
Utility and Mechanism of SHetA2 and Paclitaxel for Treatment of Endometrial Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Culture Conditions, and Chemicals
2.2. Colony Formation Assay
2.3. Wound Healing Assay
2.4. Invasion Assay
2.5. Cell Cycle Assay
2.6. Western Blot Analysis
2.7. Mitochondrial Membrane Potential (MMP) Assay
2.8. MTT Cell Viability Assay
2.9. ATP Assay
2.10. Mitochondrial-Stress and Glycolytic Rate Analysis
2.11. Annexin-V/PI Apoptosis Flow Cytometry Assay
2.12. Caspase-3 Activity Assay
2.13. Mass Spectrometry Sample Preparation and LC-MS/MS Measurement
2.14. Co-Immunoprecipitation Assay
2.15. Drug Interaction Analysis
2.16. Tumor Xenograft Model
2.17. Immunofluorescence/Immunocytochemistry
2.18. Data and Statistical Analysis
3. Results
3.1. Cancerous Features
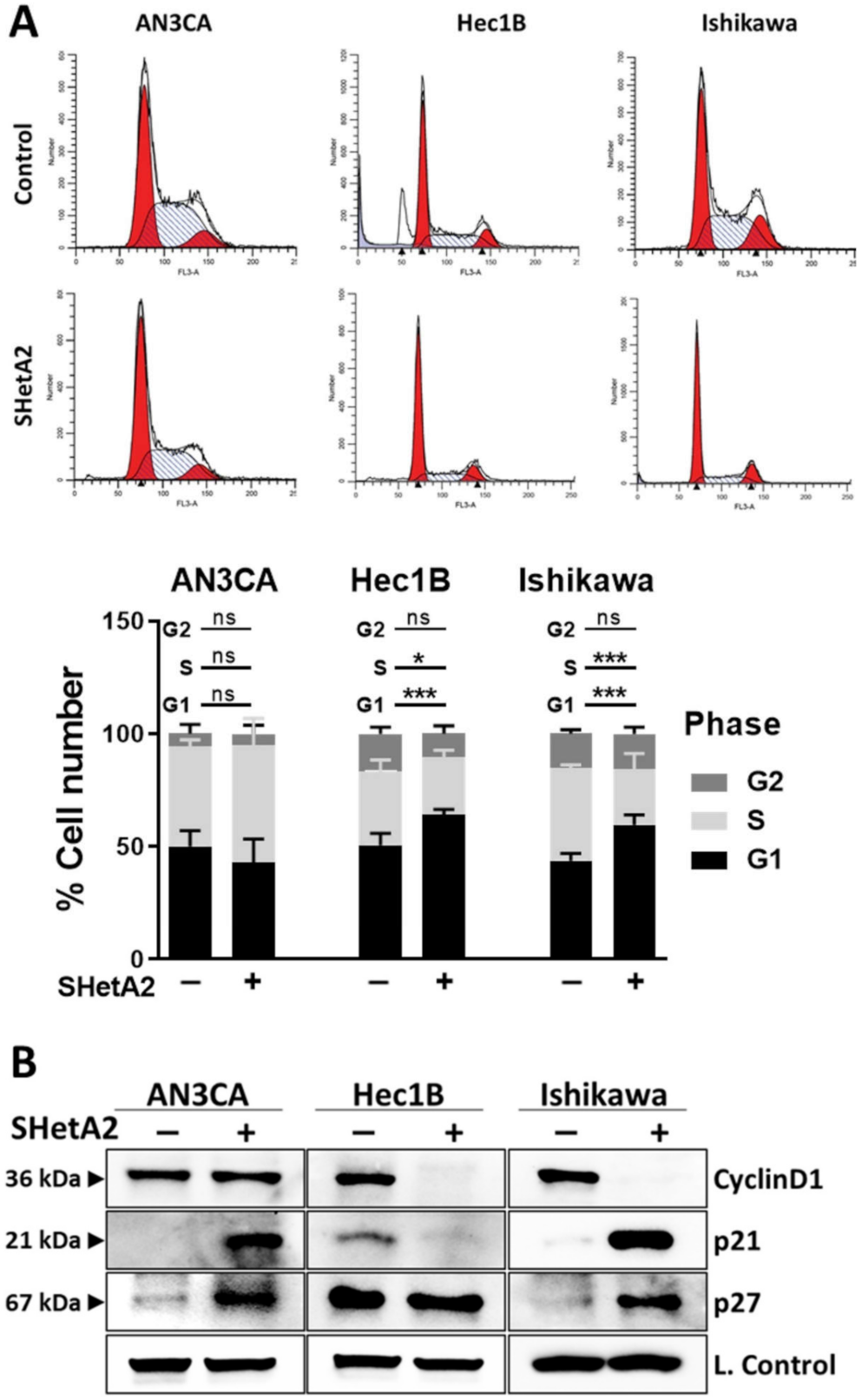
3.2. Cell Cycle
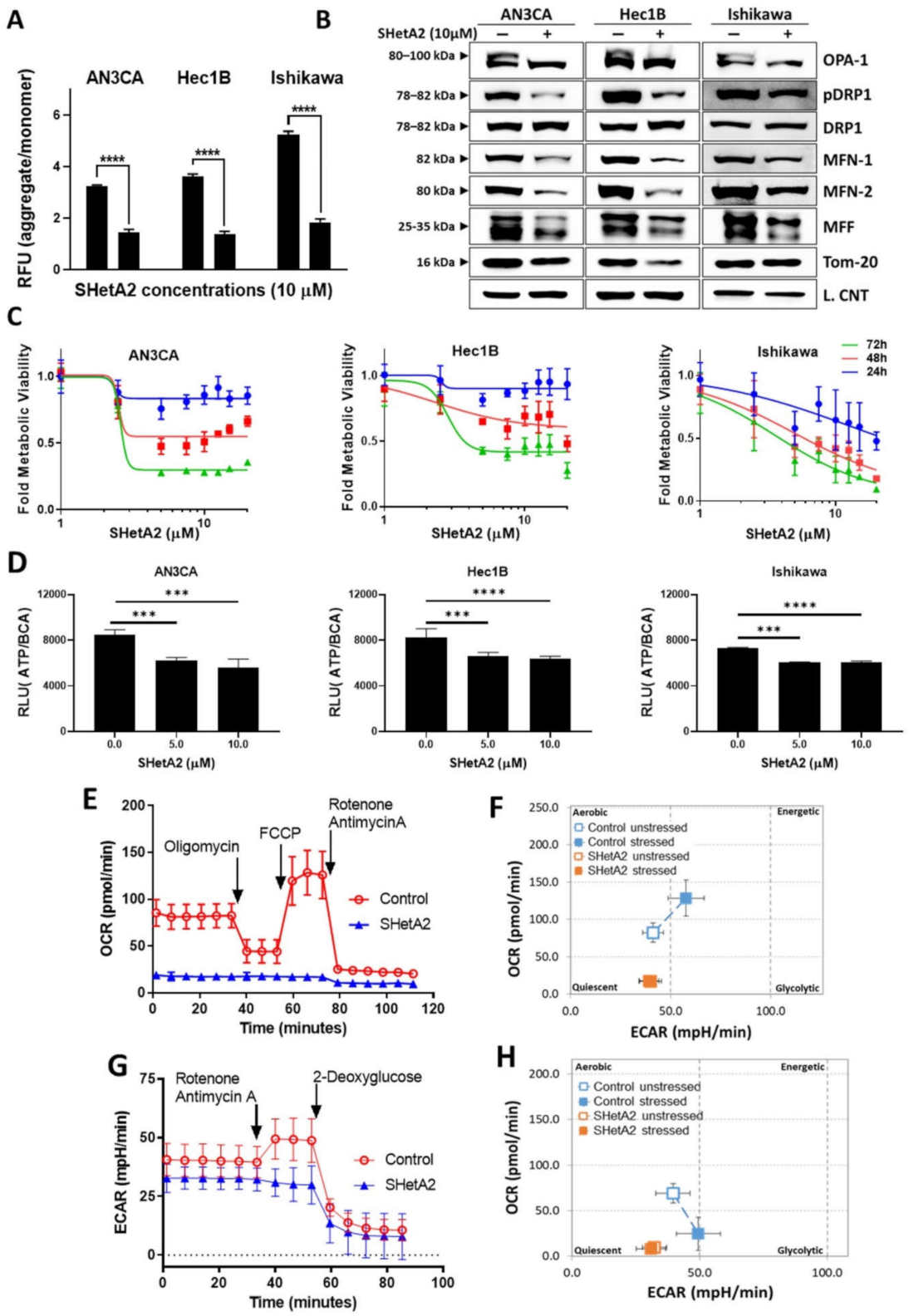
3.3. Mitochondria and Metabolism
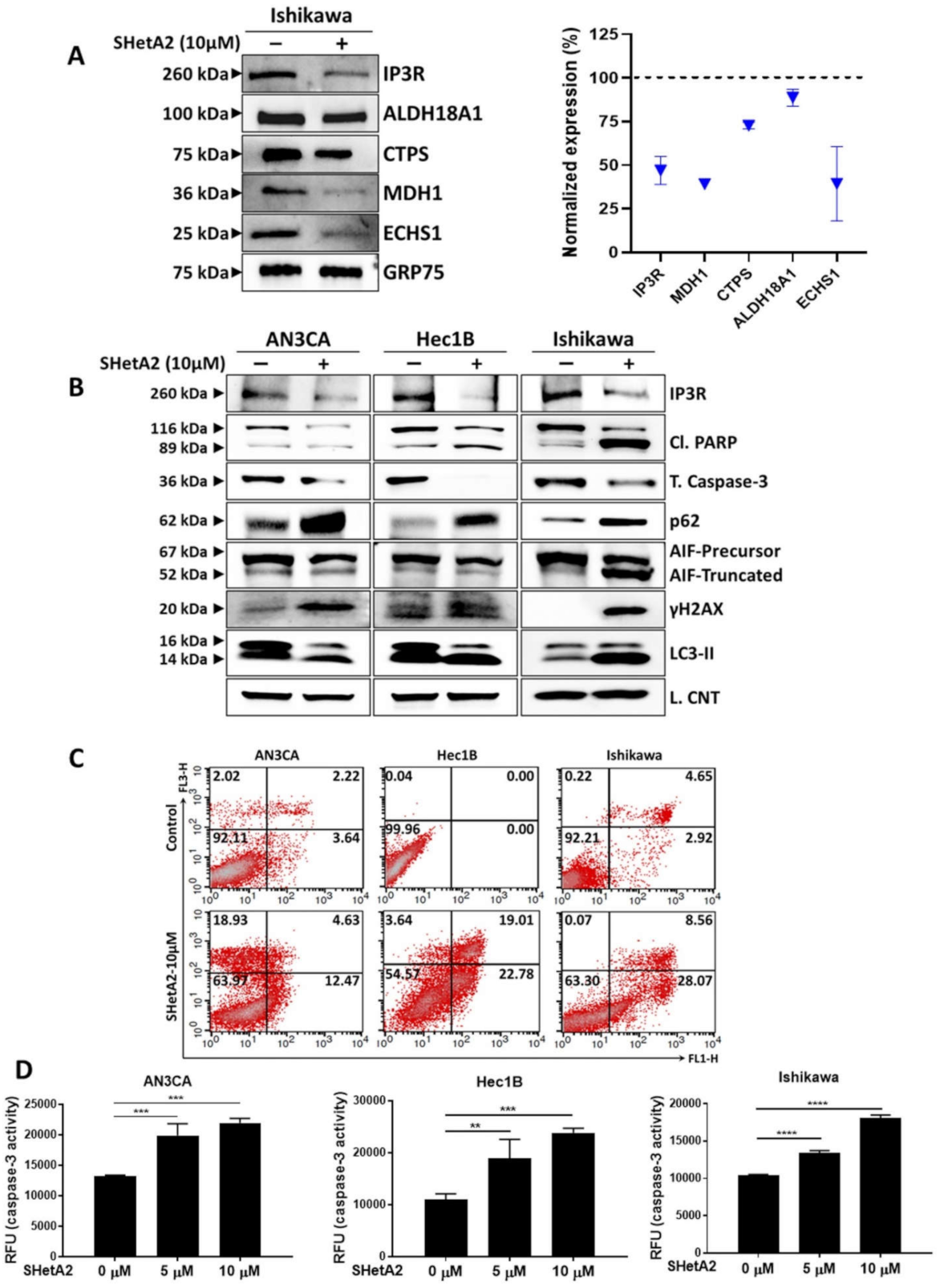
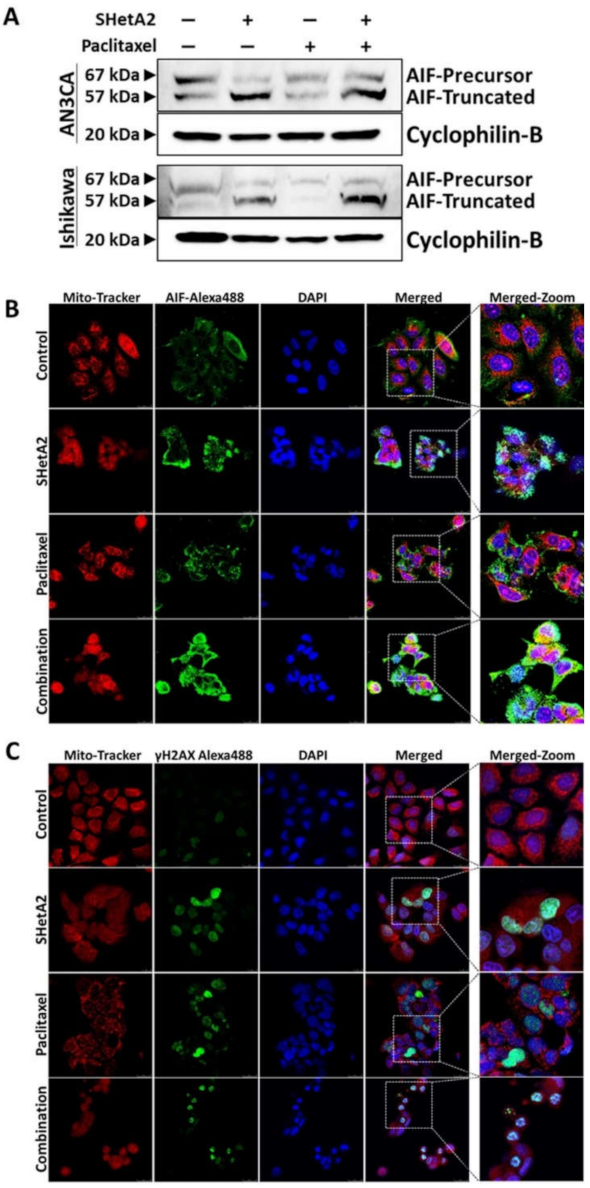
3.4. Mechanisms of SHetA2
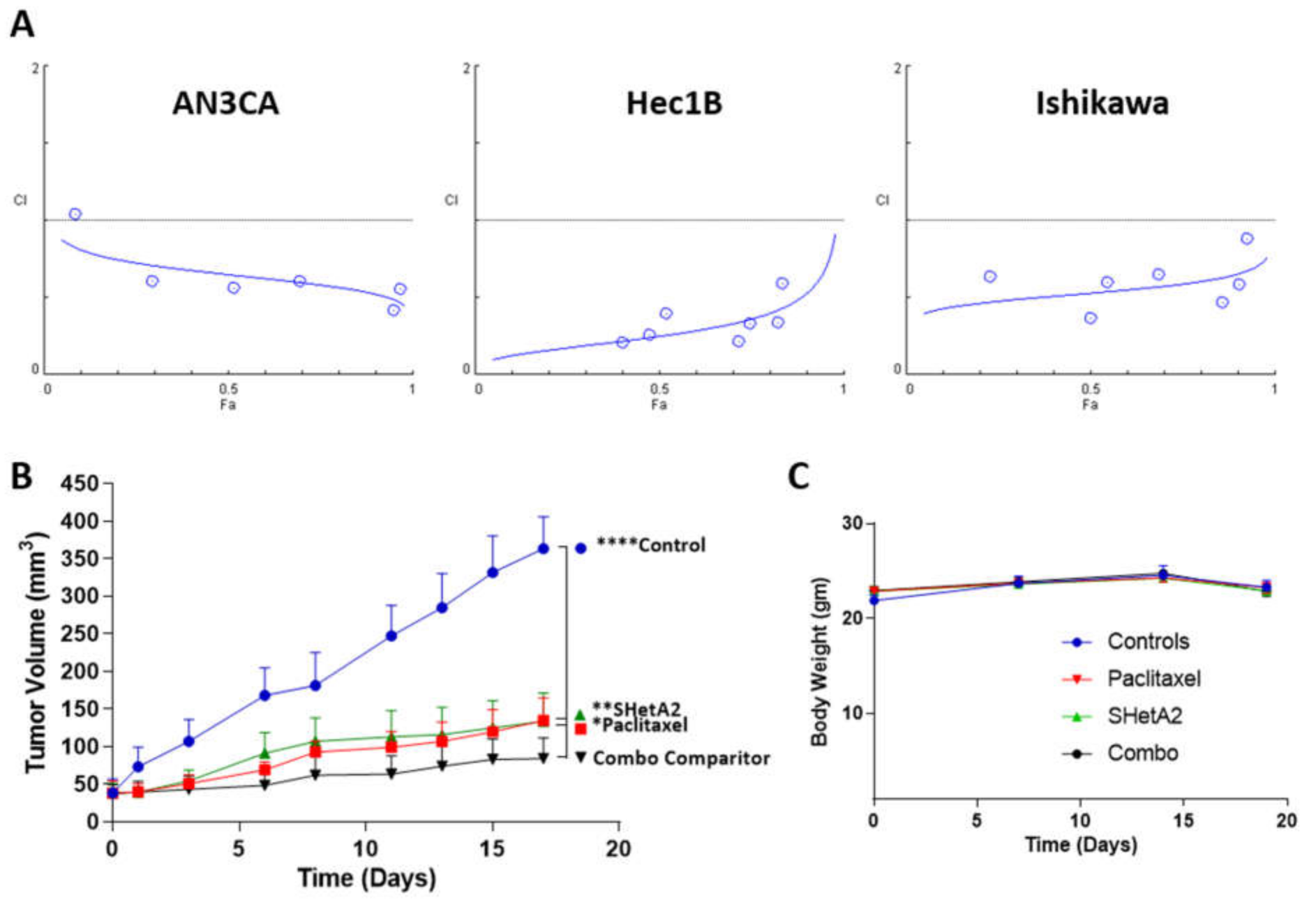
3.5. Synergy with Paclitaxel in Cell Culture and In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cancer Facts and Figures. Available online: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2020.html (accessed on 1 April 2021).
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aune, D.; Navarro Rosenblatt, D.A.; Chan, D.S.; Vingeliene, S.; Abar, L.; Vieira, A.R.; Greenwood, D.C.; Bandera, E.V.; Norat, T. Anthropometric factors and endometrial cancer risk: A systematic review and dose-response meta-analysis of prospective studies. Ann. Oncol. 2015, 26, 1635–1648. [Google Scholar] [CrossRef]
- Arem, H.; Pfeiffer, R.M.; Moore, S.C.; Irwin, M.L.; LaMonte, M.J.; Sarto, G.E.; Nassir, R.; Luo, J.; Chlebowski, R.T.; Brinton, L.A.; et al. Post-diagnosis body mass index and mortality among women diagnosed with endometrial cancer: Results from the Women’s Health Initiative. PLoS ONE 2017, 12, e0171250. [Google Scholar] [CrossRef]
- Amant, F.; Moerman, P.; Neven, P.; Timmerman, D.; Van Limbergen, E.; Vergote, I. Endometrial cancer. Lancet 2005, 366, 491–505. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucl. Acids Res. 2014, 43, D512–D520. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Bifulco, G.; Miele, C.; Di Jeso, B.; Beguinot, F.; Nappi, C.; Di Carlo, C.; Capuozzo, S.; Terrazzano, G.; Insabato, L.; Ulianich, L. Endoplasmic reticulum stress is activated in endometrial adenocarcinoma. Gynecol. Oncol. 2012, 125, 220–225. [Google Scholar] [CrossRef]
- Luvsandagva, B.; Nakamura, K.; Kitahara, Y.; Aoki, H.; Murata, T.; Ikeda, S.; Minegishi, T. GRP78 induced by estrogen plays a role in the chemosensitivity of endometrial cancer. Gynecol. Oncol. 2012, 126, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Cali, G.; Insabato, L.; Conza, D.; Bifulco, G.; Parrillo, L.; Mirra, P.; Fiory, F.; Miele, C.; Raciti, G.A.; Di Jeso, B.; et al. GRP78 mediates cell growth and invasiveness in endometrial cancer. J. Cell. Physiol. 2014, 229, 1417–1426. [Google Scholar] [CrossRef]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef] [Green Version]
- Eritja, N.; Chen, B.-J.; Rodriguez-Barrueco, R.; Santacana, M.; Gatius, S.; Vidal, A.; Marti, M.D.; Ponce, J.; Bergada, L.; Yeramian, A.; et al. Autophagy orchestrates adaptive responses to targeted therapy in endometrial cancer. Autophagy 2017, 13, 608–624. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Vishwanathan, V.; Birje, A.; Sinha, D.; D’Silva, P. Evolving paradigms on the interplay of mitochondrial Hsp70 chaperone system in cell survival and senescence. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, R.; Kaul, S.C.; Sugimoto, Y.; Mitsui, Y. Induction of cellular senescence by transfection of cytosolic mortalin cDNA in NIH 3T3 cells. J. Biol. Chem. 1993, 268, 22239–22242. [Google Scholar] [CrossRef]
- Kaul, S.C.; Yaguchi, T.; Taira, K.; Reddel, R.R.; Wadhwa, R. Overexpressed mortalin (mot-2)/mthsp70/GRP75 and hTERT cooperate to extend the in vitro lifespan of human fibroblasts. Exp. Cell Res. 2003, 286, 96–101. [Google Scholar] [CrossRef]
- Wadhwa, R.; Takano, S.; Kaur, K.; Deocaris, C.C.; Pereira-Smith, O.M.; Reddel, R.R.; Kaul, S.C. Upregulation of mortalin/mthsp70/Grp75 contributes to human carcinogenesis. Int. J. Cancer 2006, 118, 2973–2980. [Google Scholar] [CrossRef]
- Ramraj, S.K.; Elayapillai, S.P.; Pelikan, R.C.; Zhao, Y.D.; Isingizwe, Z.R.; Kennedy, A.L.; Lightfoot, S.A.; Benbrook, D.M. Novel ovarian cancer maintenance therapy targeted at mortalin and mutant p53. Int. J. Cancer 2019, 147, 1086–1097. [Google Scholar] [CrossRef]
- Rozenberg, P.; Kocsis, J.; Saar, M.; Prohaszka, Z.; Fust, G.; Fishelson, Z. Elevated levels of mitochondrial mortalin and cytosolic HSP70 in blood as risk factors in patients with colorectal cancer. Int. J. Cancer 2013, 133, 514–518. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Xu, B.; Li, H.; Yang, L.; Zuo, J.; Liu, W.; Liu, C. Expression of mortalin detected in human liver cancer by tissue microarrays. Anat. Rec. 2011, 294, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Zhang, B.; Zikeliyar, M.; Wang, J.; Jian, H.; Wu, K.; Zhang, Y.; Ding, J. Elevated Mortalin correlates with poor outcome in hepatocellular carcinoma. Ann. Diagn. Pathol. 2019, 42, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Ji, M.; Chen, L.; Liu, Q.; Che, S.; Xu, M.; Lin, Z. The clinicopathological significance of Mortalin overexpression in invasive ductal carcinoma of breast. J. Exp. Clin. Cancer Res. 2016, 35, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Che, S.L.; Piao, J.J.; Xu, M.; Chen, L.Y.; Lin, Z.H. Mortalin overexpression predicts poor prognosis in early stage of non-small cell lung cancer. Tumour Biol. 2017, 39, 1010428317695918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Jin, T.; Chen, L.; Zhang, X.; Zhu, G.; Wang, Q.; Lin, Z. Mortalin is a distinct bio-marker and prognostic factor in serous ovarian carcinoma. Gene 2019, 696, 63–71. [Google Scholar] [CrossRef]
- Yi, X.; Luk, J.M.; Lee, N.P.; Peng, J.; Leng, X.; Guan, X.-Y.; Lau, G.K.; Beretta, L.; Fan, S.-T. Association of mortalin (HSPA9) with liver cancer metastasis and prediction for early tumor recurrence. Mol. Cell. Proteom. 2008, 7, 315–325. [Google Scholar] [CrossRef] [Green Version]
- Benbrook, D.M.; Nammalwar, B.; Long, A.; Matsumoto, A.; Singh, A.; Bunce, R.A.; Berlin, K.D. SHetA2 interference with mortalin binding to p66shc and p53 identified using drug-conjugated magnetic microspheres. Investig. New Drugs 2013, 32, 412–423. [Google Scholar] [CrossRef] [Green Version]
- Benbrook, D.; Kamelle, S.; Guruswamy, S.; Lightfoot, S.; Rutledge, T.; Gould, N.; Hannafon, B.; Dunn, S.T.; Berlin, K.D. Flexible heteroarotinoids (Flex-Hets) exhibit improved therapeutic ratios as anti-cancer agents over retinoic acid receptor agonists. Investig. New Drugs 2005, 23, 417–428. [Google Scholar] [CrossRef]
- Mic, F.A.; Molotkov, A.; Benbrook, D.M.; Duester, G. Retinoid activation of retinoic acid receptor but not retinoid X receptor is sufficient to rescue lethal defect in retinoic acid synthesis. Proc. Natl. Acad. Sci. USA 2003, 100, 7135–7140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doppalapudi, R.S.; Riccio, E.S.; Davis, Z.; Menda, S.; Wang, A.; Du, N.; Green, C.; Kopelovich, L.; Rao, C.V.; Benbrook, D.M.; et al. Genotoxicity of the cancer chemopreventive drug candidates CP-31398, SHetA2, and phospho-ibuprofen. Mutat. Res. 2012, 746, 78–88. [Google Scholar] [CrossRef] [Green Version]
- Benbrook, D.M.; Guruswamy, S.; Wang, Y.; Sun, Z.; Mohammed, A.; Zhang, Y.; Li, Q.; Rao, C.V. Chemoprevention of Colon and Small Intestinal Tumorigenesis in APCmin/+ Mice By SHetA2 (NSC721689) without Toxicity. Cancer Prev. Res. 2013, 6, 908–916. [Google Scholar] [CrossRef] [Green Version]
- Kabirov, K.K.; Kapetanovic, I.M.; Benbrook, D.M.; Dinger, N.; Mankovskaya, I.; Zakharov, A.; Detrisac, C.; Pereira, M.; Martín-Jiménez, T.; Onua, E.; et al. Oral toxicity and pharmacokinetic studies of SHetA2, a new chemopreventive agent, in rats and dogs. Drug Chem. Toxicol. 2012, 36, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Masamha, C.P.; Chengedza, S.; Berlin, K.D.; Lightfoot, S.; He, F.; Benbrook, D.M. Development of flexible-heteroarotinoids for kidney cancer. Mol. Cancer Ther. 2009, 8, 1227–1238. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, A.L.; Rai, R.; Isingizwe, Z.R.; Zhao, Y.D.; Lightfoot, S.A.; Benbrook, D.M. Complementary Targeting of Rb Phosphorylation and Growth in Cervical Cancer Cell Cultures and a Xenograft Mouse Model by SHetA2 and Palbociclib. Cancers 2020, 12, 1269. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, Y.; Hua, Y.F.; Covey, J.M.; Benbrook, D.M.; Chan, K.K. Metabolism of a sulfur-containing heteroarotionoid antitumor agent, SHetA2, using liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 3371–3381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hua, Y.; Benbrook, D.M.; Covey, J.M.; Dai, G.; Liu, Z.; Chan, K.K. High performance liquid chromatographic analysis and preclinical pharmacokinetics of the heteroarotinoid antitumor agent, SHetA2. Cancer Chemother. Pharmacol. 2006, 58, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Benbrook, D.M.; Woo, S. Pharmacokinetics and interspecies scaling of a novel, orally-bioavailable anti-cancer drug, SHetA2. PLoS ONE 2018, 13, e0194046. [Google Scholar] [CrossRef]
- Sharma, A.; Li, M.; Thavathiru, E.; Ibrahim, M.; Garcia-Contreras, L.; Benbrook, D.M.; Woo, S. Physiologically Based Pharmacokinetic Modeling and Tissue Distribution Characteristics of SHetA2 in Tumor Bearing Mice. AAPS J. 2020, in press. [Google Scholar] [CrossRef]
- Sharma, A.; Thavathiru, E.; Benbrook, D.M.; Woo, S. Bioanalytical method development and validation of HPLCUV assay for the quantification of SHetA2 in mouse and human plasma: Application to pharmacokinetics study. J. Pharm. Technol. Drug Res. 2017, 6, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, M.; Hatipoglu, M.K.; Garcia-Contreras, L. Cryogenic Fabrication of Dry Powders to Enhance the Solubility of a Promising Anticancer Drug, SHetA2, for Oral Administration. AAPS Pharmscitech 2019, 20, 20. [Google Scholar] [CrossRef] [PubMed]
- Day, D.; Siu, L.L. Approaches to modernize the combination drug development paradigm. Genome Med. 2016, 8, 115. [Google Scholar] [CrossRef] [Green Version]
- Bestvina, C.M.; Fleming, G.F. Chemotherapy for Endometrial Cancer in Adjuvant and Advanced Disease Settings. Oncologist 2016, 21, 1250–1259. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Sun, X. Regulation of paclitaxel activity by microtubule-associated proteins in cancer chemotherapy. Cancer Chemother. Pharmacol. 2017, 80, 909–917. [Google Scholar] [CrossRef]
- Fukasawa, K. P53, cyclin-dependent kinase and abnormal amplification of centrosomes. Biochim. Biophys. Acta 2008, 1786, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Fisk, H.A.; Mattison, C.P.; Winey, M. Human Mps1 protein kinase is required for centrosome duplication and normal mitotic progression. Proc. Natl. Acad. Sci. USA 2003, 100, 14875–14880. [Google Scholar] [CrossRef] [Green Version]
- Kanai, M.; Ma, Z.; Izumi, H.; Kim, S.H.; Mattison, C.P.; Winey, M.; Fukasawa, K. Physical and functional interaction between mortalin and Mps1 kinase. Genes Cells 2007, 12, 797–810. [Google Scholar] [CrossRef]
- Ma, Z.; Izumi, H.; Kanai, M.; Kabuyama, Y.; Ahn, N.G.; Fukasawa, K. Mortalin controls centrosome duplication via modulating centrosomal localization of p53. Oncogene 2006, 25, 5377–5390. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Brown, C.W.; Berlin, K.D.; Dhar, A.; Guruswamy, S.; Brown, D.; Gardner, G.J.; Birrer, M.J.; Benbrook, D.M. Synthesis of flexible sulfur-containing heteroarotinoids that induce apoptosis and reactive oxygen species with discrimination between malignant and benign cells. J. Med. Chem. 2004, 47, 999–1007. [Google Scholar]
- Wisniewski, J.R.; Zougman, Z.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Van Nyen, T.; Moiola, C.P.; Colas, E.; Annibali, D.; Amant, F. Modeling Endometrial Cancer: Past, Present, and Future. Int. J. Mol. Sci. 2018, 19, 2348. [Google Scholar] [CrossRef] [Green Version]
- CCLE: Cancer Cell Line Encyclopedia. Available online: https://portals.broadinstitute.org/ccle (accessed on 21 April 2021).
- Schneider, H.C.; Berthold, J.; Bauer, M.F.; Dietmeier, K.; Guiard, B.; Brunner, M.; Neupert, W. Mitochondrial Hsp70/MIM44 complex facilitates protein import. Nature 1994, 371, 768–774. [Google Scholar] [CrossRef]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014, 204, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Guan, N.; Ren, Y.L.; Wei, Q.J.; Tao, Y.H.; Yang, G.S.; Liu, X.Y.; Bu, D.F.; Zhang, Y.; Zhu, S.N. IP(3)R-Grp75-VDAC1-MCU calcium regulation axis antagonists protect podocytes from apoptosis and decrease proteinuria in an Adriamycin nephropathy rat model. BMC Nephrol. 2018, 19, 140. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 2021, 17, 1–382. [Google Scholar]
- Lyakhovich, A.; Surrallés, J. Constitutive activation of caspase-3 and Poly ADP ribose polymerase cleavage in fanconi anemia cells. Mol. Cancer Res. 2010, 8, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Bano, D.; Prehn, J.H.M. Apoptosis-Inducing Factor (AIF) in Physiology and Disease: The Tale of a Repented Natural Born Killer. EBioMedicine 2018, 30, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. γH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Cella, D.; Huang, H.; Homesley, H.D.; Montag, A.; Salani, R.; De Geest, K.; Lee, R.; Spirtos, N.M. Patient-reported peripheral neuropathy of doxorubicin and cisplatin with and without paclitaxel in the treatment of advanced endometrial cancer: Results from GOG 184. Gynecol. Oncol. 2010, 119, 538–542. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandra, V.; Rai, R.; Benbrook, D.M. Utility and Mechanism of SHetA2 and Paclitaxel for Treatment of Endometrial Cancer. Cancers 2021, 13, 2322. https://doi.org/10.3390/cancers13102322
Chandra V, Rai R, Benbrook DM. Utility and Mechanism of SHetA2 and Paclitaxel for Treatment of Endometrial Cancer. Cancers. 2021; 13(10):2322. https://doi.org/10.3390/cancers13102322
Chicago/Turabian StyleChandra, Vishal, Rajani Rai, and Doris Mangiaracina Benbrook. 2021. "Utility and Mechanism of SHetA2 and Paclitaxel for Treatment of Endometrial Cancer" Cancers 13, no. 10: 2322. https://doi.org/10.3390/cancers13102322
APA StyleChandra, V., Rai, R., & Benbrook, D. M. (2021). Utility and Mechanism of SHetA2 and Paclitaxel for Treatment of Endometrial Cancer. Cancers, 13(10), 2322. https://doi.org/10.3390/cancers13102322