
Disruption of YY1-EZH2 Interaction Using Synthetic Peptides Inhibits Breast Cancer Development


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Plasmids
2.3. Antibodies
2.4. Synthetic Peptide Treatment and Quantitative PCR (qPCR)
2.5. Surface Plasmon Resonance (SPR)
2.6. Co-Immunoprecipitation (Co-IP) and Western Blot
2.7. GST Pull-Down Assay
2.8. Immunostaining Assay
2.9. Cell Viability Assay
2.10. Wound Healing Assay
2.11. Cell Apoptosis Assay
2.12. Mouse Xenograft Study
2.13. Immunohistochemistry
2.14. ChIP-Seq Library Preparation and Sequencing
2.15. Statistical Analysis
3. Results
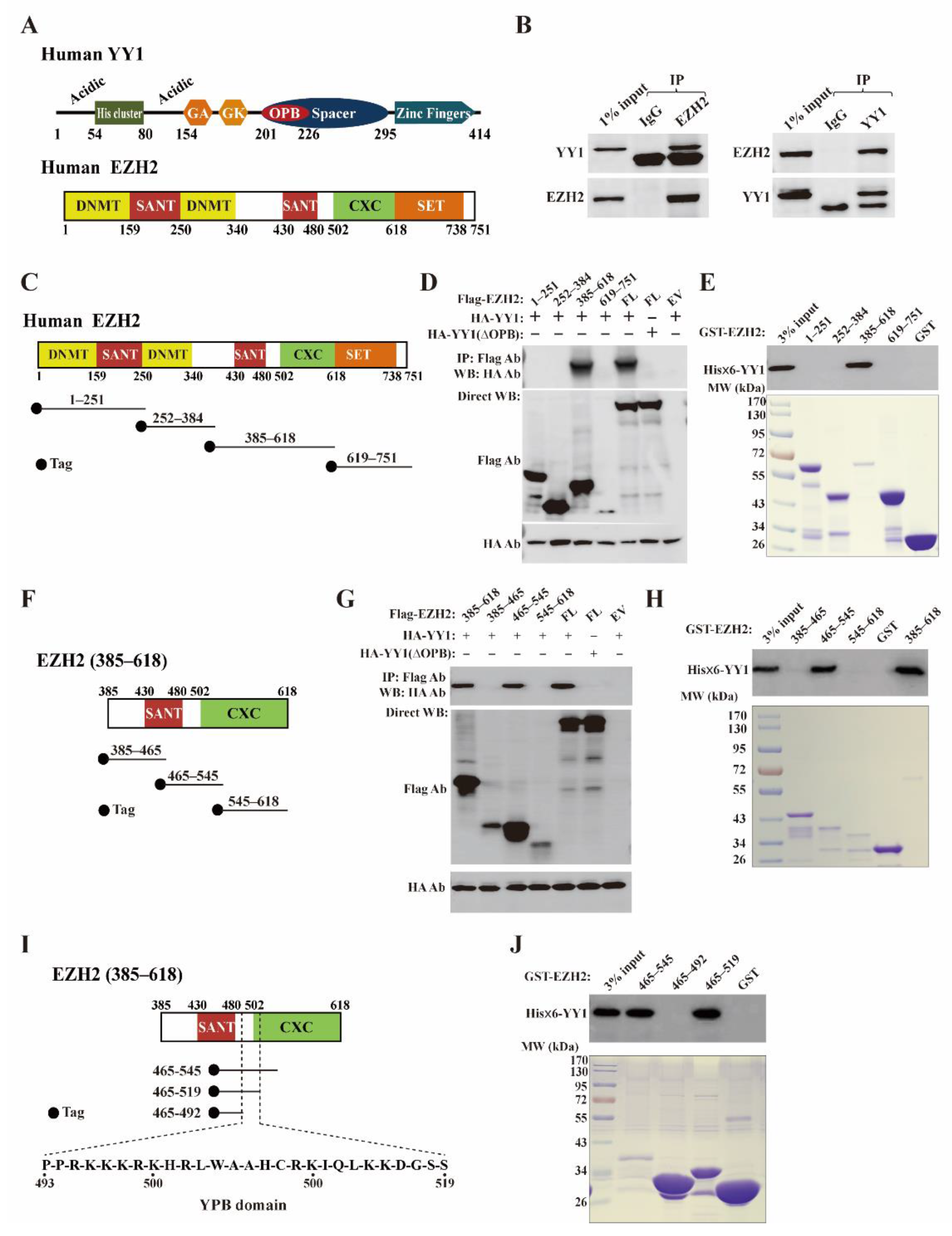
3.1. YY1 Associates with EZH2 in Breast Cancer Cells
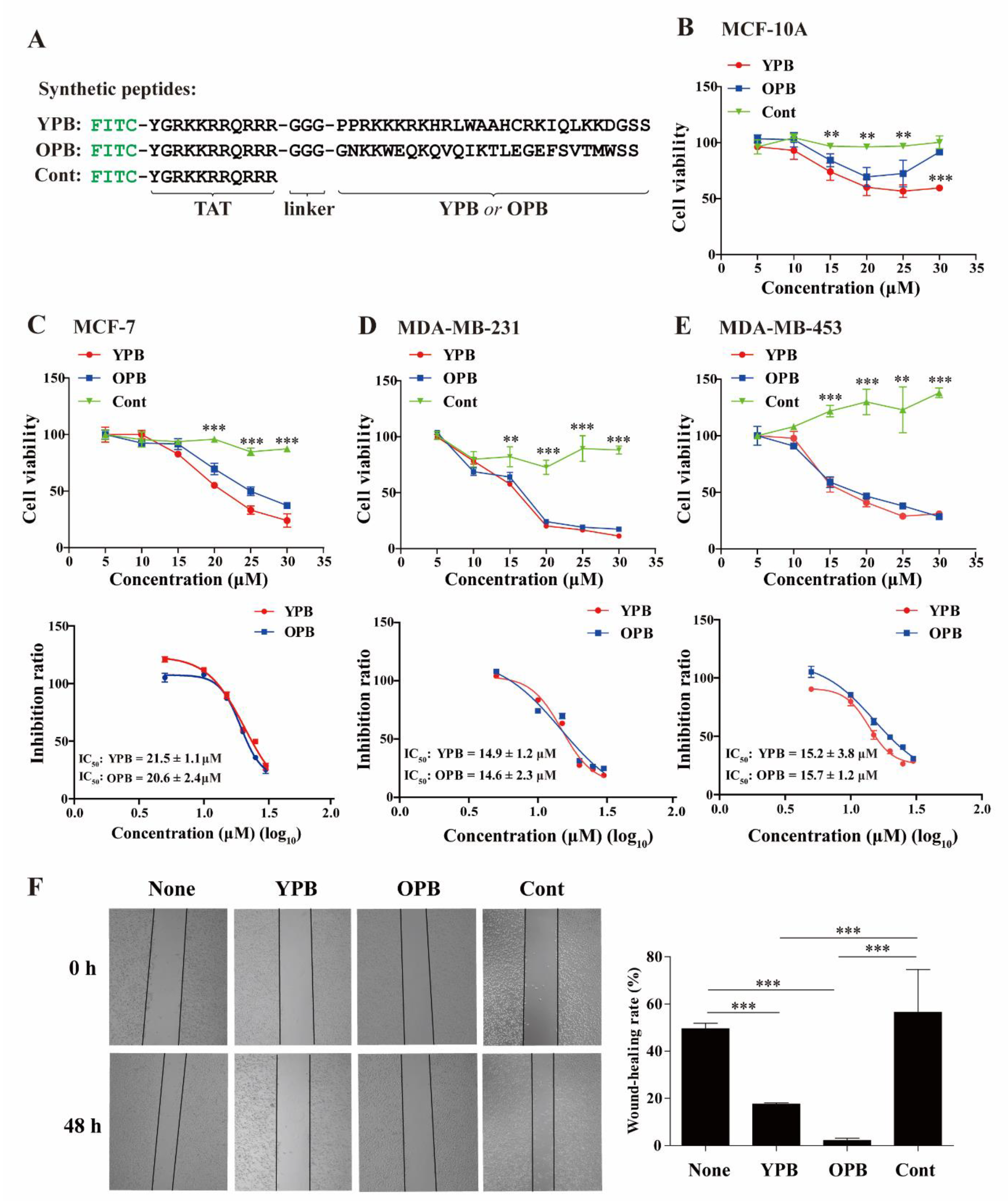
3.2. YPB and OPB Peptides Reduced Breast Cancer Cell Viability and Migration
3.3. The YPB and OPB Peptides Induced Breast Cancer Cell Apoptosis
3.4. The YPB and OPB Peptides Disrupted YY1-EZH2 Interaction and Mostly Stayed in Nuclei
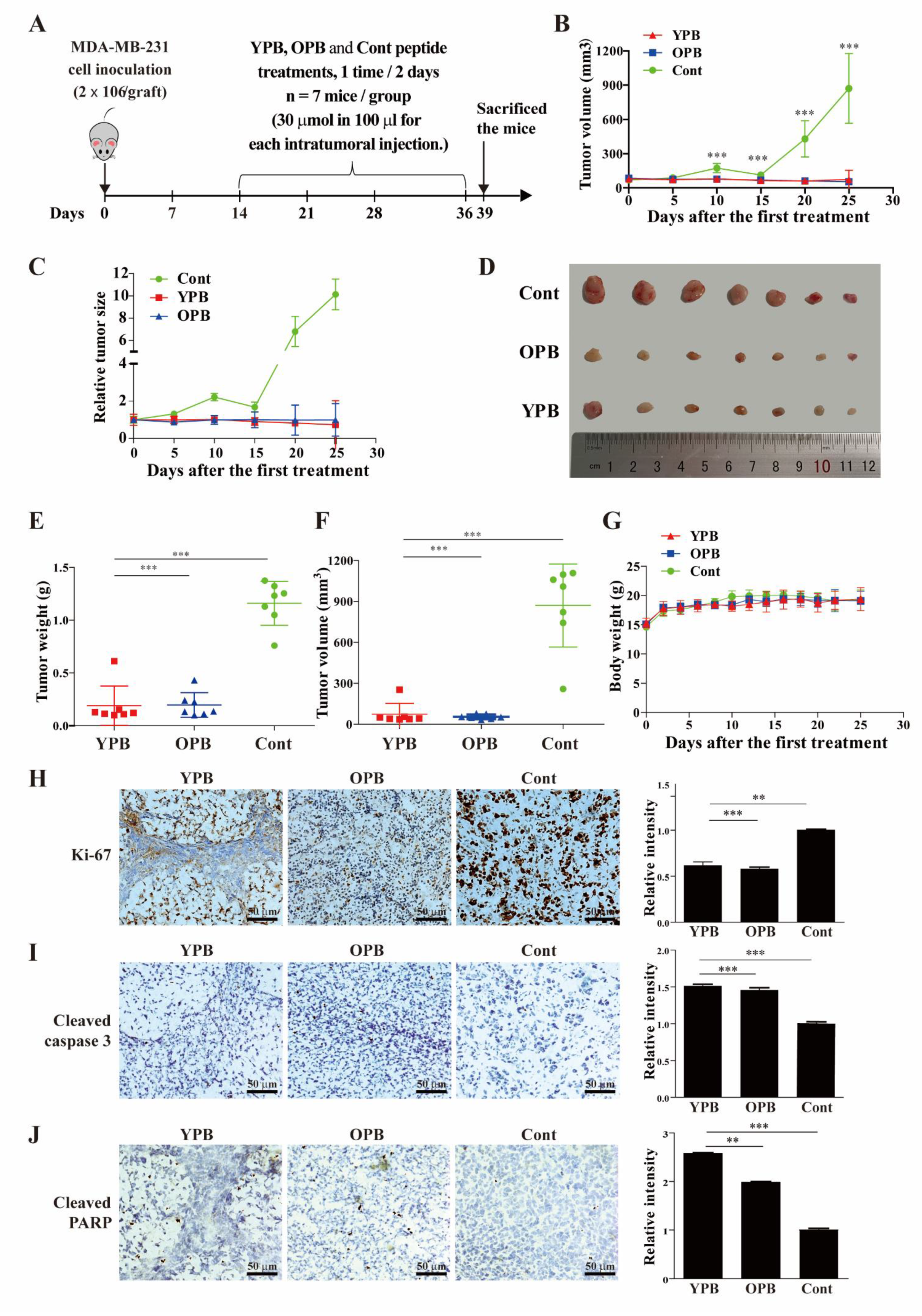
3.5. The YPB and OPB Peptides Inhibited Xenograft Tumor Growth in Nude Mice
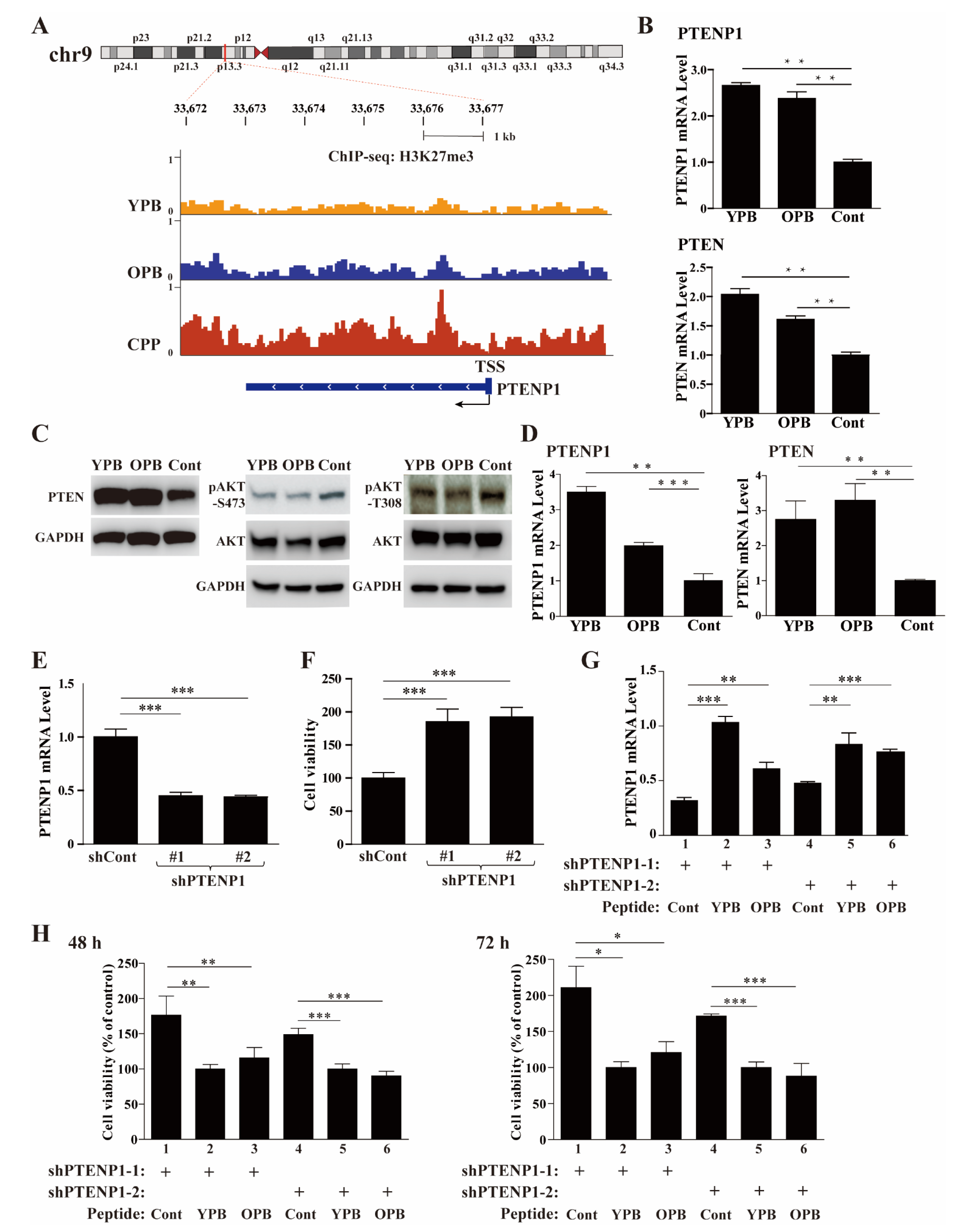
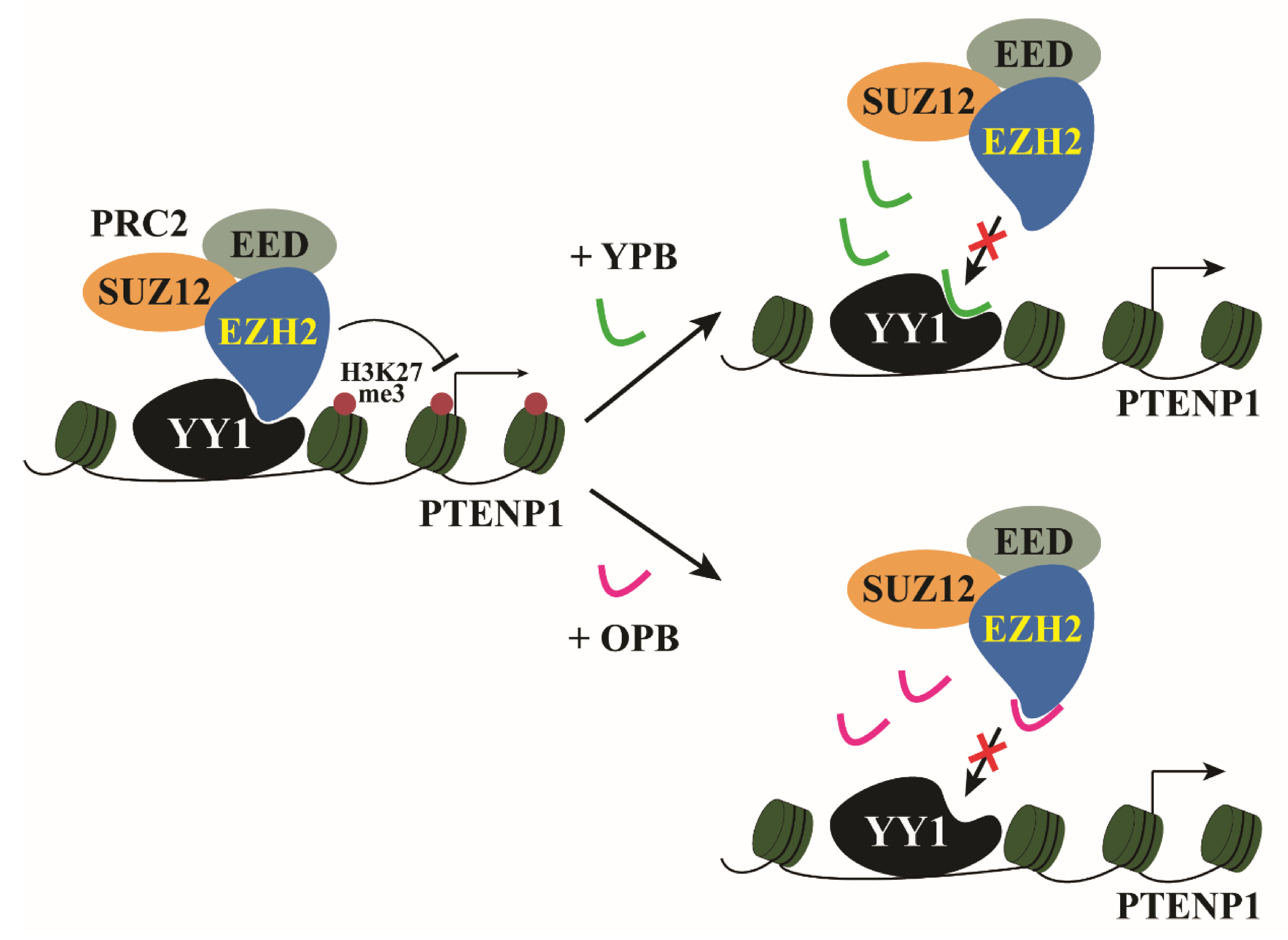
3.6. ChIP-Seq Studies Revealed the PTENP1 Gene as a Primary Target of YPB and OPB Peptides
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lund, A.H.; van Lohuizen, M. Epigenetics and cancer. Genes Dev. 2004, 18, 2315–2335. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.; Strathdee, G. Epigenomics and epigenetic therapy of cancer. Trends Mol. Med. 2002, 8, S43–S48. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajasekhar, V.K.; Begemann, M. Concise review: Roles of polycomb group proteins in development and disease: A stem cell perspective. Stem. Cells 2007, 25, 2498–2510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Carroll, D.; Erhardt, S.; Pagani, M.; Barton, S.C.; Surani, M.A.; Jenuwein, T. The polycomb-group gene Ezh2 is required for early mouse development. Mol. Cell Biol. 2001, 21, 4330–4336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chase, A.; Cross, N.C. Aberrations of EZH2 in cancer. Clin. Cancer Res. 2011, 17, 2613–2618. [Google Scholar] [CrossRef] [Green Version]
- You, B.H.; Yoon, J.H.; Kang, H.; Lee, E.K.; Lee, S.K.; Nam, J.W. HERES, a lncRNA that regulates canonical and noncanonical Wnt signaling pathways via interaction with EZH2. Proc. Natl. Acad. Sci. USA 2019, 116, 24620–24629. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Kim, W.; Howard, T.P.; Vazquez, F.; Tsherniak, A.; Wu, J.N.; Wang, W.; Haswell, J.R.; Walensky, L.D.; Hahn, W.C.; et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat. Med. 2015, 21, 1491–1496. [Google Scholar] [CrossRef] [Green Version]
- Braun, L.; Brenier-Pinchart, M.P.; Hammoudi, P.M.; Cannella, D.; Kieffer-Jaquinod, S.; Vollaire, J.; Josserand, V.; Touquet, B.; Couté, Y.; Tardieux, I.; et al. The Toxoplasma effector TEEGR promotes parasite persistence by modulating NF-κB signalling via EZH2. Nat. Microbiol. 2019, 4, 1208–1220. [Google Scholar] [CrossRef] [PubMed]
- Zhao, E.; Maj, T.; Kryczek, I.; Li, W.; Wu, K.; Zhao, L.; Wei, S.; Crespo, J.; Wan, S.; Vatan, L.; et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat. Immunol. 2016, 17, 95–103. [Google Scholar] [CrossRef]
- Chang, C.J.; Hung, M.C. The role of EZH2 in tumour progression. Br. J. Cancer 2012, 106, 243–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, M.E.; DuPrie, M.L.; Krueger, H.; Merajver, S.D.; Ventura, A.C.; Toy, K.A.; Kleer, C.G. Histone Methyltransferase EZH2 Induces Akt-Dependent Genomic Instability and BRCA1 Inhibition in Breast Cancer. Cancer Res. 2011, 71, 2360–2370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, H.M.; Gonzalez, M.E.; Toy, K.A.; Cimino-Mathews, A.; Argani, P.; Kleer, C.G. EZH2 inhibition decreases p38 signaling and suppresses breast cancer motility and metastasis. Breast Cancer Res. Treat. 2013, 138, 741–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Huang, S.; Wang, Z.; Huang, J.; Xu, L.; Tang, X.; Wan, Y.Y.; Li, Q.J.; Symonds, A.L.J.; Long, H.; et al. Targeting EZH2 histone methyltransferase activity alleviates experimental intestinal inflammation. Nat. Commun. 2019, 10, 2427. [Google Scholar] [CrossRef]
- Fioravanti, R.; Stazi, G.; Zwergel, C.; Valente, S.; Mai, A. Six Years (2012-2018) of Researches on Catalytic EZH2 Inhibitors: The Boom of the 2-Pyridone Compounds. Chem. Rec. 2018, 18, 1818–1832. [Google Scholar] [CrossRef] [PubMed]
- Caretti, G.; Di Padova, M.; Micales, B.; Lyons, G.E.; Sartorelli, V. The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev. 2004, 18, 2627–2638. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.; Zhu, W.; Meng, H.; Tong, L.; Li, X.; Luo, P.; Yi, L.; Zhang, X.; Guo, L.; Wei, T.; et al. CDYL promotes the chemoresistance of small cell lung cancer by regulating H3K27 trimethylation at the CDKN1C promoter. Theranostics 2019, 9, 4717–4729. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zeng, Z.; Xing, S.; Gullicksrud, J.A.; Shan, Q.; Choi, J.; Badovinac, V.P.; Crotty, S.; Peng, W.; Xue, H.H. Ezh2 programs TFH differentiation by integrating phosphorylation-dependent activation of Bcl6 and polycomb-dependent repression of p19Arf. Nat. Commun. 2018, 9, 5452. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long Noncoding RNA as Modular Scaffold of Histone Modification Complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Liu, G.; Yang, H.; Guo, S.; Wang, H.; Dong, Z.; Li, X.; Bai, Y.; Cheng, Y. MALAT1-mediated recruitment of the histone methyltransferase EZH2 to the microRNA-22 promoter leads to cardiomyocyte apoptosis in diabetic cardiomyopathy. Sci. Total Environ. 2020, 142191. [Google Scholar] [CrossRef]
- Li, Z.T.; Zhang, X.; Wang, D.W.; Xu, J.; Kou, K.J.; Wang, Z.W.; Yong, G.; Liang, D.S.; Sun, X.Y. Overexpressed lncRNA GATA6-AS1 Inhibits LNM and EMT via FZD4 through the Wnt/beta-Catenin Signaling Pathway in GC. Mol. Ther. Nucleic Acids 2020, 19, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Yang, S.; Han, Y.; Sun, J.; Xv, L.; Wu, L.; Ming, L. HOXD-AS1 confers cisplatin resistance in gastric cancer through epigenetically silencing PDCD4 via recruiting EZH2. Open Biol. 2019, 9, 190068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; He, C.; Xiao, M.; Wei, B.; Zhu, Y.; Zhang, G.; Zhou, H.; Yuan, J.; Hu, X.; Yi, Y. LncRNA FOXC2 antisense transcript accelerates non-small-cell lung cancer tumorigenesis via silencing p15. Am. J. Transl. Res. 2019, 11, 4552–4560. [Google Scholar] [PubMed]
- Wan, L.; Sun, M.; Liu, G.J.; Wei, C.C.; Zhang, E.B.; Kong, R.; Xu, T.P.; Huang, M.D.; Wang, Z.X. Long Noncoding RNA PVT1 Promotes Non-Small Cell Lung Cancer Cell Proliferation through Epigenetically Regulating LATS2 Expression. Mol. Cancer Ther. 2016, 15, 1082–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Stovall, D.B.; Inoue, K.; Sui, G. The oncogenic role of Yin Yang 1. Crit. Rev. Oncog. 2011, 16, 163–197. [Google Scholar] [CrossRef]
- Sui, G.; Affar, E.B.; Shi, Y.; Brignone, C.; Wall, N.R.; Yin, P.; Donohoe, M.; Luke, M.P.; Calvo, D.; Grossman, S.R.; et al. Yin Yang 1 is a negative regulator of p53. Cell 2004, 117, 859–872. [Google Scholar] [CrossRef] [Green Version]
- Bheda, A.; Creek, K.E.; Pirisi, L. Loss of p53 induces epidermal growth factor receptor promoter activity in normal human keratinocytes. Oncogene 2008, 27, 4315–4323. [Google Scholar] [CrossRef] [Green Version]
- Bain, M.; Sinclair, J. Targeted inhibition of the transcription factor YY1 in an embryonal carcinoma cell line results in retarded cell growth, elevated levels of p53 but no increase in apoptotic cell death. Eur. J. Cell Biol. 2005, 84, 543–553. [Google Scholar] [CrossRef]
- Gronroos, E.; Terentiev, A.A.; Punga, T.; Ericsson, J. YY1 inhibits the activation of the p53 tumor suppressor in response to genotoxic stress. Proc. Natl. Acad. Sci. USA 2004, 101, 12165–12170. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wan, M.M.; Shi, J.M.; Horita, D.A.; Miller, L.D.; Kute, T.E.; Kridel, S.J.; Kulik, G.; Sui, G.C. Yin Yang 1 promotes mTORC2-mediated AKT phosphorylation. J. Mol. Cell Biol. 2016, 8, 232–243. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; See, R.H.; Galvin, K.M.; Wang, J.; Shi, Y. Functional interactions between YY1 and adenovirus E1A. Nucleic Acids Res. 1995, 23, 925–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Zeng, Q.; Wang, M.; Cheng, A.; Pan, K.; Zhu, D.; Liu, M.; Jia, R.; Yang, Q.; Wu, Y.; et al. DHAV-1 2A1 Peptide—A Newly Discovered Co-expression Tool That Mediates the Ribosomal "Skipping" Function. Front Microbiol. 2018, 9, 2727. [Google Scholar] [CrossRef] [PubMed]
- Sui, G.; Soohoo, C.; Affar, E.B.; Gay, F.; Shi, Y.; Forrester, W.C.; Shi, Y. A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc. Natl. Acad. Sci. USA 2002, 99, 5515–5520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stovall, D.B.; Wan, M.; Zhang, Q.; Dubey, P.; Sui, G. DNA vector-based RNA interference to study gene function in cancer. J. Vis. Exp. 2012. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Regan, K.M.; Lou, Z.; Chen, J.; Tindall, D.J. CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science 2006, 314, 294–297. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, F.H.; Park, K.; Atchison, M.L. Polycomb recruitment to DNA in vivo by the YY1 REPO domain. Proc. Natl. Acad. Sci. USA 2006, 103, 19296–19301. [Google Scholar] [CrossRef] [Green Version]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef]
- Jin, X.; Yang, C.; Fan, P.; Xiao, J.; Zhang, W.; Zhan, S.; Liu, T.; Wang, D.; Wu, H. CDK5/FBW7-dependent ubiquitination and degradation of EZH2 inhibits pancreatic cancer cell migration and invasion. J. Biol. Chem. 2017, 292, 6269–6280. [Google Scholar] [CrossRef] [Green Version]
- Boyer, L.A.; Latek, R.R.; Peterson, C.L. The SANT domain: A unique histone-tail-binding module? Nat. Rev. Mol. Cell Biol. 2004, 5, 158–163. [Google Scholar] [CrossRef]
- Pan, X.; Papasani, M.; Hao, Y.; Calamito, M.; Wei, F.; Quinn Iii, W.J.; Basu, A.; Wang, J.; Hodawadekar, S.; Zaprazna, K.; et al. YY1 controls Igκ repertoire and B-cell development, and localizes with condensin on the Igκ locus. EMBO J. 2013, 32, 1168–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Y.; Yan, T.; Chen, L.; Zhang, Q.; Wang, W.; Han, X.; Li, D.; Shi, J.; Sui, G. Characterization of YY1 OPB Peptide for its Anticancer Activity. Curr. Cancer Drug Targets 2019, 19, 504–511. [Google Scholar] [CrossRef]
- Schlesinger, Y.; Straussman, R.; Keshet, I.; Farkash, S.; Hecht, M.; Zimmerman, J.; Eden, E.; Yakhini, Z.; Ben-Shushan, E.; Reubinoff, B.E.; et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2007, 39, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef] [Green Version]
- Wan, M.; Huang, W.; Kute, T.E.; Miller, L.D.; Zhang, Q.; Hatcher, H.; Wang, J.; Stovall, D.B.; Russell, G.B.; Cao, P.D.; et al. Yin Yang 1 plays an essential role in breast cancer and negatively regulates p27. Am. J. Pathol. 2012, 180, 2120–2133. [Google Scholar] [CrossRef] [Green Version]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef]
- Li, Z.; Li, M.; Wang, D.; Hou, P.; Chen, X.; Chu, S.; Chai, D.; Zheng, J.; Bai, J. Post-translational modifications of EZH2 in cancer. Cell Biosci. 2020, 10, 143. [Google Scholar] [CrossRef]
- Palacios, D.; Mozzetta, C.; Consalvi, S.; Caretti, G.; Saccone, V.; Proserpio, V.; Marquez, V.E.; Valente, S.; Mai, A.; Forcales, S.V.; et al. TNF/p38alpha/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell 2010, 7, 455–469. [Google Scholar] [CrossRef] [Green Version]
- Satijn, D.P.; Hamer, K.M.; den Blaauwen, J.; Otte, A.P. The polycomb group protein EED interacts with YY1, and both proteins induce neural tissue in Xenopus embryos. Mol. Cell Biol. 2001, 21, 1360–1369. [Google Scholar] [CrossRef] [Green Version]
- Ko, C.Y.; Hsu, H.C.; Shen, M.R.; Chang, W.C.; Wang, J.M. Epigenetic silencing of CCAAT/enhancer-binding protein delta activity by YY1/polycomb group/DNA methyltransferase complex. J. Biol. Chem. 2008, 283, 30919–30932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, J.A.; Lange, C.A. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 2008, 647, 21–29. [Google Scholar] [CrossRef]
- Stasik, S.; Middeke, J.M.; Kramer, M.; Rollig, C.; Kramer, A.; Scholl, S.; Hochhaus, A.; Crysandt, M.; Brummendorf, T.H.; Naumann, R.; et al. EZH2 mutations and impact on clinical outcome: An analysis in 1,604 patients with newly diagnosed acute myeloid leukemia. Haematologica 2020, 105, e228–e231. [Google Scholar] [CrossRef] [Green Version]
- Souroullas, G.P.; Jeck, W.R.; Parker, J.S.; Simon, J.M.; Liu, J.Y.; Paulk, J.; Xiong, J.; Clark, K.S.; Fedoriw, Y.; Qi, J.; et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat. Med. 2016, 22, 632–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Qin, T.; Mao, J.; Zhang, J.; Fan, S.; Lu, Y.; Sun, Z.; Zhang, Q.; Song, B.; Li, L. PTENP1/miR-20a/PTEN axis contributes to breast cancer progression by regulating PTEN via PI3K/AKT pathway. J. Exp. Clin. Cancer Res. 2019, 38, 256. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Tang, X.; Su, L. Overexpression of Long Noncoding RNA PTENP1 Inhibits Cell Proliferation and Migration via Suppression of miR-19b in Breast Cancer Cells. Oncol. Res. 2018, 26, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Li, R.K.; Gao, J.; Guo, L.H.; Huang, G.Q.; Luo, W.H. PTENP1 acts as a ceRNA to regulate PTEN by sponging miR-19b and explores the biological role of PTENP1 in breast cancer. Cancer Gene. Ther. 2017, 24, 309–315. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO Enrichment | |||
|---|---|---|---|
| Peptide (Gene Numbers) | Biological Process (BP) | Cell Component (CC) | Molecular Function (MF) |
| YPB (465) | Sensory (eye and ear) system development, neuron migration. | Synaptic membrane and other membrane structures. | Voltage-gated ion channel activity. |
| OPB (1137) | Neural and reproduction system development. | Cell-cell junction, adhesion, neuronal cell development, microvillus. | Membrane channel activities, including ion and lipid transmembrane transportation. |
| KEGG Pathway Enrichment | |||
| YPB (465) | cAMP, MAPK, adrenergic, calcium, oxytocin signaling and cGMP−PKG pathways; regulations related to cardiac muscle, cholinergic synapse, and protein digestion and absorption. | ||
| OPB (1137) | RAP1, WNT, HIPPO and thyroid hormone signaling pathways; breast cancer, gastric cancer, liver cancer, and stem cell regulations. | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yi, C.; Li, G.; Wang, W.; Sun, Y.; Zhang, Y.; Zhong, C.; Stovall, D.B.; Li, D.; Shi, J.; Sui, G. Disruption of YY1-EZH2 Interaction Using Synthetic Peptides Inhibits Breast Cancer Development. Cancers 2021, 13, 2402. https://doi.org/10.3390/cancers13102402
Yi C, Li G, Wang W, Sun Y, Zhang Y, Zhong C, Stovall DB, Li D, Shi J, Sui G. Disruption of YY1-EZH2 Interaction Using Synthetic Peptides Inhibits Breast Cancer Development. Cancers. 2021; 13(10):2402. https://doi.org/10.3390/cancers13102402
Chicago/Turabian StyleYi, Cheng, Guangyue Li, Wenmeng Wang, Yixuan Sun, Yueling Zhang, Chen Zhong, Daniel B. Stovall, Dangdang Li, Jinming Shi, and Guangchao Sui. 2021. "Disruption of YY1-EZH2 Interaction Using Synthetic Peptides Inhibits Breast Cancer Development" Cancers 13, no. 10: 2402. https://doi.org/10.3390/cancers13102402
APA StyleYi, C., Li, G., Wang, W., Sun, Y., Zhang, Y., Zhong, C., Stovall, D. B., Li, D., Shi, J., & Sui, G. (2021). Disruption of YY1-EZH2 Interaction Using Synthetic Peptides Inhibits Breast Cancer Development. Cancers, 13(10), 2402. https://doi.org/10.3390/cancers13102402