
Targeting Reactive Oxygen Species Metabolism to Induce Myeloma Cell Death



Abstract
:Simple Summary
Abstract
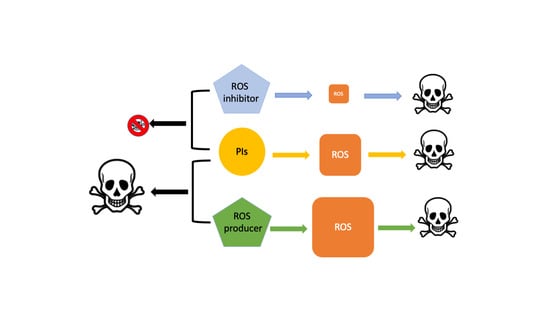
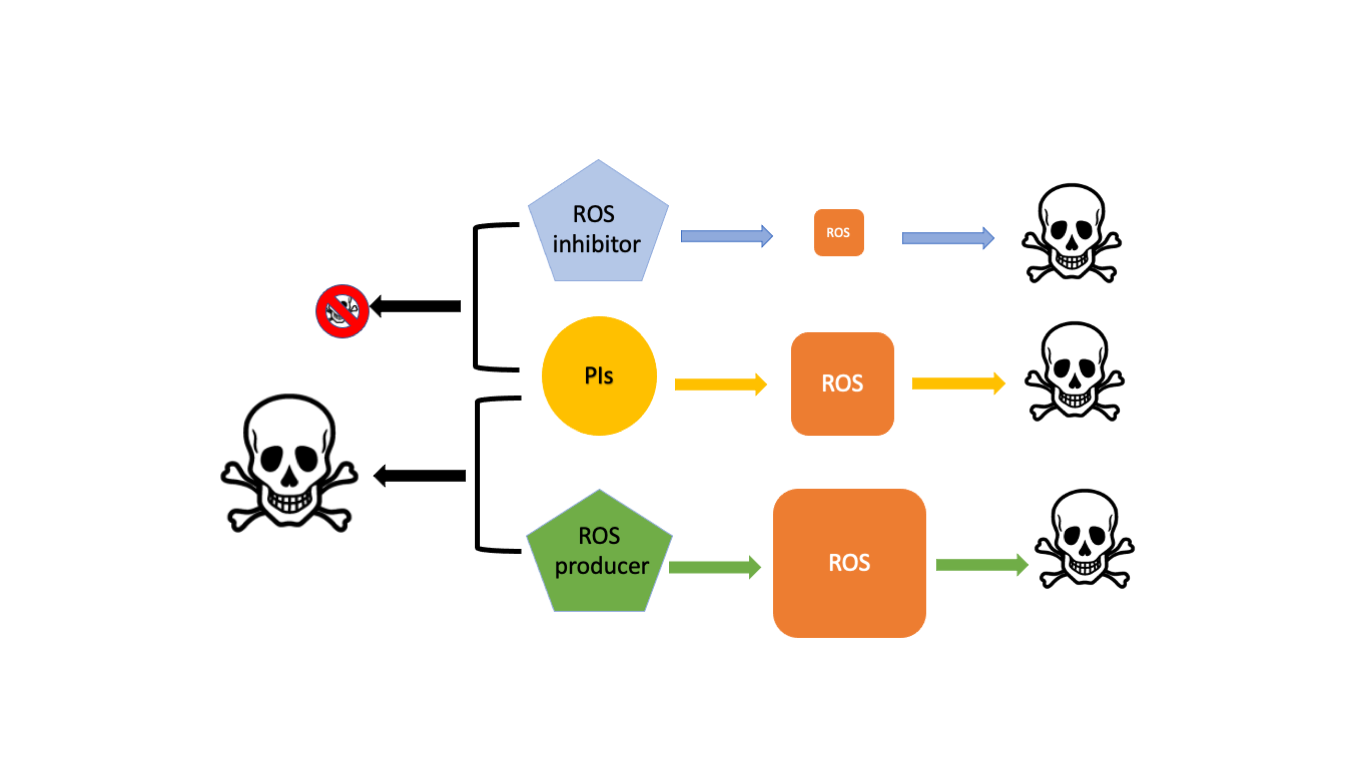
1. Introduction
2. Cellular ROS and Their Production
2.1. Generalities
2.2. ROS Homeostasis
2.3. ROS Targets
2.4. Physiological ROS Functions
2.4.1. Cell Signaling
2.4.2. Hypoxia Adaptation
2.4.3. Retro-Control and Regulation of Antioxidants
2.5. Oxidants
2.5.1. Mitochondria
2.5.2. NADPH Oxidases
2.6. Antioxidants

3. The Redox Landscape in Multiple Myeloma
3.1. Activation of Antioxidant Transcription Factors
3.2. Activation of Oncoproteins That Stimulate ROS Production
3.3. Increase of Antioxidant Defenses
3.4. Metabolism Rewiring

3.5. Impact of ROS Production on the Immune Tumor Microenvironment
4. Unbalancing ROS in Multiple Myeloma
4.1. Drugs That Target Glutathione Metabolism

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Effects on ROS Level | Drug/Combination | Effects | Preclinical Model | Reference |
|---|---|---|---|---|
| Decreasing | VAS3947 | NOX2i 1 | HMCL | [81] |
| Increasing | APR-246 | GSH depletion | HCML Primary cell In vivo | [163] |
| CAPE | GSH | HCML | [166] | |
| APR-246/BSO | GSH depletion/γGCSi | In vivo | [161] | |
| APR-246/auranofin | GSHdepletion/TXNRD1i r | HCMLs Primary cells | [164] | |
| Auranofin | TXNRD1i | HCMLs | [167] | |
| Auranofin/ZnPP IX | TXNRD1i/HO1i | HCMLs | [127] | |
| PX-12 | TXNi | HCMLs | [168] | |
| Auranofin/BTZ | TXNi/PI | HCMLs | [81] | |
| Lenalidomide/BTZ | TXNi/PI | HCMLs | [169] | |
| LCS-1 | SOD1i | HCMLs Primary cells In vivo | [130] | |
| 2-methoxyestradiol/BTZ | SOD2i/PI | HCMLs | [129] | |
| Disulfiram/BTZ | SOD1i/PI | HCMLs | [128] | |
| Scutellarein/BTZ | SOD2i/PI | HCMLs | [170] | |
| CCF642/BTZ | PDIi/PI | HCMLs In vivo | [171] | |
| E64FC26 | PDIi | HCMLs In vivo | [172,173] | |
| L-asparaginase/CFZ | AA depletion/PI | HCMLs | [174] | |
| DPI/HK2-ASO/PER | Mitochondria complex I/HK2i/FAOi | HCMLs In vivo | [175] | |
| SR18292 | PCG-1αi | HCMLs Primary cells | [176] |
4.2. Drugs That Target Antioxidant Enzymes
4.2.1. Thioredoxin System Inhibitors (Auranofin and Other Gold Compounds)
4.2.2. Superoxide Dismutase (SOD1/2) Inhibitors
4.3. Other ROS-Inducing Drugs
5. Global Alteration of MM Metabolism
6. Autophagy and Ferroptosis Are Forms of Death Controlled by MM Metabolism
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Vené, R.; Delfino, L.; Castellani, P.; Balza, E.; Bertolotti, M.; Sitia, R.; Rubartelli, A. Redox remodeling allows and controls B-cell activation and differentiation. Antioxid. Redox Signal. 2010, 13, 1145–1155. [Google Scholar] [CrossRef]
- Kumar, S.K.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Pandey, S.; Kapoor, P.; Dingli, D.; Hayman, S.R.; Leung, N.; et al. Continued improvement in survival in multiple myeloma: Changes in early mortality and outcomes in older patients. Leukemia 2014, 28, 1122–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, H.; Miguel, J.S.; Dimopoulos, M.A.; Palumbo, A.; Garcia-Sanz, R.; Powles, R.; Lentzsch, S.; Ming Chen, W.; Hou, J.; Jurczyszyn, A.; et al. International Myeloma Working Group recommendations for global myeloma care. Leukemia 2014, 28, 981–992. [Google Scholar] [CrossRef]
- Gay, F.; Engelhardt, M.; Terpos, E.; Wäsch, R.; Giaccone, L.; Auner, H.W.; Caers, J.; Gramatzki, M.; van de Donk, N.; Oliva, S.; et al. From transplant to novel cellular therapies in multiple myeloma: European Myeloma Network guidelines and future perspectives. Haematologica 2018, 103, 197–211. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Jakubowiak, A.J.; McCarthy, P.L.; Orlowski, R.Z.; Attal, M.; Bladé, J.; Goldschmidt, H.; Weisel, K.C.; Ramasamy, K.; Zweegman, S.; et al. Developments in continuous therapy and maintenance treatment approaches for patients with newly diagnosed multiple myeloma. Blood Cancer J. 2020, 10, 17. [Google Scholar] [CrossRef] [Green Version]
- Bobin, A.; Liuu, E.; Moya, N.; Gruchet, C.; Sabirou, F.; Lévy, A.; Gardeney, H.; Nsiala, L.; Cailly, L.; Guidez, S.; et al. Multiple myeloma: An overview of the current and novel therapeutic approaches in 2020. Cancers 2020, 12, 2885. [Google Scholar] [CrossRef]
- Ito, S. Proteasome inhibitors for the treatment of multiple myeloma. Cancers 2020, 12, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [Green Version]
- Kawabata, S.; Gills, J.J.; Mercado-Matos, J.R.; LoPiccolo, J.; Wilson, W.; Hollander, M.C.; Dennis, P.A. Synergistic effects of nelfinavir and bortezomib on proteotoxic death of NSCLC and multiple myeloma cells. Cell Death Dis. 2012, 3, e353. [Google Scholar] [CrossRef]
- Mimura, N.; Fulciniti, M.; Gorgun, G.; Tai, Y.T.; Cirstea, D.; Santo, L.; Hu, Y.; Fabre, C.; Minami, J.; Ohguchi, H.; et al. Blockade of XBP1 splicing by inhibition of IRE1α is a promising therapeutic option in multiple myeloma. Blood 2012, 119, 5772–5781. [Google Scholar] [CrossRef] [PubMed]
- Lipchick, B.C.; Fink, E.E.; Nikiforov, M.A. Oxidative stress and proteasome inhibitors in multiple myeloma. Pharmacol. Res. 2016, 105, 210–215. [Google Scholar] [CrossRef] [Green Version]
- Nerini-Molteni, S.; Ferrarini, M.; Cozza, S.; Caligaris-Cappio, F. Redox homeostasis modulates the sensitivity of myeloma cells to bortezomib. Br. J. Haematol. 2008, 141, 494–503. [Google Scholar] [CrossRef]
- Pinto, V.; Bergantim, R.; Caires, H.R.; Seca, H.; Guimarães, J.E.; Vasconcelos, M.H. Multiple myeloma: Available therapies and causes of drug resistance. Cancers 2020, 12, 407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rückrich, T.; Kraus, M.; Gogel, J.; Beck, A.; Ovaa, H.; Verdoes, M.; Overkleeft, H.S.; Kalbacher, H.; Driessen, C. Characterization of the ubiquitin-proteasome system in bortezomib-adapted cells. Leukemia 2009, 23, 1098–1105. [Google Scholar] [CrossRef] [Green Version]
- Balsas, P.; Galán-Malo, P.; Marzo, I.; Naval, J. Bortezomib resistance in a myeloma cell line is associated to PSMβ5 overexpression and polyploidy. Leuk. Res. 2012, 36, 212–218. [Google Scholar] [CrossRef]
- Acosta-Alvear, D.; Cho, M.Y.; Wild, T.; Buchholz, T.J.; Lerner, A.G.; Simakova, O.; Hahn, J.; Korde, N.; Landgren, O.; Maric, I.; et al. Paradoxical resistance of multiple myeloma to proteasome inhibitors by decreased levels of 19S proteasomal subunits. Elife 2015, 4, e08153. [Google Scholar] [CrossRef] [PubMed]
- Besse, A.; Stolze, S.C.; Rasche, L.; Weinhold, N.; Morgan, G.J.; Kraus, M.; Bader, J.; Overkleeft, H.S.; Besse, L.; Driessen, C. Carfilzomib resistance due to ABCB1/MDR1 overexpression is overcome by nelfinavir and lopinavir in multiple myeloma. Leukemia 2017, 32, 391–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiso, P.; Huynh, D.; Moschetta, M.; Sacco, A.; Aljawai, Y.; Mishima, Y.; Asara, J.M.; Roccaro, A.M.; Kimmelman, A.C.; Ghobrial, I.M. Metabolic signature identifies novel targets for drug resistance in multiple myeloma. Cancer Res. 2015, 75, 2071–2082. [Google Scholar] [CrossRef] [Green Version]
- Zaal, E.A.; Wu, W.; Jansen, G.; Zweegman, S.; Cloos, J.; Berkers, C.R. Bortezomib resistance in multiple myeloma is associated with increased serine synthesis. Cancer Metab. 2017, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Besse, L.; Besse, A.; Mendez-Lopez, M. A metabolic switch in proteasome inhibitor-resistant multiple myeloma ensures higher mitochondrial metabolism, protein folding and sphingomyelin synthesis. Haematologica 2019, 104, e415–e429. [Google Scholar] [CrossRef] [Green Version]
- Soriano, G.P.; Besse, L.; Li, N.; Kraus, M.; Besse, A.; Meeuwenoord, N.; Bader, J.; Everts, B.; den Dulk, H.; Overkleeft, H.S.; et al. Proteasome inhibitor-adapted myeloma cells are largely independent from proteasome activity and show complex proteomic changes, in particular in redox and energy metabolism. Leukemia 2016, 30, 2198–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méndez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef]
- Wu, S.; Kuang, H.; Ke, J.; Pi, M.; Yang, D.H. Metabolic reprogramming induces immune cell dysfunction in the tumor microenvironment of multiple myeloma. Front. Oncol. 2020, 10, 591342. [Google Scholar] [CrossRef]
- Masciarelli, S.; Sitia, R. Building and operating an antibody factory: Redox control during B to plasma cell terminal differentiation. Biochim. Biophys. Acta 2008, 1783, 578–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sbarra, A.J.; Karnovsky, M.L. Biochemical basis of phagocytosis. I. Metabolic changes during the ingestion of particles by polymorphonuclear leukocytes. J. Biol. Chem. 1959, 234, 1355–1362. [Google Scholar] [CrossRef]
- Iyer, G.Y.N.; Islam, M.F.; Quastel, J.H. Biochemical aspects of phagocytosis. Nature 1961, 192, 535–541. [Google Scholar] [CrossRef]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Babior, B.M.; Kipnes, R.S.; Curnutte, J.T. Biological defense mechanisms—Production by leukocytes of superoxide a potential bactericidal agent. J. Clin. Investig. 1973, 52, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, S.J. Oxygen-metabolism and the toxic properties of phagocytes. Ann. Intern. Med. 1980, 93, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Royer-Pokora, B.; Kunkel, L.M.; Monaco, A.P.; Goff, S.C.; Newburger, P.E.; Baehner, R.L.; Cole, F.S.; Curnutte, J.T.; Orkin, S.H. Cloning the gene for an inherited human disorder—Chronic granulomatous-disease—On the basis of its chromosomal location. Nature 1986, 322, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Segal, A.W.; Jones, O.T.G. Novel cytochrome-b system in phagocytic vacuoles of human granulocytes. Nature 1978, 276, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen peroxide sensing and signaling. Mol. Cell. 2007, 26, 1–14. [Google Scholar] [CrossRef]
- Jiang, F.; Zhang, Y.; Dusting, G.J. NADPH oxidase-mediated redox signaling: Roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 2011, 63, 218–242. [Google Scholar] [CrossRef] [Green Version]
- Zuo, L.; Zhou, T.; Pannell, B.K.; Ziegler, A.C.; Best, T.M. Biological and physiological role of reactive oxygen species—The good, the bad and the ugly. Acta Physiol. 2015, 214, 329–348. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Nathan, C. Specificity of a third kind: Reactive oxygen and nitrogen intermediates in cell signaling. J. Clin. Investig. 2003, 111, 769–778. [Google Scholar] [CrossRef]
- Imlay, J.A. Cellular defenses against superoxide and hydrogen peroxide. Annu. Rev. Biochem. 2008, 77, 755–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, U.; Bialy, L.P.; Ebstein, F.; Bech-Otschir, D.; Voigt, A.; Schroter, F.; Prozorovski, T.; Lange, N.; Steffen, J.; Rieger, M.; et al. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell 2010, 142, 613–624. [Google Scholar] [CrossRef] [Green Version]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef]
- Nathan, C.; Cunningham-Bussel, A. Beyond oxidative stress: An immunologist’s guide to reactive oxygen species. Nat. Rev. Immunol. 2013, 13, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Karisch, R.; Fernandez, M.; Taylor, P.; Virtanen, C.; St-Germain, J.R.; Jin, L.L.; Harris, I.S.; Mori, J.; Mak, T.W.; Senis, Y.A.; et al. Global proteomic assessment of the classical protein-tyrosine phosphatome and “redoxome”. Cell 2011, 146, 826–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truong, T.H.; Carroll, K.S. Redox regulation of epidermal growth factor receptor signaling through cysteine oxidation. Biochemistry 2012, 51, 9954–9965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denu, J.M.; Tanner, K.G. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: Evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 1998, 37, 5633–5642. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, C.E.; Truong, T.H.; Garcia, F.J.; Homann, A.; Gupta, V.; Leonard, S.E.; Carroll, K.S. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol. 2012, 8, 57–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 2003, 300, 650–653. [Google Scholar] [CrossRef]
- Brunelle, J.K.; Bell, E.L.; Quesada, N.M.; Vercauteren, K.; Tiranti, V.; Zeviani, M.; Scarpulla, R.C.; Chandel, N.S. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005, 1, 409–414. [Google Scholar] [CrossRef] [Green Version]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.P.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansfield, K.D.; Guzy, R.D.; Pan, Y.; Young, R.M.; Cash, T.P.; Schumacker, P.T.; Simon, M.C. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab. 2005, 1, 393–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruchko, M.V.; Gorodnya, O.M.; Pastukh, V.M.; Swiger, B.M.; Middleton, N.S.; Wilson, G.L.; Gillespie, M.N. Hypoxia-induced oxidative base modifications in the VEGF hypoxia-response element are associated with transcriptionally active nucleosomes. Free Radic. Biol. Med. 2009, 46, 352–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Vrishni, S.; Singh, B.K.; Rahman, I.; Kakkar, P. Nrf2-ARE stress response mechanism: A control point in oxidative stress-mediated dysfunctions and chronic inflammatory diseases. Free Radic. Res. 2010, 44, 1267–1288. [Google Scholar] [CrossRef]
- de Keizer, P.L.J.; Burgering, B.M.T.; Dansen, T.B. Forkhead box O as a sensor, mediator, and regulator of redox signaling. Antioxid. Redox Signal. 2011, 14, 1093–1106. [Google Scholar] [CrossRef]
- Cosentino, C.; Grieco, D.; Costanzo, V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011, 30, 546–555. [Google Scholar] [CrossRef] [Green Version]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Yoo, Y.D. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 2011, 32, 491–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Morselli, E.; Kepp, O.; Vitale, I.; Rigoni, A.; Vacchelli, E.; Michaud, M.; Zischka, H.; Castedo, M.; Kroemer, G. Mitochondrial gateways to cancer. Mol. Asp. Med. 2010, 31, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Poyton, R.O.; Ball, K.A.; Castello, P.R. Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrinol. Metabol. 2009, 20, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Kudin, A.P.; Bimpong-Buta, N.Y.B.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. Characterization of superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem. 2004, 279, 4127–4135. [Google Scholar] [CrossRef] [Green Version]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Fisher-Wellman, K.H.; Gilliam, L.A.A.; Lin, C.-T.; Cathey, B.L.; Lark, D.S.; Neufer, P.D. Mitochondrial glutathione depletion reveals a novel role for the pyruvate dehydrogenase complex as a key H2O2-emitting source under conditions of nutrient overload. Free Radic. Biol. Med. 2013, 65, 1201–1208. [Google Scholar] [CrossRef] [Green Version]
- Lo, Y.Y.C.; Cruz, T.F. Involvement of reactive oxygen species in cytokine and growth-factor induction of c-fos expression in chondrocytes. J. Biol. Chem. 1995, 270, 11727–11730. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, S.; Kurz, S.; Münzel, T.; Tarpey, M.; Freeman, B.A.; Griendling, K.K.; Harrison, D.G. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J. Clin. Investig. 1996, 97, 1916–1923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth-factor signal-transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef] [Green Version]
- Suh, Y.A.; Arnold, R.S.; Lassegue, B.; Shi, J.; Xu, X.X.; Sorescu, D.; Chung, A.B.; Griendling, K.K.; Lambeth, J.D. Cell transformation by the superoxide-generating oxidase Mox1. Nature 1999, 401, 79–82. [Google Scholar] [CrossRef]
- Cheng, G.; Cao, Z.; Xu, X.; van Meir, E.G.; Lambeth, J.D. Homologs of gp91phox: Cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 2001, 269, 131–140. [Google Scholar] [CrossRef]
- De Deken, X.; Wang, D.; Many, M.C.; Costagliola, S.; Libert, F.; Vassart, G.; Dumont, J.E.; Miot, F. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J. Biol. Chem. 2000, 275, 23227–23233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuy, C.; Ohayon, R.; Valent, A.; Noël-Hudson, M.S.; Dème, D.; Virion, A. Purification of a novel flavoprotein involved in the thyroid NADPH oxidase. Cloning of the porcine and human cDNAs. J. Biol. Chem. 1999, 274, 37265–37269. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, J.; Lambeth, J.D. Nox enzymes from fungus to fly to fish and what they tell us about Nox function in mammals. Free Radic. Biol. Med. 2010, 49, 1342–1353. [Google Scholar] [CrossRef] [Green Version]
- Drummond, G.R.; Selemidis, S.; Griendling, K.K.; Sobey, C.G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 453–471. [Google Scholar] [CrossRef] [Green Version]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [Green Version]
- van den Bosch, H.; Schutgens, R.B.H.; Wanders, R.J.A.; Tager, J.M. Biochemistry of peroxisomes. Ann. Rev. Biochem. 1992, 61, 157–197. [Google Scholar] [CrossRef]
- Shingu, M.; Yoshioka, K.; Nobunaga, M.; Yoshida, K. Human vascular smooth-muscle cells and endothelial-cells lack catalase activity and are susceptible to hydrogen-peroxide. Inflammation 1985, 9, 309–320. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Flohé, L.; Toppo, S.; Cozza, G.; Ursini, F. A Comparison of Thiol Peroxidase Mechanisms. Antioxid. Redox Signal. 2011, 15, 763–780. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Woo, H.A.; Kil, I.S.; Bae, S.H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 2012, 287, 4403–4410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnér, E.S.J.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef]
- Sun, Q.A.; Kirnarsky, L.; Sherman, S.; Gladyshev, V.N. Selenoprotein oxidoreductase with specificity for thioredoxin and glutathione systems. Proc. Natl. Acad. Sci. USA 2001, 98, 3673–3678. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Lillig, C.H.; Berndt, C.; Holmgren, A. Glutaredoxin systems. Biochim. Biophys. Acta 2008, 1780, 1304–1317. [Google Scholar] [CrossRef]
- Caillot, M.; Zylbersztejn, F.; Maitre, E.; Bourgeais, J.; Hérault, O.; Sola, B. ROS overproduction sensitises myeloma cells to bortezomib-induced apoptosis and alleviates tumour microenvironment-mediated cell resistance. Cells 2020, 9, 2357. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative stress in cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Rizzieri, D.; Paul, B.; Kang, Y. Metabolic alterations and the potential for targeting metabolic pathways in the treatment of multiple myeloma. J. Cancer Metastasis Treat. 2019, 5, 26. [Google Scholar] [CrossRef] [Green Version]
- Samanta, D.; Semenza, G.L. Maintenance of redox homeostasis by hypoxia-inducible factors. Redox Biol. 2017, 13, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sattler, M.; Tonon, G.; Grabher, C.; Lababidi, S.; Zimmerhackl, A.; Raab, M.S.; Vallet, S.; Zhou, Y.; Cartron, M.A.; et al. Targeting angiogenesis via a c-Myc/hypoxia-inducible factor-1-dependent pathway in multiple myeloma. Cancer Res. 2009, 69, 5082–5090. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.K.; Diamond, P.; Gronthos, S.; Peet, D.J.; Zannettino, A.C.W. The emerging role of hypoxia, HIF-1 and HIF-2 in multiple myeloma. Leukemia 2011, 25, 1533–1542. [Google Scholar] [CrossRef] [Green Version]
- Storti, P.; Bolzoni, M.; Donofrio, G.; Airoldi, I.; Guasco, D.; Toscani, D.; Martella, E.; Lazzaretti, M.; Mancini, C.; Agnelli, L.; et al. Hypoxia-inducible factor (HIF)-1α suppression in myeloma cells blocks tumoral growth in vivo inhibiting angiogenesis and bone destruction. Leukemia 2013, 27, 1697–1706. [Google Scholar] [CrossRef] [Green Version]
- Borsi, E.; Perrone, G.; Terragna, C.; Martello, M.; Dico, A.F.; Solaini, G.; Baracca, A.; Sgarbi, G.; Pasquinelli, G.; Valente, S.; et al. Hypoxia inducible factor-1 alpha as a therapeutic target in multiple myeloma. Oncotarget 2014, 5, 1779–1792. [Google Scholar] [CrossRef] [Green Version]
- Fok, J.H.L.; Hedayat, S.; Zhang, L.; Aronson, L.I.; Mirabella, F.; Pawlyn, C.; Bright, M.D.; Wardell, C.P.; Keats, J.J.; De Billy, E.; et al. HSF1 is essential for myeloma cell survival and a promising therapeutic target. Clin. Cancer Res. 2018, 24, 2395–2407. [Google Scholar] [CrossRef] [Green Version]
- Bustany, S.; Cahu, J.; Descamps, G.; Pellat-Deceunynck, C.; Sola, B. Heat shock factor 1 is a potent therapeutic target for enhancing the efficacy of treatments for multiple myeloma with adverse prognosis. J. Hematol. Oncol. 2015, 8, 40. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, N.; Wu, H.J.; Bennett, R.L.; Troche, C.; Licht, J.D.; Weber, J.D.; Maggi, L.B., Jr.; Tomasson, M.H. Sabotaging of the oxidative stress response by an oncogenic noncoding RNA. FASEB J. 2016, 31, 482–490. [Google Scholar] [CrossRef] [Green Version]
- Bergsagel, P.L.; Kuehl, W.M. Molecular pathogenesis and a consequent classification of multiple myeloma. J. Clin. Oncol. 2005, 23, 6333–6338. [Google Scholar] [CrossRef]
- Starheim, K.K.; Holien, T.; Misund, K.; Johansson, I.; Baranowska, K.A.; Sponaas, A.M.; Hella, H.; Buene, G.; Waage, A.; Sundan, A.; et al. Intracellular glutathione determines bortezomib cytotoxicity in multiple myeloma cells. Blood Cancer J. 2016, 6, e446. [Google Scholar] [CrossRef] [Green Version]
- Riz, I.; Hawley, T.S.; Marsal, J.W.; Hawley, R.G. Noncanonical SQSTM1/p62-Nrf2 pathway activation mediates proteasome inhibitor resistance in multiple myeloma cells via redox, metabolic and translational reprogramming. Oncotarget 2016, 7, 66360–66385. [Google Scholar] [CrossRef] [Green Version]
- Zucker, S.N.; Fink, E.E.; Bagati, A.; Mannava, S.; Bianchi-Smiraglia, A.; Bogner, P.N.; Wawrzyniak, J.A.; Foley, C.; Leonova, K.I.; Grimm, M.J.; et al. Nrf2 amplifies oxidative stress via induction of Klf9. Mol. Cell 2014, 53, 916–928. [Google Scholar] [CrossRef] [Green Version]
- Fink, E.E.; Mannava, S.; Bagati, A.; Bianchi-Smiraglia, A.; Nair, J.R.; Moparthy, K.; Lipchick, B.C.; Drokov, M.; Utley, A.; Ross, J.; et al. Mitochondrial thioredoxin reductase regulates major cytotoxicity pathways of proteasome inhibitors in multiple myeloma cells. Leukemia 2016, 30, 104–111. [Google Scholar] [CrossRef] [Green Version]
- Zhan, F.; Zhan, F.; Huang, Y.; Colla, S.; Stewart, J.P.; Hanamura, I.; Gupta, S.; Epstein, J.; Yaccoby, S.; Sawyer, J.; et al. The molecular classification of multiple myeloma. Blood 2006, 108, 2020–2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, F.; Bashari, M.H.; Morelli, E.; Tonon, G.; Malvestiti, S.; Vallet, S.; Jarahian, M.; Seckinger, A.; Hose, D.; Bakiri, L.; et al. The AP-1 transcription factor JunB is essential for multiple myeloma cell proliferation and drug resistance in the bone marrow microenvironment. Leukemia 2017, 31, 1570–1581. [Google Scholar] [CrossRef]
- Vrábel, D.; Pour, L.; Ševčíková, S. The impact of NF-κB signaling on pathogenesis and current treatment strategies in multiple myeloma. Blood Rev. 2019, 34, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.J.; Van Wier, S.; Tiedemann, R.; Shi, C.X.; Sebag, M.; et al. Promiscuous mutations activate the noncanonical NF-κB pathway in multiple myeloma. Cancer Cell 2007, 12, 131–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W.; et al. Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007, 12, 115–130. [Google Scholar] [CrossRef] [Green Version]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Bruno, T.; De Nicola, F.; Corleone, G.; Catena, V.; Goeman, F.; Pallocca, M.; Sorino, C.; Bossi, G.; Amadio, B.; Cigliana, G.; et al. Che-1/AATF-induced transcriptionally active chromatin promotes cell proliferation in multiple myeloma. Blood Adv. 2020, 4, 5616–5630. [Google Scholar] [CrossRef]
- Ordoñez, R.; Kulis, M.; Russiñol, N.; Chapaprieta, V.; Carrasco-Leon, A.; García-Torre, B.; Charalampopoulou, S.; Clot, G.; Beekman, R.; Meydan, C.; et al. Chromatin activation as a unifying principle underlying pathogenic mechanisms in multiple myeloma. Genome Res. 2020, 30, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Pfarr, N.; Endris, V.; Mai, E.K.; Hanafiah, N.H.M.; Lehners, N.; Penzel, R.; Weichert, W.; Ho, A.D.; Schirmacher, P.; et al. Molecular signaling in multiple myeloma: Association of RAS/RAF mutations and MEK/ERK pathway activation. Oncogenesis 2017, 6, e337. [Google Scholar] [CrossRef] [Green Version]
- Shirazi, F.; Jones, R.J.; Singh, R.K.; Zou, J.; Kuiatse, I.; Berkova, Z.; Wang, H.; Lee, H.C.; Hong, S.; Dick, L.; et al. Activating KRAS, NRAS, and BRAF mutants enhance proteasome capacity and reduce endoplasmic reticulum stress in multiple myeloma. Proc. Natl. Acad. Sci. USA 2020, 117, 20004–20014. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [Green Version]
- Tchakarska, G.; Sola, B. The double dealing of cyclin D1. Cell Cycle 2020, 19, 163–178. [Google Scholar] [CrossRef] [Green Version]
- Bustany, S.; Bourgeais, J.; Tchakarska, G.; Body, S.; Hérault, O.; Gouilleux, F.; Sola, B. Cyclin D1 unbalances the redox status controlling cell adhesion, migration, and drug resistance in myeloma cells. Oncotarget 2016, 7, 45214–45224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caillot, M.; Bourgeais, J.; Dakik, H.; Costé, É.; Mazure, N.M.; Lelièvre, É.; Coqueret, O.; Hérault, O.; Mazurier, F.; Sola, B. Cyclin D1 targets hexokinase 2 to control aerobic glycolysis in myeloma cells. Oncogenesis 2020, 9, 1–13. [Google Scholar] [CrossRef]
- Tchakarska, G.; Roussel, M.; Troussard, X.; Sola, B. Cyclin D1 inhibits mitochondrial activity in B cells. Cancer Res. 2011, 71, 1690–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronca, R.; Ghedini, G.C.; Maccarinelli, F.; Sacco, A.; Locatelli, S.L.; Foglio, E.; Taranto, S.; Grillo, E.; Matarazzo, S.; Castelli, R.; et al. FGF trapping inhibits multiple myeloma growth through c-Myc degradation-induced mitochondrial oxidative stress. Cancer Res. 2020, 80, 2340–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuehl, W.M.; Bergsagel, P.L. MYC addiction: A potential therapeutic target in MM. Blood 2012, 120, 2351–2352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovanović, K.K.; Roche-Lestienne, C.; Ghobrial, I.M.; Facon, T.; Quesnel, B.; Manier, S. Targeting MYC in multiple myeloma. Leukemia 2018, 32, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Punnoose, E.A.; Leverson, J.D.; Peale, F.; Boghaert, E.R.; Belmont, L.D.; Tan, N.; Young, A.; Mitten, M.; Ingalla, E.; Darbonne, W.C.; et al. Expression profile of BCL-2, BCL-X L, and MCL-1 predicts pharmacological response to the BCL-2 selective antagonist Venetoclax in multiple myeloma models. Mol. Cancer Ther. 2016, 15, 1132–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slomp, A.; Peperzak, V. Role and regulation of pro-survival BCL-2 proteins in multiple myeloma. Front. Oncol. 2018, 8, 533. [Google Scholar] [CrossRef]
- Chong, S.J.F.; Lai, J.X.H.; Eu, J.Q.; Bellot, G.L.; Pervaiz, S. Reactive oxygen species and oncoprotein signaling-A dangerous liaison. Antioxid. Redox Signal. 2018, 29, 1553–1588. [Google Scholar] [CrossRef]
- Touzeau, C.; Dousset, C.; Le Gouill, S.; Sampath, D.; Leverson, J.D.; Souers, A.J.; Maïga, S.; Béné, M.C.; Moreau, P.; Pellat-Deceunynck, C.; et al. The Bcl-2 specific BH3 mimetic ABT-199: A promising targeted therapy for t(11;14) multiple myeloma. Leukemia 2013, 28, 210–212. [Google Scholar] [CrossRef]
- Bajpai, R.; Sharma, A.; Achreja, A.; Edgar, C.L.; Wei, C.; Siddiqa, A.A.; Gupta, V.A.; Matulis, S.M.; McBrayer, S.K.; Mittal, A.; et al. Electron transport chain activity is a predictor and target for venetoclax sensitivity in multiple myeloma. Nat. Commun. 2020, 11, 1228. [Google Scholar] [CrossRef]
- Lin, L.; Cao, L.; Liu, Y.; Wang, K.; Zhang, X.; Qin, X.; Zhao, D.; Hao, J.; Chang, Y.; Huang, X.; et al. B7-H3 promotes multiple myeloma cell survival and proliferation by ROS-dependent activation of Src/STAT3 and c-Cbl-mediated degradation of SOCS3. Leukemia 2019, 33, 1475–1486. [Google Scholar] [CrossRef]
- Stroopinsky, D.; Kufe, D.; Avigan, D. MUC1 in hematological malignancies. Leuk. Lymphoma 2016, 57, 2489–2498. [Google Scholar] [CrossRef]
- Yin, L.; Kosugi, M.; Kufe, D. Inhibition of the MUC1-C oncoprotein induces multiple myeloma cell death by down-regulating TIGAR expression and depleting NADPH. Blood 2012, 119, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Kufe, T.; Avigan, D.; Kufe, D. Targeting MUC1-C is synergistic with bortezomib in downregulating TIGAR and inducing ROS-mediated myeloma cell death. Blood 2014, 123, 2997–3006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, M.F.; Johnson, D.C.; Wu, P.; Walker, B.A.; Brioli, A.; Mirabella, F.; Wardell, C.P.; Melchor, L.; Davies, F.E.; Morgan, G.J. Global methylation analysis identifies prognostically important epigenetically inactivated tumor suppressor genes in multiple myeloma. Blood 2013, 122, 219–226. [Google Scholar] [CrossRef]
- Raninga, P.V.; Di Trapani, G.; Vuckovic, S.; Bhatia, M.; Tonissen, K.F. Inhibition of thioredoxin 1 leads to apoptosis in drug-resistant multiple myeloma. Oncotarget 2015, 6, 15410–15424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dytfeld, D.; Luczak, M.; Wrobel, T.; Usnarska-Zubkiewicz, L.; Brzezniakiewicz, K.; Jamroziak, K.; Giannopoulos, K.; Przybylowicz-Chalecka, A.; Ratajczak, B.; Czerwinska-Rybak, J.; et al. Comparative proteomic profiling of refractory/relapsed multiple myeloma reveals biomarkers involved in resistance to bortezomib-based therapy. Oncotarget 2016, 7, 56726–56736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raninga, P.V.; Di Trapani, G.; Vuckovic, S.; Tonissen, K.F. TrxR1 inhibition overcomes both hypoxia-induced and acquired bortezomib resistance in multiple myeloma through NF-κβ inhibition. Cell Cycle 2016, 15, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Salem, K.; McCormick, M.L.; Wendlandt, E.; Zhan, F.; Goel, A. Copper–zinc superoxide dismutase-mediated redox regulation of bortezomib resistance in multiple myeloma. Redox Biol. 2015, 4, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Song, I.S.; Kim, H.K.; Lee, S.R.; Jeong, S.H.; Kim, N.; Ko, K.S.; Rhee, B.D.; Han, J. Mitochondrial modulation decreases the bortezomib-resistance in multiple myeloma cells. Int. J. Cancer 2013, 133, 1357–1367. [Google Scholar] [CrossRef]
- Du, T.; Song, Y.; Ray, A.; Chauhan, D.; Anderson, K.C. Proteomic analysis identifies mechanism(s) of overcoming bortezomib resistance via targeting ubiquitin receptor Rpn13. Leukemia 2021, 35, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Bloedjes, T.A.; de Wilde, G.; Guikema, J.E.J. Metabolic effects of recurrent genetic aberrations in multiple myeloma. Cancers 2021, 13, 396. [Google Scholar] [CrossRef]
- Dalva-Aydemir, S.; Bajpai, R.; Martinez, M.; Adekola, K.U.A.; Kandela, I.; Wei, C.; Singhal, S.; Koblinski, J.E.; Raje, N.S.; Rosen, S.T.; et al. Targeting the metabolic plasticity of multiple myeloma with FDA-approved ritonavir and metformin. Clin. Cancer Res. 2015, 21, 1161–1171. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, S.; Kawano, Y.; Yuki, H.; Okuno, Y.; Nosaka, K.; Mitsuya, H.; Hata, H. PDK1 inhibition is a novel therapeutic target in multiple myeloma. Br. J. Cancer 2013, 108, 170–178. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, S.; Wada, N.; Kawano, Y.; Okuno, Y.; Kikukawa, Y.; Endo, S.; Nishimura, N.; Ueno, N.; Mitsuya, H.; Hata, H. Lactate, a putative survival factor for myeloma cells, is incorporated by myeloma cells through monocarboxylate transporters 1. Exp. Hematol. Oncol. 2015, 4, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Xia, J.; Zhang, J.; Zhu, Y.; Wu, Y.; Guo, J.; Chen, S.; Lei, Q.; Meng, B.; Kuang, C.; et al. Phosphoglycerate dehydrogenase promotes proliferation and bortezomib resistance through increasing reduced glutathione synthesis in multiple myeloma. Br. J. Haematol. 2020, 190, 52–66. [Google Scholar] [CrossRef]
- Cagnetta, A.; Cea, M.; Patrone, F.; Nencioni, A.; Gobbi, M.; Anderson, K.C. Intracellular NAD+ depletion induces autophagic death in multiple myeloma cells. Autophagy 2013, 9, 410–412. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Wang, Y.; Liu, H.; Xu, X.; He, S.; Tang, J.; Huang, Y.; Miao, X.; Wu, Y.; Wang, Q.; et al. Pyruvate kinase isoform M2 (PKM2) participates in multiple myeloma cell proliferation, adhesion and chemoresistance. Leuk. Res. 2015, 39, 1428–1436. [Google Scholar] [CrossRef]
- Gu, Z.; Xia, J.; Xu, H.; Frech, I.; Tricot, G.; Zhan, F. NEK2 promotes aerobic glycolysis in multiple myeloma through regulating splicing of pyruvate kinase. J. Hematol. Oncol. 2017, 10, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolzoni, M.; Chiu, M.; Accardi, F.; Vescovini, R.; Airoldi, I.; Storti, P.; Todoerti, K.; Agnelli, L.; Missale, G.; Andreoli, R.; et al. Dependence on glutamine uptake and glutamine addiction characterize myeloma cells: A new attractive target. Blood 2016, 128, 667–679. [Google Scholar] [CrossRef]
- McBrayer, S.K.; Yarrington, M.; Qian, J.; Feng, G.; Shanmugam, M.; Gandhi, V.; Krett, N.L.; Rosen, S.T. Integrative gene expression profiling reveals G6PD-mediated resistance to RNA-directed nucleoside analogues in B-cell neoplasms. PLoS ONE 2012, 7, e41455. [Google Scholar] [CrossRef] [PubMed]
- Marlein, C.R.; Piddock, R.E.; Mistry, J.J.; Zaitseva, L.; Hellmich, C.; Horton, R.H.; Zhou, Z.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. CD38-driven mitochondrial trafficking promotes bioenergetic plasticity in multiple myeloma. Cancer Res. 2019, 79, 2285–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbato, A.; Scandura, G.; Puglisi, F.; Cambria, D.; La Spina, E.; Palumbo, G.A.; Lazzarino, G.; Tibullo, D.; Di Raimondo, F.; Giallongo, C.; et al. Mitochondrial bioenergetics at the onset of drug resistance in hematological malignancies: An overview. Front. Oncol. 2020, 10, 604143. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Yu, W.; Franqui-Machin, R.; Bates, M.L.; Nadiminti, K.; Cao, H.; Amendt, B.A.; Jethava, Y.; Frech, I.; Zhan, F.; et al. Alteration of mitochondrial biogenesis promotes disease progression in multiple myeloma. Oncotarget 2017, 8, 111213–111224. [Google Scholar] [CrossRef]
- Tibullo, D.; Giallongo, C.; Romano, A.; Vicario, N.; Barbato, A.; Puglisi, F.; Parenti, R.; Amorini, A.M.; Saab, M.W.; Tavazzi, B.; et al. Mitochondrial functions, energy metabolism and protein glycosylation are interconnected processes mediating resistance to bortezomib in multiple myeloma cells. Biomolecules 2020, 10, 696. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Smyth, M.J.; Martinet, L. Cancer immunoediting and immune dysregulation in multiple myeloma. Blood 2020, 136, 2731–2740. [Google Scholar] [CrossRef] [PubMed]
- Lomas, O.C.; Tahri, S.; Ghobria, I.M. The microenvironment in myeloma. Curr. Opin. Oncol. 2020, 32, 170–175. [Google Scholar] [CrossRef]
- Capp, J.P.; Bataille, R. Multiple myeloma as a bone disease? The tissue disruption-induced cell stochasticity (TiDiS) theory. Cancers 2020, 12, 2158. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Nishiwaki, K.; Yano, S. Treatment strategies considering micro-environment and clonal evolution in multiple myeloma. Cancers 2021, 13, 215. [Google Scholar] [CrossRef]
- Morris, E.V.; Edwards, M. Morphogens and growth factor signalling in the myeloma bone-lining niche. Cell. Mol. Life Sci. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Beider, K.; Bitner, H.; Leiba, M.; Gutwein, O.; Koren-Michowitz, M.; Ostrovsky, O.; Abraham, M.; Wald, H.; Galun, E.; Peled, A.; et al. Multiple myeloma cells recruit tumor-supportive macrophages through the CXCR4/CXCL12 axis and promote their polarization toward the M2 phenotype. Oncotarget 2014, 5, 11283–11896. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Li, Y.; Yan, H.; Zhang, E.; Huang, X.; Chen, Q.; Chen, J.; Qu, J.; Liu, Y.; He, J.; et al. CCL2 promotes macrophages-associated chemoresistance via MCPIP1 dual catalytic activities in multiple myeloma. Cell Death Dis. 2019, 10, 781. [Google Scholar] [CrossRef] [PubMed]
- Maj, T.; Wang, W.; Crespo, J.; Zhang, H.; Wang, W.; Wei, S.; Zhao, L.; Vatan, L.; Shao, I.; Szeliga, W.; et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol. 2017, 18, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Gunes, E.G.; Rosen, S.T.; Querfeld, C. The role of myeloid-derived suppressor cells in hematologic malignancies. Curr. Opin. Oncol. 2020, 32, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, I.R.; Martner, A.; Pisklakova, A.; Condamine, T.; Chase, T.; Vogl, T.; Roth, J.; Gabrilovich, D.; Nefedova, Y. Myeloid-derived suppressor cells regulate growth of multiple myeloma by inhibiting T cells in bone marrow. J. Immunol. 2013, 190, 3815–3823. [Google Scholar] [CrossRef] [Green Version]
- Corzo, C.A.; Cotter, M.J.; Cheng, P.; Cheng, F.; Kusmartsev, S.; Sotomayor, E.; Padhya, T.; McCaffrey, T.V.; McCaffrey, J.C.; Gabrilovich, D.I. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J. Immunol. 2009, 182, 5693–5701. [Google Scholar] [CrossRef]
- D’Souza, C.; Prince, H.M.; Neeson, P.J. Understanding the role of T-cells in the antimyeloma effect of immunomodulatory drugs. Front. Immunol. 2021, 12, 632399. [Google Scholar] [CrossRef] [PubMed]
- Zelle-Rieser, C.; Thangavadivel, S.; Biedermann, R.; Brunner, A.; Stoitzner, P.; Willenbacher, E.; Greil, R.; Jöhrer, K. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J. Hematol. Oncol. 2016, 9, 116. [Google Scholar] [CrossRef] [Green Version]
- Fauriat, C.; Mallet, F.; Olive, D.; Costello, R.T. Impaired activating receptor expression pattern in natural killer cells from patients with multiple myeloma. Leukemia 2006, 20, 732–733. [Google Scholar] [CrossRef]
- Kotsafti, A.; Scarpa, M.; Castagliuolo, I.; Scarpa, M. Reactive oxygen species and antitumor immunity-From surveillance to evasion. Cancers 2020, 12, 1748. [Google Scholar] [CrossRef]
- Goel, A.; Spitz, D.R.; Weiner, G.J. Manipulation of cellular redox parameters for improving therapeutic responses in B-cell lymphoma and multiple yeloma. J. Biol. Chem. 2012, 113, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Tessoulin, B.; Descamps, G.; Moreau, P.; Maïga, S.; Lode, L.; Godon, C.; Marionneau-Lambot, S.; Oullier, T.; Le Gouill, S.; Amiot, M.; et al. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood 2014, 124, 1626–1636. [Google Scholar] [CrossRef] [Green Version]
- Saha, M.N.; Abdi, J.; Yang, Y.; Chang, H. miRNA-29a as a tumor suppressor mediates PRIMA-1Met-induced anti-myeloma activity by targeting c-Myc. Oncotarget 2016, 7, 7149–7160. [Google Scholar] [CrossRef]
- Saha, M.N.; Jiang, H.; Yang, Y.; Reece, D.; Chang, H. PRIMA-1Met/APR-246 displays high antitumor activity in multiple myeloma by induction of p73 and Noxa. Mol. Cancer Ther. 2013, 12, 2331–2341. [Google Scholar] [CrossRef] [Green Version]
- Tessoulin, B.; Descamps, G.; Dousset, C.; Amiot, M.; Pellat-Deceunynck, C. Targeting oxidative stress with auranofin or Prima-1Met to circumvent p53 or Bax/Bak deficiency in myeloma cells. Front. Oncol. 2019, 9, 128. [Google Scholar] [CrossRef] [PubMed]
- Teoh, P.J.; Bi, C.; Sintosebastian, C.; Tay, L.S.; Fonseca, R.; Chng, W.J. PRIMA-1 targets the vulnerability of multiple myeloma of deregulated protein homeostasis through the perturbation of ER stress via p73 demethylation. Oncotarget 2016, 7, 61806–61819. [Google Scholar] [CrossRef] [Green Version]
- Marin Hernandez, E.; Paek, H.; Li, M.; Ban, Y.; Karaga, M.K.; Shashidharamurthy, R.; Wang, X. Caffeic acid phenethyl ester exerts apoptotic and oxidative stress on human multiple myeloma cells. Investig. New Drugs 2018, 37, 837–848. [Google Scholar] [CrossRef]
- Raninga, P.V.; Di Trapani, G.; Vuckovic, S.; Tonissen, K.F. Cross-talk between two antioxidants, thioredoxin reductase and heme oxygenase-1, and therapeutic implications for multiple myeloma. Redox Biol. 2016, 8, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Fan, S.; Zheng, J.; Huang, W.; Gasparetto, C.; Chao, N.J.; Hu, J.; Kang, Y. Inhibition of thioredoxin activates mitophagy and overcomes adaptive bortezomib resistance in multiple myeloma. J. Hematol. Oncol. 2018, 11, 29. [Google Scholar] [CrossRef] [Green Version]
- Sebastian, S.; Zhu, Y.X.; Braggio, E.; Shi, C.X.; Panchabhai, S.C.; Van Wier, S.A.; Ahmann, G.J.; Chesi, M.; Bergsagel, P.L.; Stewart, A.K.; et al. Multiple myeloma cells’ capacity to decompose H2O2 determines lenalidomide sensitivity. Blood 2017, 129, 991–1007. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Wu, Y.; Liang, D.; Feng, L. Scutellarein selectively targets multiple myeloma cells by increasing mitochondrial superoxide production and activating intrinsic apoptosis pathway. Biomed. Pharmacol. 2019, 109, 2109–2118. [Google Scholar] [CrossRef] [PubMed]
- Vatolin, S.; Phillips, J.G.; Jha, B.K.; Govindgari, S.; Hu, J.; Grabowski, D.; Parker, Y.; Lindner, D.J.; Zhong, F.; Distelhorst, C.W.; et al. Novel protein disulfide isomerase inhibitor with anticancer activity in multiple myeloma. Cancer Res. 2016, 76, 3340–3350. [Google Scholar] [CrossRef] [Green Version]
- Robinson, R.M.; Reyes, L.; Duncan, R.M.; Bian, H.; Reitz, A.B.; Manevich, Y.; McClure, J.J.; Champion, M.M.; Chou, C.J.; Sharik, M.E.; et al. Inhibitors of the protein disulfide isomerase family for the treatment of multiple myeloma. Leukemia 2019, 33, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.M.; Reyes, L.; Duncan, R.M.; Bian, H.; Strobel, E.D.; Hyman, S.L.; Reitz, A.B.; Dolloff, N.G. Tuning isoform selectivity and bortezomib sensitivity with a new class of alkenyl indene PDI inhibitor. Eur. J. Med. Chem. 2020, 186, 111906. [Google Scholar] [CrossRef]
- Soncini, D.; Minetto, P.; Martinuzzi, C.; Becherini, P.; Fenu, V.; Guolo, F.; Todoerti, K.; Calice, G.; Contini, P.; Miglino, M.; et al. Amino acid depletion triggered by ʟ-asparaginase sensitizes MM cells to carfilzomib by inducing mitochondria ROS-mediated cell death. Blood Adv. 2020, 4, 4312–4326. [Google Scholar] [CrossRef]
- Xu, S.; Zhou, T.; Doh, H.M.; Trinh, K.R.; Catapang, A.; Lee, J.T.; Braas, D.; Bayley, N.A.; Yamada, R.E.; Vasuthasawat, A.; et al. An HK2 antisense oligonucleotide induces synthetic lethality in HK1−HK2+ multiple myeloma. Cancer Res. 2019, 79, 2748–2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Fang, B.; Liu, Y.; Yan, S.; Cao, D.; Mei, H.; Wang, Q.; Hu, Y.; Guo, T. SR18292 exerts potent antitumor effects in multiple myeloma via inhibition of oxidative phosphorylation. Life Sci. 2020, 256, 117971. [Google Scholar] [CrossRef] [PubMed]
- Sze, J.H.; Raninga, P.V.; Nakamura, K.; Casey, M.; Khanna, K.K.; Berners-Price, S.J.; Di Trapani, G.; Tonissen, K.F. Anticancer activity of a Gold(I) phosphine thioredoxin reductase inhibitor in multiple myeloma. Redox Biol. 2020, 28, 101310. [Google Scholar] [CrossRef]
- Mountjoy, L.; Sebastian, S.; Jain, T.; Hilal, T.; Gonzalez-Velez, M.; Girardo, M.; Ahmann, G.; Larsen, J.; Bergsagel, L.; Fonseca, R. Prediction of immunomodulatory drugs (IMiDs) sensitivity in myeloma via determination of baseline anti-oxidative stress capacity. Leukemia 2020, 34, 3060–3063. [Google Scholar] [CrossRef]
- Ray, A.; Song, Y.; Chauhan, D.; Anderson, K.C. Blockade of ubiquitin receptor Rpn13 in plasmacytoid dendritic cells triggers anti-myeloma immunity. Blood Cancer J. 2019, 9, 64. [Google Scholar] [CrossRef] [Green Version]
- McBrayer, S.K.; Cheng, J.C.; Singhal, S.; Krett, N.L.; Rosen, S.T.; Shanmugam, M. Multiple myeloma exhibits novel dependence on GLUT4, GLUT8, and GLUT11: Implications for glucose transporter-directed therapy. Blood 2012, 119, 4686–4697. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.; Jimi, S.; Migita, K.; Takamatsu, Y.; Hara, S. Inhibition of glucose transporter 1 induces apoptosis and sensitizes multiple myeloma cells to conventional chemotherapeutic agents. Leuk. Res. 2016, 41, 103–110. [Google Scholar] [CrossRef]
- Niedzwiecka, K.; Dylag, M.; Augustyniak, D.; Majkowska-Skrobek, G.; Cal-Bakowska, M.; Ko, Y.H.; Pedersen, P.L.; Goffeau, A.; Ułaszewski, S. Glutathione may have implications in the design of 3-bromopyruvate treatment protocols for both fungal and algal infections as well as multiple myeloma. Oncotarget 2017, 7, 65614–65626. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.; Song, Y.; Du, T.; Chauhan, D.; Anderson, K.C. Preclinical validation of alpha-enolase (ENO1) as a novel immunometabolic target in multiple myeloma. Oncogene 2020, 39, 2786–2796. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi, P.; Vandsemb, E.N.; Elsaadi, S.; Røst, L.M.; Yang, R.; Hjort, M.A.; Andreassen, T.; Misund, K.; Slørdahl, T.S.; Rø, T.B.; et al. Phosphatase of regenerating liver-3 regulates cancer cell metabolism in multiple myeloma. FASEB J. 2021, 35, e21344. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.S.Y.; Zhou, J.; Lim, J.S.L.; Hee, Y.T.; Chooi, J.Y.; Chung, T.H.; Tan, Z.T.; Zeng, Q.; Waller, D.D.; Sebag, M.; et al. IL6 promotes a STAT3-PRL3 feedforward loop via SHP2 repression in multiple myeloma. Cancer Res. 2019, 79, 4679–4688. [Google Scholar] [CrossRef]
- Effenberger, M.; Bommert, K.S.; Kunz, V.; Kruk, J.; Leich, E.; Rudelius, M.; Bargou, R.; Bommert, K. Glutaminase inhibition in multiple myeloma induces apoptosis via MYC degradation. Oncotarget 2017, 8, 85858–85867. [Google Scholar] [CrossRef] [Green Version]
- Gonsalves, W.I.; Jang, J.S.; Jessen, E.; Hitosugi, T.; Evans, L.A.; Jevremovic, D.; Pettersson, X.M.; Bush, A.G.; Gransee, J.; Anderson, E.I.; et al. In vivo assessment of glutamine anaplerosis into the TCA cycle in human pre-malignant and malignant clonal plasma cells. Cancer Metab. 2020, 8, 29. [Google Scholar] [CrossRef]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef]
- Tirado-Vélez, J.M.; Joumady, I.; Sáez-Benito, A.; Cózar-Castellano, I.; Perdomo, G. Inhibition of fatty acid metabolism reduces human myeloma cells proliferation. PLoS ONE 2012, 7, e46484. [Google Scholar] [CrossRef] [PubMed]
- Trotter, T.N.; Gibson, J.T.; Sherpa, T.L.; Gowda, P.S.; Peker, D.; Yang, Y. Adipocyte-lineage cells support growth and dissemination of multiple myeloma in bone. Am. J. Pathol. 2016, 186, 3054–3063. [Google Scholar] [CrossRef] [Green Version]
- Masarwi, M.; DeSchiffart, A.; Ham, J.; Reagan, M.R. Multiple myeloma and fatty acid metabolism. JBMR Plus 2019, 3, e10173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, L.M.; Gridley, G.; Pottern, L.M.; Baris, D.; Swanso, C.A.; Silverman, D.T.; Hayes, R.B.; Greenberg, R.S.; Swanson, G.M.; Schoenberg, J.B.; et al. Diet and nutrition as risk factors for multiple myeloma among blacks and whites in the United States. Cancer Causes Control 2001, 12, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Hosgood, H.D., 3rd; Baris, D.; Zahm, S.H.; Zheng, T.; Cross, A.J. Diet and risk of multiple myeloma in Connecticut women. Cancer Causes Control 2007, 18, 1065–1076. [Google Scholar] [CrossRef]
- Lee, D.H.; Fung, T.T.; Tabung, F.K.; Marinac, C.R.; Devore, E.E.; Rosner, B.A.; Ghobrial, I.M.; Colditz, G.A.; Giovannucci, E.L.; Birmann, B.M. Prediagnosis dietary pattern and survival in patients with multiple myeloma. Int. J. Cancer 2020, 147, 1823–1830. [Google Scholar] [CrossRef]
- Thordardottir, M.; Lindqvist, E.K.; Lund, S.H.; Costello, R.; Burton, D.; Steingrimsdottir, L.; Korde, N.; Mailankody, S.; Eiriksdottir, G.; Launer, L.J.; et al. Dietary intake is associated with risk of multiple myeloma and its precursor disease. PLoS ONE 2018, 13, e0206047. [Google Scholar] [CrossRef]
- Janker, L.; Mayer, R.L.; Bileck, A.; Kreutz, D.; Mader, J.C.; Utpatel, K.; Heudobler, D.; Agis, H.; Gerner, C.; Slany, A. Metabolic, anti-apoptotic and immune evasion strategies of primary human myeloma cells indicate adaptations to hypoxia. Mol. Cell Proteom. 2019, 18, 936–953. [Google Scholar] [CrossRef]
- Uckun, F.M. Overcoming the immunosuppressive tumor microenvironment in multiple myeloma. Cancers 2021, 13, 2018. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Tejedor, A.; Lorenzo-Mohamed, M.; Puig, N.; García-Sanz, R.; Mateos, M.V.; Garayoa, M.; Paíno, T. Immune system alterations in multiple myeloma: Molecular mechanisms and therapeutic strategies to reverse immunosuppression. Cancers 2021, 13, 1353. [Google Scholar] [CrossRef]
- Tamura, H.; Ishibashi, M.; Sunakawa-Kii, M.; Inokuchi, K. PD-L1-PD-1 pathway in the pathophysiology of multiple myeloma. Cancers 2020, 12, 924. [Google Scholar] [CrossRef]
- Tremblay-Lemay, R.; Rastgoo, N.; Chang, H. Modulating PD-L1 expression in multiple myeloma: An alternative strategy to target the PD-1/PD-L1 pathway. J. Hematol. Oncol. 2018, 11, 46. [Google Scholar] [CrossRef] [Green Version]
- Ryu, D.; Kim, S.J.; Hong, Y.; Jo, A.; Kim, N.; Kim, H.J.; Lee, H.O.; Kim, K.; Park, W.Y. Alterations in the transcriptional programs of myeloma cells and the microenvironment during extramedullary progression affect proliferation and immune evasion. Clin. Cancer Res. 2020, 26, 935–944. [Google Scholar] [CrossRef] [Green Version]
- Barsoum, I.B.; Smallwood, C.A.; Siemens, D.R.; Graham, C.H. A Mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014, 74, 665–674. [Google Scholar] [CrossRef] [Green Version]
- Görgün, G.T.; Whitehill, G.; Anderson, J.L.; Hideshima, T.; Maguire, C.; Laubach, J.; Raje, N.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood 2013, 121, 2975–2987. [Google Scholar] [CrossRef] [Green Version]
- de Beule, N.; de Veirman, K.; Maes, K.; de Bruyne, E.; Menu, E.; Breckpot, K.; de Raeve, H.; van Rampelbergh, R.; van Ginderachter, J.A.; Schots, R.; et al. Tumour-associated macrophage-mediated survival of myeloma cells through STAT3 activation. J. Pathol. 2017, 241, 534–546. [Google Scholar] [CrossRef]
- Sarkar, S.; Germeraad, W.T.V.; Rouschop, K.M.A.; Steeghs, E.M.P.; van Gelder, M.; Bos, G.M.J.; Wieten, L. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS ONE 2013, 8, e64835. [Google Scholar] [CrossRef]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; de Milito, A.; Ricupito, A.; Cova, A.; Canese, R.; Jachetti, E.; Rossetti, M.; et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012, 72, 2746–2756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohn, T.; Rapp, S.; Luther, N.; Klein, M.; Bruehl, T.J.; Kojima, N.; Aranda Lopez, P.; Hahlbrock, J.; Muth, S.; Endo, S.; et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat. Immunol. 2018, 19, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.C. Progress and paradigms in multiple myeloma. Clin. Cancer Res. 2016, 22, 5419–5427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milan, E.; Fabbri, M.; Cenci, S. Autophagy in plasma cell ontogeny and malignancy. J. Clin. Immunol. 2016, 36 (Suppl. 1), 18–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Lernia, G.; Leone, P.; Solimando, A.G.; Buonavoglia, A.; Saltarella, I.; Ria, R.; Ditonno, P.; Silvestris, N.; Crudele, L.; Vacca, A.; et al. Bortezomib treatment modulates autophagy in multiple myeloma. J. Clin. Med. 2020, 9, 552. [Google Scholar] [CrossRef] [Green Version]
- Fucci, C.; Resnati, M.; Riva, E.; Perini, T.; Ruggieri, E.; Orfanelli, U.; Paradiso, F.; Cremasco, F.; Raimondi, A.; Pasqualetto, E.; et al. The interaction of the tumor suppressor FAM46C with p62 and FNDC3 proteins integrates protein and secretory homeostasis. Cell Rep. 2020, 32, 108162. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Tian, F.; Ma, H.; Wang, H.; Yang, W.; Liu, Z.; Liao, A. FTY720 induces ferroptosis and autophagy via PP2A/AMPK pathway in multiple myeloma cells. Life Sci. 2020, 260, 118077. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Gao, M.; Kong, Y.; Yang, G.; Gao, L.; Shi, J. Multiple myeloma cancer stem cells. Oncotarget 2016, 7, 35466–35477. [Google Scholar] [CrossRef] [Green Version]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Tagde, A.; Singh, H.; Kang, M.H.; Reynolds, C.P. The glutathione synthesis inhibitor buthionine sulfoximine synergistically enhanced melphalan activity against preclinical models of multiple myeloma. Blood Cancer J. 2014, 4, e229. [Google Scholar] [CrossRef] [Green Version]
- Gourzones, C.; Bellanger, C.; Lamure, S.; Gadacha, O.; Garcia De Paco, E.; Vincent, L.; Cartron, G.; Klein, B.; Moreaux, J. Antioxidant defenses confer resistance to high dose melphalan in multiple myeloma cells. Cancers 2019, 11, 439. [Google Scholar] [CrossRef] [Green Version]
- Zub, K.A.; Mittelstedt Leal de Sousa, M.; Sarno, A.; Sharma, A.; Demirovic, A.; Rao, S.; Young, C.; Aas, P.A.; Ericsson, I.; Sundan, A.; et al. Modulation of cell metabolic pathways and oxidative stress signaling contribute to acquired melphalan resistance in multiple myeloma cells. PLoS ONE 2015, 10, e0119857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, P.G.; Oriol, A.; Larocca, A.; Bladé, J.; Cavo, M.; Rodriguez-Otero, P.; Leleu, X.; Nadeem, O.; Hiemenz, J.W.; Hassoun, H.; et al. HORIZON (OP-106) investigators. Melflufen and dexamethasone in heavily pretreated relapsed and refractory multiple myeloma. J. Clin. Oncol. 2021, 39, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Ray, A.; Viktorsson, K.; Spira, J.; Paba-Prada, C.; Munshi, N.; Richardson, P.; Lewensohn, R.; Anderson, K.C. In vitro and in vivo antitumor activity of a novel alkylating agent, melphalan-flufenamide, against multiple myeloma cells. Clin. Cancer Res. 2013, 19, 3019–3031. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Lu, W.; Chen, G.; Zhang, H.; Jia, Y.; Wei, Y.; Yang, H.; Zhang, W.; Fiskus, W.; Bhalla, K.; et al. Overcoming resistance to histone deacetylase inhibitors in human leukemia with the redox modulating compound β-phenylethyl isothiocyanate. Blood 2010, 116, 2732–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, Y.; Hirano, M.; Kobayashi, M.; Futami, M.; Tojo, A. HDAC inhibitors exert anti-myeloma effects through multiple modes of action. Cancers 2019, 11, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caillot, M.; Dakik, H.; Mazurier, F.; Sola, B. Targeting Reactive Oxygen Species Metabolism to Induce Myeloma Cell Death. Cancers 2021, 13, 2411. https://doi.org/10.3390/cancers13102411
Caillot M, Dakik H, Mazurier F, Sola B. Targeting Reactive Oxygen Species Metabolism to Induce Myeloma Cell Death. Cancers. 2021; 13(10):2411. https://doi.org/10.3390/cancers13102411
Chicago/Turabian StyleCaillot, Mélody, Hassan Dakik, Frédéric Mazurier, and Brigitte Sola. 2021. "Targeting Reactive Oxygen Species Metabolism to Induce Myeloma Cell Death" Cancers 13, no. 10: 2411. https://doi.org/10.3390/cancers13102411
APA StyleCaillot, M., Dakik, H., Mazurier, F., & Sola, B. (2021). Targeting Reactive Oxygen Species Metabolism to Induce Myeloma Cell Death. Cancers, 13(10), 2411. https://doi.org/10.3390/cancers13102411