetics 2019, 14, 1164–1176. [Google Scholar] [CrossRef]
- Tse, J.W.; Jenkins, L.J.; Chionh, F.; Mariadason, J.M. Aberrant DNA Methylation in Colorectal Cancer: What Should We Target? Trends Cancer 2017, 3, 698–712. [Google Scholar] [CrossRef]
- Okugawa, Y.; Grady, W.M.; Goel, A. Epigenetic Alterations in Colorectal Cancer: Emerging Biomarkers. Gastroenterology 2015, 149, 1204–1225.e12. [Google Scholar] [CrossRef] [Green Version]
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer—The stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, Y.; Zumwalt, T.J.; Goel, A. DNA methylation patterns as noninvasive biomarkers and targets of epigenetic therapies in colorectal cancer. Epigenomics 2016, 8, 685–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, M.; Gao, X.; Zhang, Y.; Hoffmeister, M.; Brenner, H. Different definitions of CpG island methylator phenotype and outcomes of colorectal cancer: A systematic review. Clin. Epigenet. 2016, 8, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.-H.; Lin, L.-I.; Tseng, L.-H.; Lin, Y.-L.; Liau, J.-Y.; Tsai, J.-H.; Liang, J.-T.; Lin, B.-R.; Cheng, A.-L.; Yeh, K.-H. CpG Island Methylator Phenotype May Predict Poor Overall Survival of Patients with Stage IV Colorectal Cancer. Oncology 2019, 96, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Lee, K.-D.; Pai, M.-Y.; Chu, P.-Y.; Hsu, C.-C.; Chiu, C.-C.; Chen, L.-T.; Chang, J.-Y.; Hsiao, S.-H.; Leu, Y.-W. Changes in DNA methylation are associated with the development of drug resistance in cervical cancer cells. Cancer Cell Int. 2015, 15, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, T.; Li, H.-T.; Wei, J.; Li, M.; Hsieh, T.-C.; Lu, Y.-T.; Lakshminarasimhan, R.; Xu, R.; Hodara, E.; Morrison, G.; et al. Epigenetic plasticity potentiates a rapid cyclical shift to and from an aggressive cancer phenotype. Int. J. Cancer 2020, 146, 3065–3076. [Google Scholar] [CrossRef]
- Testa, U.; Pelosi, E.; Castelli, G. Colorectal Cancer: Genetic Abnormalities, Tumor Progression, Tumor Heterogeneity, Clonal Evolution and Tumor-Initiating Cells. Med. Sci. 2018, 6, 31. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, H.; Zaanan, A.; Sinicrope, F.A. Microsatellite Instability Testing and Its Role in the Management of Colorectal Cancer. Curr. Treat. Options Oncol. 2015, 16, 30. [Google Scholar] [CrossRef]
- Zeinalian, M.; Hashemzadeh-Chaleshtori, M.; Salehi, R.; Emami, M. Clinical Aspects of Microsatellite Instability Testing in Colorectal Cancer. Adv. Biomed. Res. 2018, 7, 28. [Google Scholar] [CrossRef]
- Battaglin, F.; Naseem, M.; Lenz, H.-J.; Salem, M.E. Microsatellite Instability in Colorectal Cancer: Overview of Its Clinical Significance and Novel Perspectives. Clin. Adv. Hematol. Oncol. 2018, 16, 735–745. [Google Scholar]
- Hu, W.; Yang, Y.; Qi, L.; Chen, J.; Ge, W.; Zheng, S. Subtyping of microsatellite instability-high colorectal cancer. Cell Commun. Signal. 2019, 17, 79. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Li, F.; Zhou, X.; Ma, Y.; Fu, W. Is microsatellite instability-high really a favorable prognostic factor for advanced colorectal cancer? A meta-analysis. World J. Surg. Oncol. 2019, 17, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Freeman, G.J. The Microsatellite Instable Subset of Colorectal Cancer Is a Particularly Good Candidate for Checkpoint Blockade Immunotherapy. Cancer Discov. 2015, 5, 16–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, S.; Roepman, P.; Popovici, V.; Michaut, M.; Majewski, I.; Salazar, R.; Santos, C.; Rosenberg, R.; Nitsche, U.; Mesker, W.E.; et al. A robust genomic signature for the detection of colorectal cancer patients with microsatellite instability phenotype and high mutation frequency. J. Pathol. 2012, 228, 586–595. [Google Scholar] [CrossRef] [Green Version]
- Visone, R.; Bacalini, M.G.; Di Franco, S.; Ferracin, M.; Colorito, M.L.; Pagotto, S.; Laprovitera, N.; Licastro, D.; Di Marco, M.; Scavo, E.; et al. DNA methylation of shelf, shore and open sea CpG positions distinguish high microsatellite instability from low or stable microsatellite status colon cancer stem cells. Epigenomics 2019, 11, 587–604. [Google Scholar] [CrossRef] [Green Version]
- Huisman, C.; Wisman, G.B.A.; Kazemier, H.G.; Van Vugt, M.A.; Van Der Zee, A.G.J.; Schuuring, E.; Rots, M.G. Functional validation of putative tumor suppressor gene C13ORF18 in cervical cancer by Artificial Transcription Factors. Mol. Oncol. 2013, 7, 669–679. [Google Scholar] [CrossRef]
- Liu, H.; Brannon, A.R.; Reddy, A.; Alexe, G.; Seiler, M.W.; Arreola, A.; Oza, J.H.; Yao, M.; Juan, D.; Liou, L.S.; et al. Identifying mRNA targets of microRNA dysregulated in cancer: With application to clear cell Renal Cell Carcinoma. BMC Syst. Biol. 2010, 4, 51. [Google Scholar] [CrossRef] [Green Version]
- Hermsen, M.; Postma, C.; Baak, J.; Weiss, M.; Rapallo, A.; Sciutto, A.; Roemen, G.; Arends, J.; Williams, R.; Giaretti, W.; et al. Colorectal adenoma to carcinoma progression follows multiple pathways of chromosomal instability. Gastroenterology 2002, 123, 1109–1119. [Google Scholar] [CrossRef] [Green Version]
- Todoerti, K.; Calice, G.; Trino, S.; Simeon, V.; Lionetti, M.; Manzoni, M.; Fabris, S.; Barbieri, M.; Pompa, A.; Baldini, L.; et al. Global methylation patterns in primary plasma cell leukemia. Leuk. Res. 2018, 73, 95–102. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, Y.; Zhang, B. Efficient Test and Visualization of Multi-Set Intersections. Sci. Rep. 2015, 5, 16923. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.; Han, Y.; He, Q. ClusterProfier: An R package for comparing biological themes among gene cluster. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Condelli, V.; Calice, G.; Cassano, A.; Basso, M.; Rodriquenz, M.G.; Zupa, A.; Maddalena, F.; Crispo, F.; Pietrafesa, M.; Aieta, M.; et al. Novel Epigenetic Eight-Gene Signature Predictive of Poor Prognosis and MSI-Like Phenotype in Human Metastatic Colorectal Carcinomas. Cancers 2021, 13, 158. https://doi.org/10.3390/cancers13010158
Condelli V, Calice G, Cassano A, Basso M, Rodriquenz MG, Zupa A, Maddalena F, Crispo F, Pietrafesa M, Aieta M, et al. Novel Epigenetic Eight-Gene Signature Predictive of Poor Prognosis and MSI-Like Phenotype in Human Metastatic Colorectal Carcinomas. Cancers. 2021; 13(1):158. https://doi.org/10.3390/cancers13010158
Chicago/Turabian StyleCondelli, Valentina, Giovanni Calice, Alessandra Cassano, Michele Basso, Maria Grazia Rodriquenz, Angela Zupa, Francesca Maddalena, Fabiana Crispo, Michele Pietrafesa, Michele Aieta, and et al. 2021. "Novel Epigenetic Eight-Gene Signature Predictive of Poor Prognosis and MSI-Like Phenotype in Human Metastatic Colorectal Carcinomas" Cancers 13, no. 1: 158. https://doi.org/10.3390/cancers13010158
APA StyleCondelli, V., Calice, G., Cassano, A., Basso, M., Rodriquenz, M. G., Zupa, A., Maddalena, F., Crispo, F., Pietrafesa, M., Aieta, M., Sgambato, A., Tortora, G., Zoppoli, P., & Landriscina, M. (2021). Novel Epigenetic Eight-Gene Signature Predictive of Poor Prognosis and MSI-Like Phenotype in Human Metastatic Colorectal Carcinomas. Cancers, 13(1), 158. https://doi.org/10.3390/cancers13010158