BCL2L10 Is Overexpressed in Melanoma Downstream of STAT3 and Promotes Cisplatin and ABT-737 Resistance

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
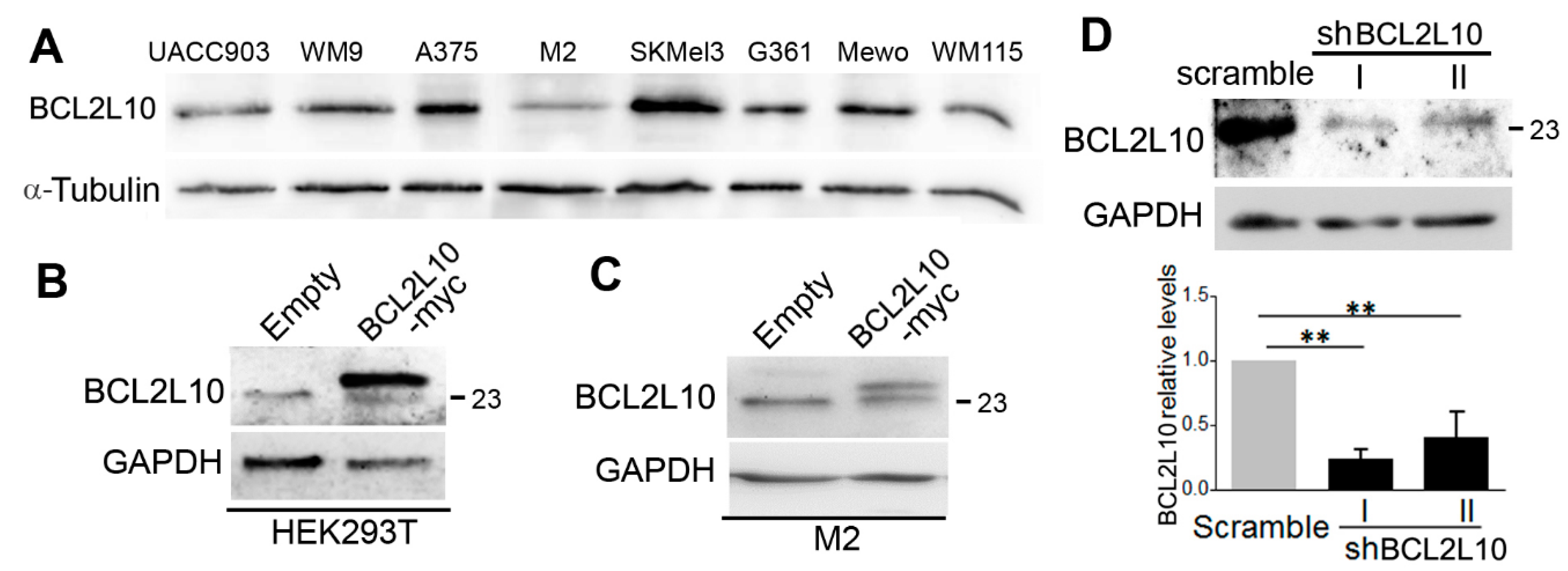
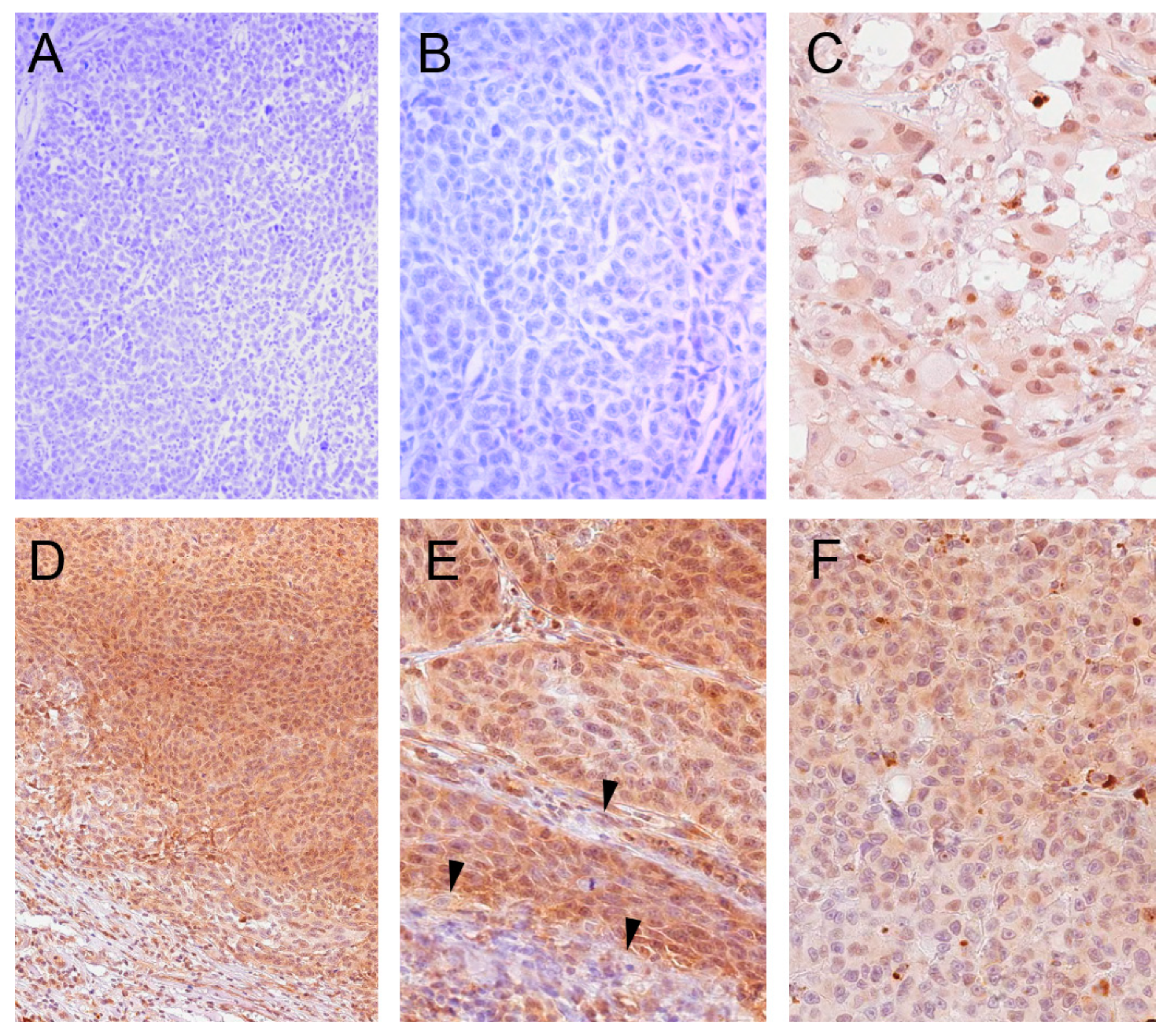
2.1. BCL2L10 Expression in Melanoma
2.2. STAT3 Regulates BCL2L10 Expression
2.3. STAT3 Binds the Human BCL2L10 Promoter and Activates BCL2L10 Transcription
2.4. BCL2L10 Contributes to Melanoma Cell Survival
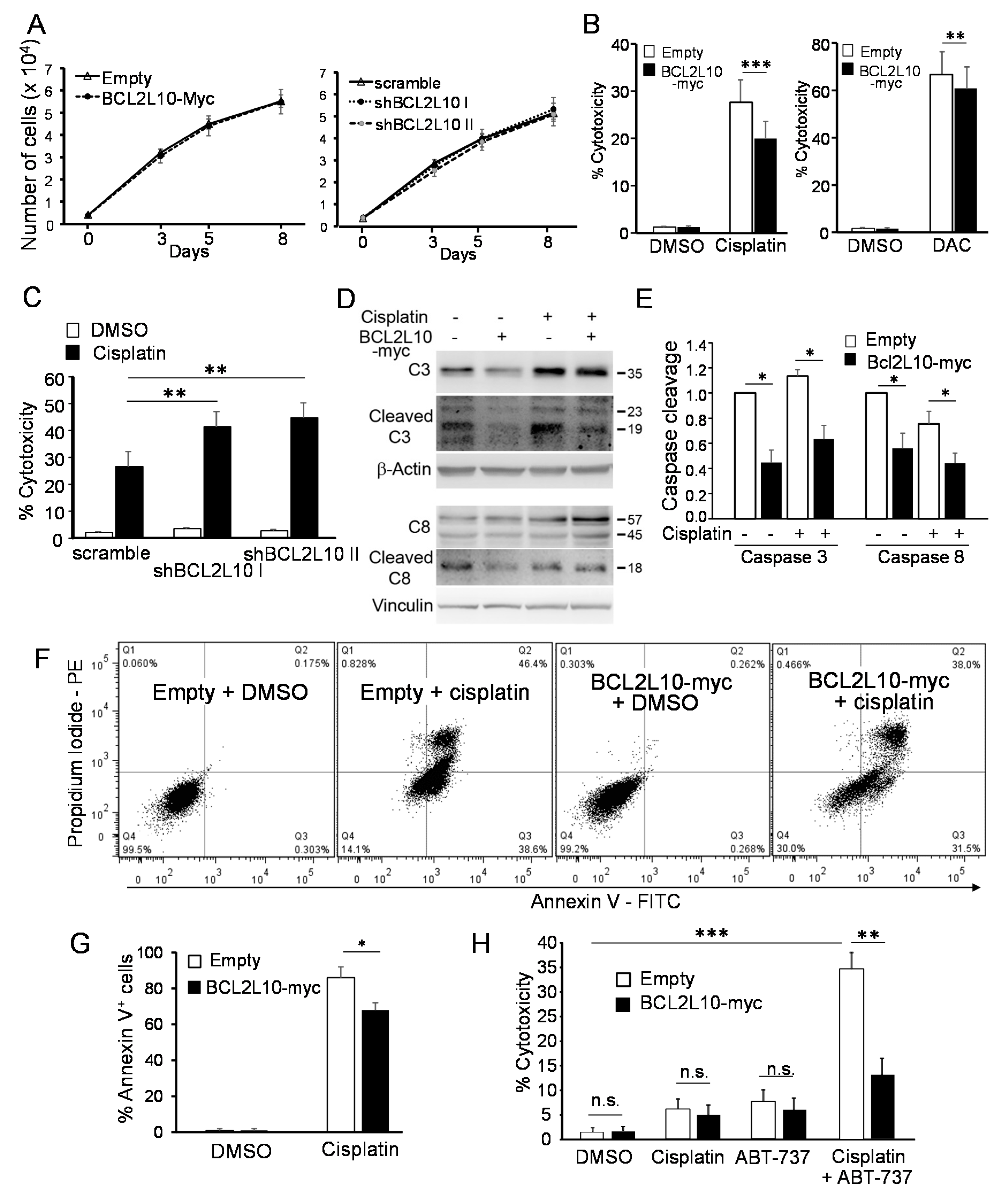
2.4.1. BCL2L10 Enhances the Survival of Melanoma Cells Treated with DNA-Damaging Agents
2.4.2. BCL2L10 Promotes Resistance to ABT-737
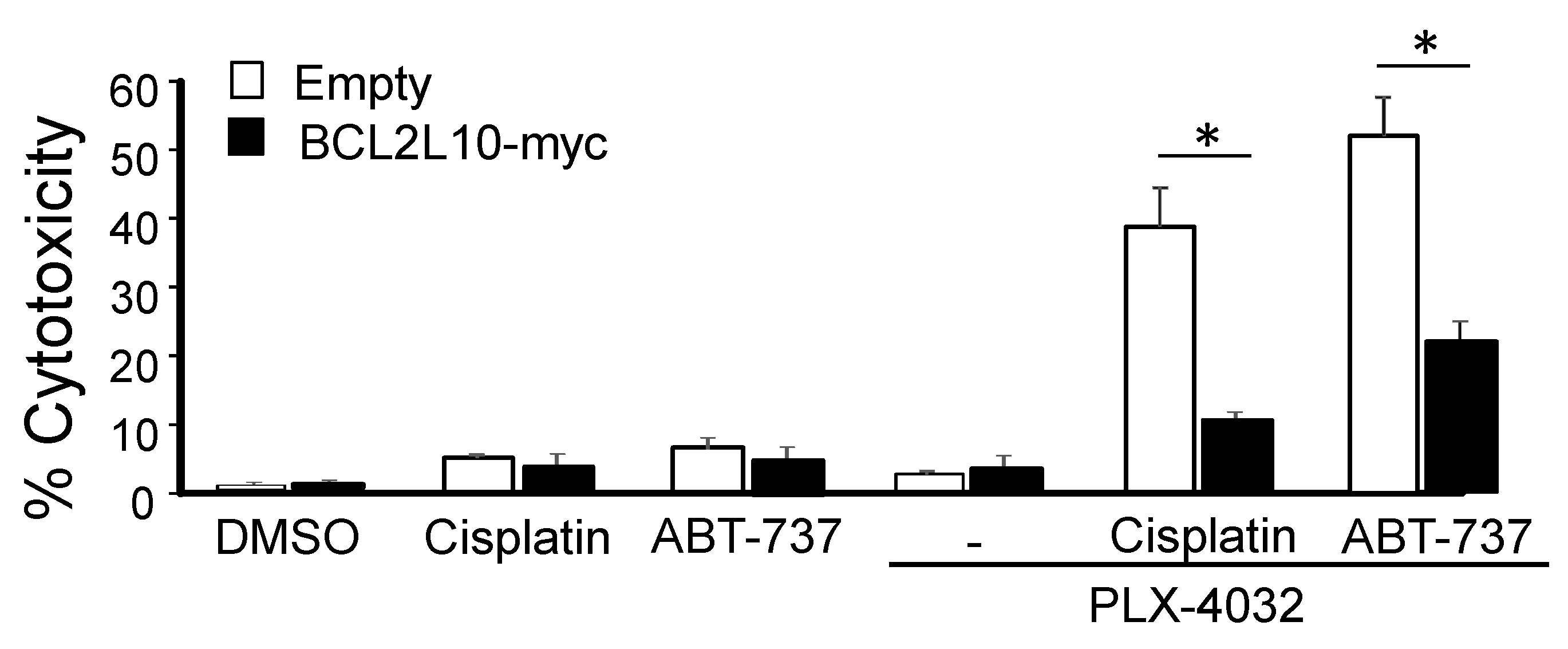
2.4.3. BCL2L10 Enhances the Survival of Melanoma Cells Treated with a Combination of PLX-4032 and cisplatin or ABT-737
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmids and Viral Constructs
4.3. Real-Time PCR
4.4. Chromatin Immunoprecipitation (ChIP)
4.5. Luciferase Assays
4.6. Proliferation Assays
4.7. Immunohistochemistry
4.8. Crystal Violet Cytotoxicity Assay
4.9. Quantification of Apoptotic Cell Death
4.10. Western Blotting
4.11. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| STAT3 | Signal transducer and activator of transcription 3 |
| BCL2 | B-cell lymphoma 2 |
| BCL2L10 | BCL2 Like 10 |
| SRE | STAT3 responsive elements |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| PI3K | Phosphoinositide-3-kinase |
| shRNA | Short hairpin RNA |
| RNPII | RNA polymerase II |
| MOMP | Mitochondrial outer membrane permeability |
| IHC | Immunohistochemistry |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA A Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Bergami, P.; Ronai, Z. Requirements for PKC-augmented JNK activation by MKK4/7. Int. J. Biochem. Cell Biol. 2008, 40, 1055–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paluncic, J.; Kovacevic, Z.; Jansson, P.J.; Kalinowski, D.; Merlot, A.M.; Huang, M.L.-H.; Lok, H.C.; Sahni, S.; Lane, D.J.R.; Richardson, D.R. Roads to melanoma: Key pathways and emerging players in melanoma progression and oncogenic signaling. Biochim. Biophys. Acta BBA Mol. Cell Res. 2016, 1863, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.-Y.; Lopez-Bergami, P.; Goydos, J.S.; Yip, D.; Walker, A.M.; Pasquale, E.B.; Ethell, I.M. The EphB4 receptor promotes the growth of melanoma cells expressing the ephrin-B2 ligand. Pigment. Cell Melanoma Res. 2010, 23, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Bergami, P.; Fitchman, B.; Ronai, Z. Understanding signaling cascades in melanoma. Photochem. Photobiol. 2008, 84, 289–306. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, S.; Tykodi, S.S.; Thompson, J.A. Treatment of metastatic melanoma: An overview. Oncology 2009, 23, 488–496. [Google Scholar]
- Lee, C.; Collichio, F.; Ollila, D.; Moschos, S. Historical review of melanoma treatment and outcomes. Clin. Dermatol. 2013, 31, 141–147. [Google Scholar] [CrossRef]
- Soengas, M.S.; Lowe, S.W. Apoptosis and melanoma chemoresistance. Oncogene 2003, 22, 3138–3151. [Google Scholar] [CrossRef] [Green Version]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Shamas-Din, A.; Kale, J.; Leber, B.; Andrews, D.W. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008714. [Google Scholar] [CrossRef] [Green Version]
- D’Aguanno, S.; Del Bufalo, D. Inhibition of Anti-Apoptotic Bcl-2 Proteins in Preclinical and Clinical Studies: Current Overview in Cancer. Cells 2020, 9, 1287. [Google Scholar] [CrossRef] [PubMed]
- Ke, N.; Godzik, A.; Reed, J.C. Bcl-B, a novel Bcl-2 family member that differentially binds and regulates Bax and Bak. J. Biol. Chem. 2001, 276, 12481–12484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, D.; Ke, N.; Zhang, H.; Ladror, U.; Joseph, M.; Eichinger, A.; Godzik, A.; Ng, S.-C.; Reed, J.C. Characterization of the anti-apoptotic mechanism of Bcl-B. Biochem. J. 2003, 376, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikata, R.; Fukai, K.; Imazeki, F.; Arai, M.; Fujiwara, K.; Yonemitsu, Y.; Zhang, K.; Nabeya, Y.; Ochiai, T.; Yokosuka, O. BCL2L10 is frequently silenced by promoter hypermethylation in gastric cancer. Oncol. Rep. 2010, 23, 1701–1708. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Wang, J.; Han, J.; Xie, X.-L.; Ji, C.-G.; Yin, J.; Chen, L.; Wang, C.-K.; Jiang, X.-Y.; Qi, W.; et al. BCL2L10 inhibits growth and metastasis of hepatocellular carcinoma both in vitro and in vivo. Mol. Carcinog. 2017, 56, 1137–1149. [Google Scholar] [CrossRef]
- Lee, R.; Chen, J.; Matthews, C.P.; McDougall, J.K.; Neiman, P.E. Characterization of NR13-related human cell death regulator, Boo/Diva, in normal and cancer tissues. Biochim. Biophys. Acta 2001, 1520, 187–194. [Google Scholar] [CrossRef]
- Liu, X.; Hu, X.; Kuang, Y.; Yan, P.; Li, L.; Li, C.; Tao, Q.; Cai, X. BCLB, methylated in hepatocellular carcinoma, is a starvation stress sensor that induces apoptosis and autophagy through the AMPK-mTOR signaling cascade. Cancer Lett. 2017, 395, 63–71. [Google Scholar] [CrossRef]
- Xu, J.D.; Cao, X.X.; Long, Z.W.; Liu, X.P.; Furuya, T.; Xu, J.W.; Liu, X.L.; De Xu, Z.; Sasaki, K.; Li, Q.Q. BCL2L10 protein regulates apoptosis/proliferation through differential pathways in gastric cancer cells. J. Pathol. 2011, 223, 400–409. [Google Scholar] [CrossRef]
- Inohara, N.; Gourley, T.S.; Carrio, R.; Muñiz, M.; Merino, J.; Garcia, I.; Koseki, T.; Hu, Y.; Chen, S.; Núñez, G. Diva, a Bcl-2 Homologue that Binds Directly to Apaf-1 and Induces BH3-independent Cell Death. J. Biol. Chem. 1998, 273, 32479–32486. [Google Scholar] [CrossRef] [Green Version]
- Aouacheria, A.; Arnaud, E.; Venet, S.; Lalle, P.; Gouy, M.; Rigal, D.; Gillet, G. Nrh, a human homologue of Nr-13 associates with Bcl-Xs and is an inhibitor of apoptosis. Oncogene 2001, 20, 5846–5855. [Google Scholar] [CrossRef] [Green Version]
- Song, Q. Boo, a novel negative regulator of cell death, interacts with Apaf-1. EMBO J. 1999, 18, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Holzgreve, W.; De Geyter, C. BCL2-L-10, a novel anti-apoptotic member of the Bcl-2 family, blocks apoptosis in the mitochondria death pathway but not in the death receptor pathway. Hum. Mol. Genet. 2001, 10, 2329–2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-Y.; Kwon, J.; Woo, J.H.; Kim, K.-H.; Lee, K.-A. BCL2L10 mediates the proliferation, invasion and migration of ovarian cancer cells. Int. J. Oncol. 2020, 56, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Mikata, R.; Yokosuka, O.; Fukai, K.; Imazeki, F.; Arai, M.; Tada, M.; Kurihara, T.; Zhang, K.; Kanda, T.; Saisho, H. Analysis of genes upregulated by the demethylating agent 5-aza-2′-deoxycytidine in gastric cancer cell lines. Int. J. Cancer 2006, 119, 1616–1622. [Google Scholar] [CrossRef]
- Fabiani, E.; Leone, G.; Giachelia, M.; D’alo’, F.; Greco, M.; Criscuolo, M.; Guidi, F.; Rutella, S.; Hohaus, S.; Teresa voso, M. Analysis of genome-wide methylation and gene expression induced by 5-aza-2′-deoxycytidine identifies BCL2L10 as a frequent methylation target in acute myeloid leukemia. Leuk. Lymphoma 2010, 51, 2275–2284. [Google Scholar] [CrossRef]
- Xu, J.D.; Furuya, T.; Cao, X.X.; Liu, X.L.; Li, Q.Q.; Wang, W.J.; Xu, J.W.; Xu, Z.D.; Sasaki, K.; Liu, X.P. Loss of BCL2L10 protein expression as prognostic predictor for poor clinical outcome in gastric carcinoma. Histopathology 2010, 57, 814–824. [Google Scholar] [CrossRef]
- Krajewska, M.; Kitada, S.; Winter, J.N.; Variakojis, D.; Lichtenstein, A.; Zhai, D.; Cuddy, M.; Huang, X.; Luciano, F.; Baker, C.H.; et al. Bcl-B expression in human epithelial and nonepithelial malignancies. Clin. Cancer Res. 2008, 14, 3011–3021. [Google Scholar] [CrossRef] [Green Version]
- Hamouda, M.-A.; Jacquel, A.; Robert, G.; Puissant, A.; Richez, V.; Cassel, R.; Fenouille, N.; Roulland, S.; Gilleron, J.; Griessinger, E.; et al. BCL-B (BCL2L10) is overexpressed in patients suffering from multiple myeloma (MM) and drives an MM-like disease in transgenic mice. J. Exp. Med. 2016, 213, 1705–1722. [Google Scholar] [CrossRef] [Green Version]
- Nougarede, A.; Popgeorgiev, N.; Kassem, L.; Omarjee, S.; Borel, S.; Mikaelian, I.; Lopez, J.; Gadet, R.; Marcillat, O.; Treilleux, I.; et al. Breast Cancer Targeting through Inhibition of the Endoplasmic Reticulum-Based Apoptosis Regulator Nrh/BCL2L10. Cancer Res. 2018, 78, 1404–1417. [Google Scholar] [CrossRef] [Green Version]
- Cluzeau, T.; Robert, G.; Mounier, N.; Karsenti, J.M.; Dufies, M.; Puissant, A.; Jacquel, A.; Renneville, A.; Preudhomme, C.; Cassuto, J.-P.; et al. BCL2L10 is a predictive factor for resistance to azacitidine in MDS and AML patients. Oncotarget 2012, 3, 490–501. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Hurley, D.G.; Watkins, W.; Araki, H.; Tamada, Y.; Muthukaruppan, A.; Ranjard, L.; Derkac, E.; Imoto, S.; Miyano, S.; et al. Cell cycle gene networks are associated with melanoma prognosis. PLoS ONE 2012, 7, e34247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, Y.M.; Kim, J.K.; Choi, Y.; Choi, S.; Yoo, J.-Y. Prediction and Experimental Validation of Novel STAT3 Target Genes in Human Cancer Cells. PLoS ONE 2009, 4, e6911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aslantürk, Ö.S. In Vitro Cytotoxicity and Cell Viability Assays: Principles, Advantages, and Disadvantages. In Genotoxicity—A Predictable Risk to Our Actual World; Larramendy, M.L., Soloneski, S., Eds.; InTech: London, UK, 2018; ISBN 978-1-78923-418-3. [Google Scholar]
- Martin, A.; Clynes, M. Comparison of 5 microplate colorimetric assays forin vitro cytotoxicity testing and cell proliferation assays. Cytotechnology 1993, 11, 49–58. [Google Scholar] [CrossRef]
- Papadimitriou, M.; Hatzidaki, E.; Papasotiriou, I. Linearity Comparison of Three Colorimetric Cytotoxicity Assays. JCT 2019, 10, 580–590. [Google Scholar] [CrossRef] [Green Version]
- Śliwka, L.; Wiktorska, K.; Suchocki, P.; Milczarek, M.; Mielczarek, S.; Lubelska, K.; Cierpiał, T.; Łyżwa, P.; Kiełbasiński, P.; Jaromin, A.; et al. The Comparison of MTT and CVS Assays for the Assessment of Anticancer Agent Interactions. PLoS ONE 2016, 11, e0155772. [Google Scholar] [CrossRef] [Green Version]
- Eichhorn, J.M.; Alford, S.E.; Sakurikar, N.; Chambers, T.C. Molecular analysis of functional redundancy among anti-apoptotic Bcl-2 proteins and its role in cancer cell survival. Exp. Cell Res. 2014, 322, 415–424. [Google Scholar] [CrossRef] [Green Version]
- Campbell, K.J.; Tait, S.W.G. Targeting BCL-2 regulated apoptosis in cancer. Open Biol. 2018, 8, 180002. [Google Scholar] [CrossRef]
- Carrington, E.M.; Zhan, Y.; Brady, J.L.; Zhang, J.-G.; Sutherland, R.M.; Anstee, N.S.; Schenk, R.L.; Vikstrom, I.B.; Delconte, R.B.; Segal, D.; et al. Anti-apoptotic proteins BCL-2, MCL-1 and A1 summate collectively to maintain survival of immune cell populations both in vitro and in vivo. Cell Death Differ. 2017, 24, 878–888. [Google Scholar] [CrossRef] [Green Version]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Zou, J.; Ardecky, R.; Pinkerton, A.B.; Sergienko, E.; Su, Y.; Stonich, D.; Curpan, R.F.; Simons, P.C.; Zhai, D.; Diaz, P.; et al. Selective Bcl-2 Inhibitor Probes. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information: Bethesda, MD, USA, 2010. [Google Scholar]
- Wacheck, V.; Losert, D.; Günsberg, P.; Vornlocher, H.-P.; Hadwiger, P.; Geick, A.; Pehamberger, H.; Müller, M.; Jansen, B. Small Interfering RNA Targeting Bcl-2 Sensitizes Malignant Melanoma. Oligonucleotides 2003, 13, 393–400. [Google Scholar] [CrossRef]
- Zupi, G. Antitumor Efficacy of bcl-2 and c-myc Antisense Oligonucleotides in Combination with Cisplatin in Human Melanoma Xenografts: Relevance of the Administration Sequence. Clin. Cancer Res. 2005, 11, 1990–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heere-Ress, E.; Thallinger, C.; Lucas, T.; Schlagbauer-Wadl, H.; Wacheck, V.; Monia, B.P.; Wolff, K.; Pehamberger, H.; Jansen, B. Bcl-XL is a chemoresistance factor in human melanoma cells that can be inhibited by antisense therapy. Int. J. Cancer 2002, 99, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Sinnberg, T.; Lasithiotakis, K.; Niessner, H.; Schittek, B.; Flaherty, K.T.; Kulms, D.; Maczey, E.; Campos, M.; Gogel, J.; Garbe, C.; et al. Inhibition of PI3K-AKT-mTOR Signaling Sensitizes Melanoma Cells to Cisplatin and Temozolomide. J. Investig. Dermatol. 2009, 129, 1500–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Nikolovska-Coleska, Z.; Yang, C.-Y.; Wang, R.; Tang, G.; Guo, J.; Shangary, S.; Qiu, S.; Gao, W.; Yang, D.; et al. Structure-Based Design of Potent Small-Molecule Inhibitors of Anti-Apoptotic Bcl-2 Proteins. J. Med. Chem. 2006, 49, 6139–6142. [Google Scholar] [CrossRef] [PubMed]
- Reuland, S.N.; Goldstein, N.B.; Partyka, K.A.; Smith, S.; Luo, Y.; Fujita, M.; Gonzalez, R.; Lewis, K.; Norris, D.A.; Shellman, Y.G. ABT-737 synergizes with Bortezomib to kill melanoma cells. Biol. Open 2012, 1, 92–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudin, C.M.; Hann, C.L.; Garon, E.B.; Ribeiro de Oliveira, M.; Bonomi, P.D.; Camidge, D.R.; Chu, Q.; Giaccone, G.; Khaira, D.; Ramalingam, S.S.; et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin. Cancer Res. 2012, 18, 3163–3169. [Google Scholar] [CrossRef] [Green Version]
- Mazumder, S.; Choudhary, G.S.; Al-Harbi, S.; Almasan, A. Mcl-1 Phosphorylation defines ABT-737 resistance that can be overcome by increased NOXA expression in leukemic B cells. Cancer Res. 2012, 72, 3069–3079. [Google Scholar] [CrossRef] [Green Version]
- Yecies, D.; Carlson, N.E.; Deng, J.; Letai, A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood 2010, 115, 3304–3313. [Google Scholar] [CrossRef] [Green Version]
- Geserick, P.; Wang, J.; Feoktistova, M.; Leverkus, M. The ratio of Mcl-1 and Noxa determines ABT737 resistance in squamous cell carcinoma of the skin. Cell Death Dis. 2014, 5, e1412. [Google Scholar] [CrossRef] [Green Version]
- Warren, C.F.A.; Wong-Brown, M.W.; Bowden, N.A. BCL-2 family isoforms in apoptosis and cancer. Cell Death Dis. 2019, 10, 177. [Google Scholar] [CrossRef] [Green Version]
- Eberle, J.; Hossini, A. Expression and Function of Bcl-2 Proteins in Melanoma. CG 2008, 9, 409–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, M.L.; Czyz, M. Anti-apoptotic proteins on guard of melanoma cell survival. Cancer Lett. 2013, 331, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Placzek, W.J.; Wei, J.; Kitada, S.; Zhai, D.; Reed, J.C.; Pellecchia, M. A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis. 2010, 1, e40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beverly, L.J.; Lockwood, W.W.; Shah, P.P.; Erdjument-Bromage, H.; Varmus, H. Ubiquitination, localization, and stability of an anti-apoptotic BCL2-like protein, BCL2L10/BCLb, are regulated by Ubiquilin1. Proc. Natl. Acad. Sci. USA 2012, 109, E119–E126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.; Garraway, L.A.; Fisher, D.E. Malignant melanoma: Genetics and therapeutics in the genomic era. Genes Dev. 2006, 20, 2149–2182. [Google Scholar] [CrossRef] [Green Version]
- Messina, J.L.; Yu, H.; Riker, A.I.; Munster, P.N.; Jove, R.L.; Daud, A.I. Activated stat-3 in melanoma. Cancer Control. 2008, 15, 196–201. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Fox, P.S.; Ferguson, S.D.; Bassett, R.; Kong, L.-Y.; Schacherer, C.W.; Gershenwald, J.E.; Grimm, E.A.; Fuller, G.N.; Heimberger, A.B. The expression of p-STAT3 in stage IV melanoma: Risk of CNS metastasis and survival. Oncotarget 2012, 3, 336–344. [Google Scholar] [CrossRef] [Green Version]
- Zushi, S.; Shinomura, Y.; Kiyohara, T.; Miyazaki, Y.; Kondo, S.; Sugimachi, M.; Higashimoto, Y.; Kanayama, S.; Matsuzawa, Y. STAT3 mediates the survival signal in oncogenic ras-transfected intestinal epithelial cells. Int. J. Cancer 1998, 78, 326–330. [Google Scholar] [CrossRef]
- Karni, R.; Jove, R.; Levitzki, A. Inhibition of pp60c-Src reduces Bcl-XL expression and reverses the transformed phenotype of cells overexpressing EGF and HER-2 receptors. Oncogene 1999, 18, 4654–4662. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Ma, Y.; Cole, S.M.; Zander, C.; Chen, K.-H.; Karras, J.; Pope, R.M. Serine phosphorylation of STAT3 is essential for Mcl-1 expression and macrophage survival. Blood 2003, 102, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, N.; Schwan, J.V.; Fujita, M.; Norris, D.A.; Shellman, Y.G. Alternative Treatments For Melanoma: Targeting BCL-2 Family Members to De-Bulk and Kill Cancer Stem Cells. J. Investig. Dermatol. 2015, 135, 2155–2161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rooswinkel, R.W.; van de Kooij, B.; Verheij, M.; Borst, J. Bcl-2 is a better ABT-737 target than Bcl-xL or Bcl-w and only Noxa overcomes resistance mediated by Mcl-1, Bfl-1, or Bcl-B. Cell Death Dis. 2012, 3, e366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aouacheria, A.; Brunet, F.; Gouy, M. Phylogenomics of life-or-death switches in multicellular animals: Bcl-2, BH3-Only, and BNip families of apoptotic regulators. Mol. Biol. Evol. 2005, 22, 2395–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillemin, Y.; Cornut-Thibaut, A.; Gillet, G.; Penin, F.; Aouacheria, A. Characterization of unique signature sequences in the divergent maternal protein BCL2L10. Mol. Biol. Evol. 2011, 28, 3271–3283. [Google Scholar] [CrossRef] [Green Version]
- Naumann, U.; Weit, S.; Wischhusen, J.; Weller, M. Diva/Boo is a negative regulator of cell death in human glioma cells. FEBS Lett. 2001, 505, 23–26. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Xie, X.-L.; Wang, M.-M.; Yin, J.; Tian, J.-M.; Jiang, X.-Y.; Zhang, D.; Han, J.; Bai, Y.; Cui, Z.-J.; et al. The role of the apoptosis-related protein BCL-B in the regulation of mitophagy in hepatic stellate cells during the regression of liver fibrosis. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Sale, M.J.; Cook, S.J. That which does not kill me makes me stronger; combining ERK1/2 pathway inhibitors and BH3 mimetics to kill tumour cells and prevent acquired resistance. Br. J. Pharmacol. 2013, 169, 1708–1722. [Google Scholar] [CrossRef] [Green Version]
- Cragg, M.S.; Jansen, E.S.; Cook, M.; Harris, C.; Strasser, A.; Scott, C.L. Treatment of B-RAF mutant human tumor cells with a MEK inhibitor requires Bim and is enhanced by a BH3 mimetic. J. Clin. Investig. 2008, 118, 3651–3659. [Google Scholar] [CrossRef] [Green Version]
- Wroblewski, D.; Mijatov, B.; Mohana-Kumaran, N.; Lai, F.; Gallagher, S.J.; Haass, N.K.; Zhang, X.D.; Hersey, P. The BH3-mimetic ABT-737 sensitizes human melanoma cells to apoptosis induced by selective BRAF inhibitors but does not reverse acquired resistance. Carcinogenesis 2013, 34, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Serasinghe, M.N.; Missert, D.J.; Asciolla, J.J.; Podgrabinska, S.; Wieder, S.Y.; Izadmehr, S.; Belbin, G.; Skobe, M.; Chipuk, J.E. Anti-apoptotic BCL-2 proteins govern cellular outcome following B-RAF(V600E) inhibition and can be targeted to reduce resistance. Oncogene 2015, 34, 857–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattila, K.E.; Vihinen, P.; Ramadan, S.; Skyttä, T.; Tiainen, L.; Vuoristo, M.-S.; Tyynelä-Korhonen, K.; Koivunen, J.; Kohtamäki, L.; Mäkelä, S.; et al. Combination chemotherapy with temozolomide, lomustine, vincristine and interferon-alpha (TOL-IFN) plus vemurafenib or TOL-IFN as first-line treatment for patients with advanced melanoma. Acta Oncol. 2020, 59, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Makino, E.; Gutmann, V.; Kosnopfel, C.; Niessner, H.; Forschner, A.; Garbe, C.; Sinnberg, T.; Schittek, B. Melanoma cells resistant towards MAPK inhibitors exhibit reduced TAp73 expression mediating enhanced sensitivity to platinum-based drugs. Cell Death Dis. 2018, 9, 930. [Google Scholar] [CrossRef]
- Bhatty, M.; Kato, S.; Piha-Paul, S.A.; Naing, A.; Subbiah, V.; Huang, H.J.; Karp, D.D.; Tsimberidou, A.M.; Zinner, R.G.; Hwu, W.; et al. Phase 1 study of the combination of vemurafenib, carboplatin, and paclitaxel in patients with BRAF -mutated melanoma and other advanced malignancies. Cancer 2019, 125, 463–472. [Google Scholar] [CrossRef]
- Simon, A.; Kourie, H.R.; Kerger, J. Is there still a role for cytotoxic chemotherapy after targeted therapy and immunotherapy in metastatic melanoma? A case report and literature review. Chin. J. Cancer 2017, 36, 10. [Google Scholar] [CrossRef] [Green Version]
- Borst, A.; Haferkamp, S.; Grimm, J.; Rösch, M.; Zhu, G.; Guo, S.; Li, C.; Gao, T.; Meierjohann, S.; Schrama, D.; et al. BIK is involved in BRAF/MEK inhibitor induced apoptosis in melanoma cell lines. Cancer Lett. 2017, 404, 70–78. [Google Scholar] [CrossRef]
- Sale, M.J.; Cook, S.J. The increase in BIK expression following ERK1/2 pathway inhibition is a consequence of G1 cell-cycle arrest and not a direct effect on BIK protein stability. Biochem. J. 2014, 459, 513–524. [Google Scholar] [CrossRef]
- Campos, L.S.; Rodriguez, Y.I.; Leopoldino, A.M.; Hait, N.C.; Lopez Bergami, P.; Castro, M.G.; Sanchez, E.S.; Maceyka, M.; Spiegel, S.; Alvarez, S.E. Filamin A Expression Negatively Regulates Sphingosine-1-Phosphate-Induced NF-κB Activation in Melanoma Cells by Inhibition of Akt Signaling. Mol. Cell. Biol. 2016, 36, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Picco, M.E.; Castro, M.V.; Quezada, M.J.; Barbero, G.; Villanueva, M.B.; Fernández, N.B.; Kim, H.; Lopez-Bergami, P. STAT3 enhances the constitutive activity of AGC kinases in melanoma by transactivating PDK1. Cell Biosci. 2019, 9, 3. [Google Scholar] [CrossRef]
- Guo, B.; Zhai, D.; Cabezas, E.; Welsh, K.; Nouraini, S.; Satterthwait, A.C.; Reed, J.C. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature 2003, 423, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, S.; Gazin, C.; Chamberlain, L.; Ou, J.; Zhu, X.; Tushir, J.S.; Virbasius, C.-M.; Lin, L.; Zhu, L.J.; Wajapeyee, N.; et al. TRIM37 is a new histone H2A ubiquitin ligase and breast cancer oncoprotein. Nature 2014, 516, 116–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbero, G.; Castro, M.V.; Villanueva, M.B.; Quezada, M.J.; Fernández, N.B.; DeMorrow, S.; Lopez-Bergami, P. An Autocrine Wnt5a Loop Promotes NF-κB Pathway Activation and Cytokine/Chemokine Secretion in Melanoma. Cells 2019, 8, 1060. [Google Scholar] [CrossRef] [Green Version]
- Fernández, N.B.; Lorenzo, D.; Picco, M.E.; Barbero, G.; Dergan-Dylon, L.S.; Marks, M.P.; García-Rivello, H.; Gimenez, L.; Labovsky, V.; Grumolato, L.; et al. ROR1 contributes to melanoma cell growth and migration by regulating N-cadherin expression via the PI3K/Akt pathway: ROR1 INCREASES MELANOMA CELL GROWTH AND MIGRATION. Mol. Carcinog. 2016, 55, 1772–1785. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Bergami, P.; Huang, C.; Goydos, J.S.; Yip, D.; Bar-Eli, M.; Herlyn, M.; Smalley, K.S.M.; Mahale, A.; Eroshkin, A.; Aaronson, S.; et al. Rewired ERK-JNK Signaling Pathways in Melanoma. Cancer Cell 2007, 11, 447–460. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quezada, M.J.; Picco, M.E.; Villanueva, M.B.; Castro, M.V.; Barbero, G.; Fernández, N.B.; Illescas, E.; Lopez-Bergami, P. BCL2L10 Is Overexpressed in Melanoma Downstream of STAT3 and Promotes Cisplatin and ABT-737 Resistance. Cancers 2021, 13, 78. https://doi.org/10.3390/cancers13010078
Quezada MJ, Picco ME, Villanueva MB, Castro MV, Barbero G, Fernández NB, Illescas E, Lopez-Bergami P. BCL2L10 Is Overexpressed in Melanoma Downstream of STAT3 and Promotes Cisplatin and ABT-737 Resistance. Cancers. 2021; 13(1):78. https://doi.org/10.3390/cancers13010078
Chicago/Turabian StyleQuezada, María Josefina, María Elisa Picco, María Belén Villanueva, María Victoria Castro, Gastón Barbero, Natalia Brenda Fernández, Edith Illescas, and Pablo Lopez-Bergami. 2021. "BCL2L10 Is Overexpressed in Melanoma Downstream of STAT3 and Promotes Cisplatin and ABT-737 Resistance" Cancers 13, no. 1: 78. https://doi.org/10.3390/cancers13010078
APA StyleQuezada, M. J., Picco, M. E., Villanueva, M. B., Castro, M. V., Barbero, G., Fernández, N. B., Illescas, E., & Lopez-Bergami, P. (2021). BCL2L10 Is Overexpressed in Melanoma Downstream of STAT3 and Promotes Cisplatin and ABT-737 Resistance. Cancers, 13(1), 78. https://doi.org/10.3390/cancers13010078