
Rely on Each Other: DNA Binding Cooperativity Shapes p53 Functions in Tumor Suppression and Cancer Therapy



{kind=link}
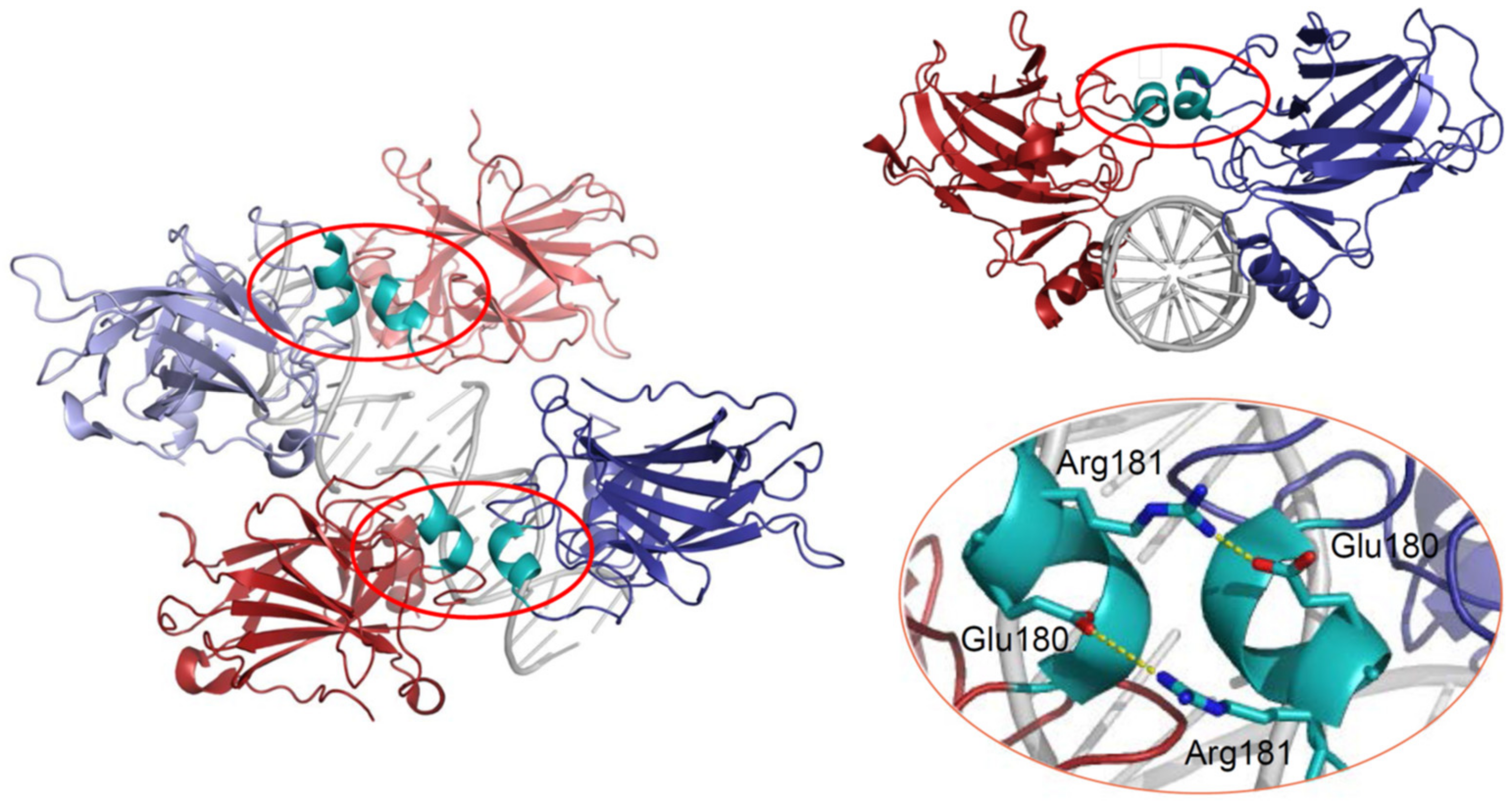{kind=link}
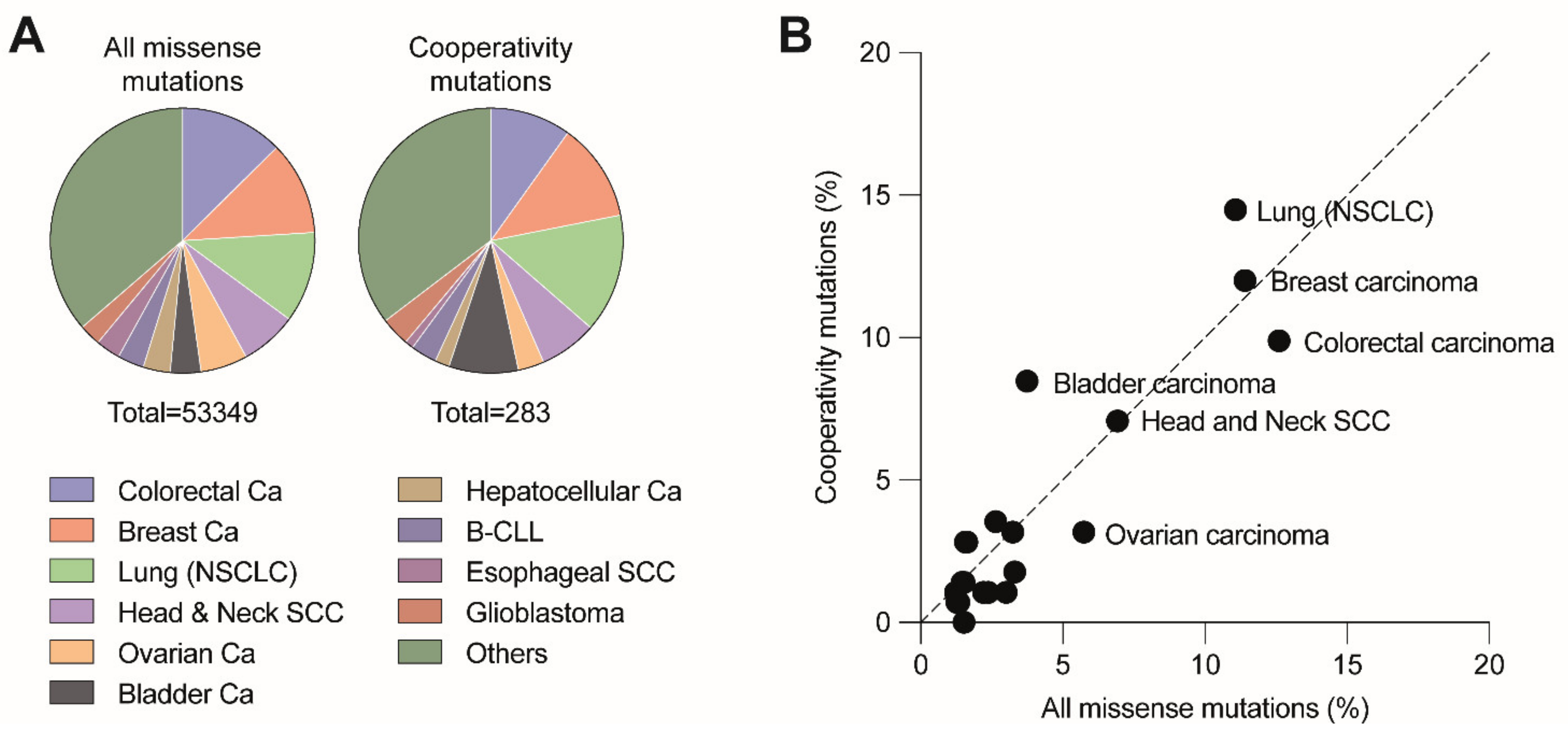{kind=link}
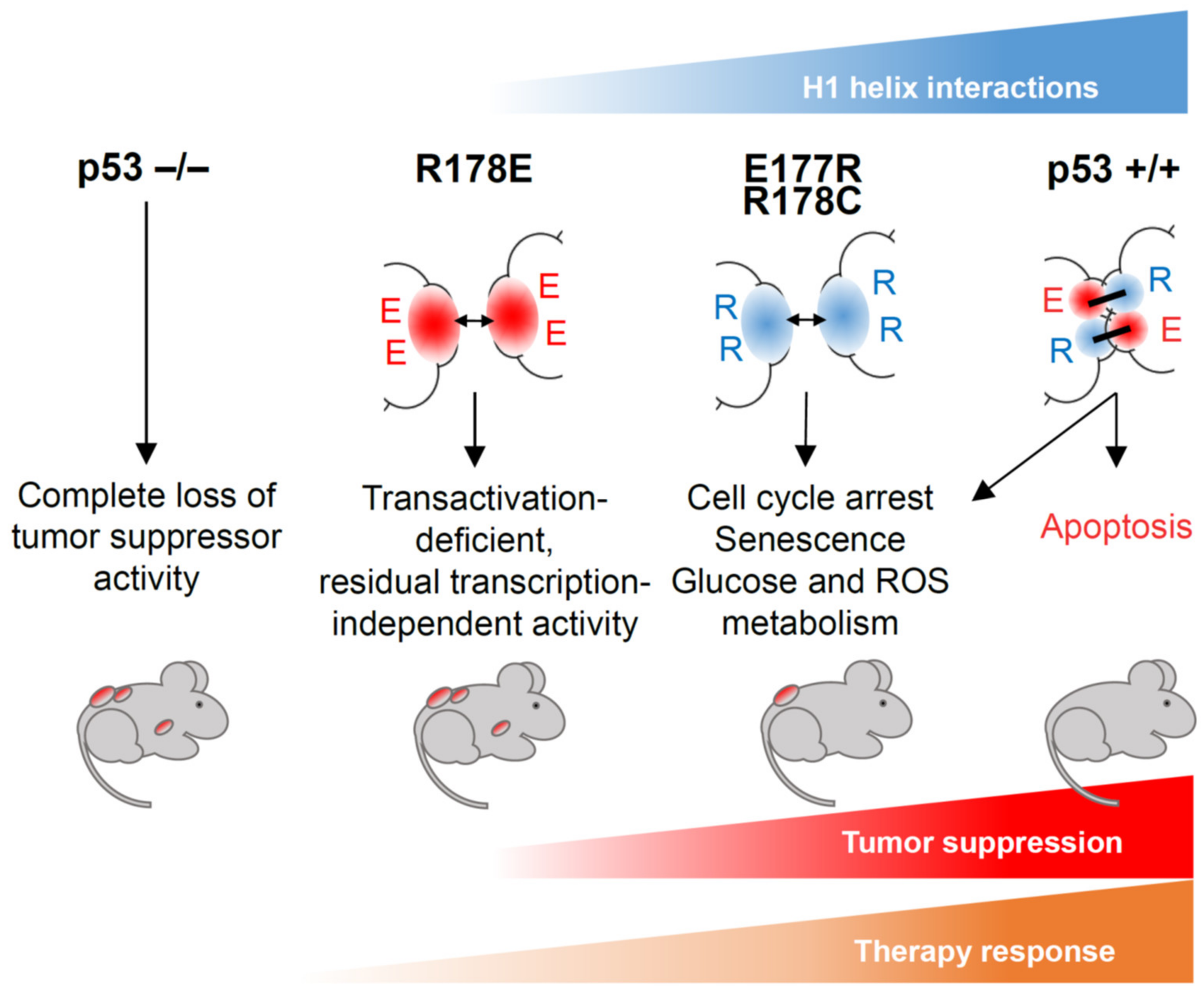{kind=link}
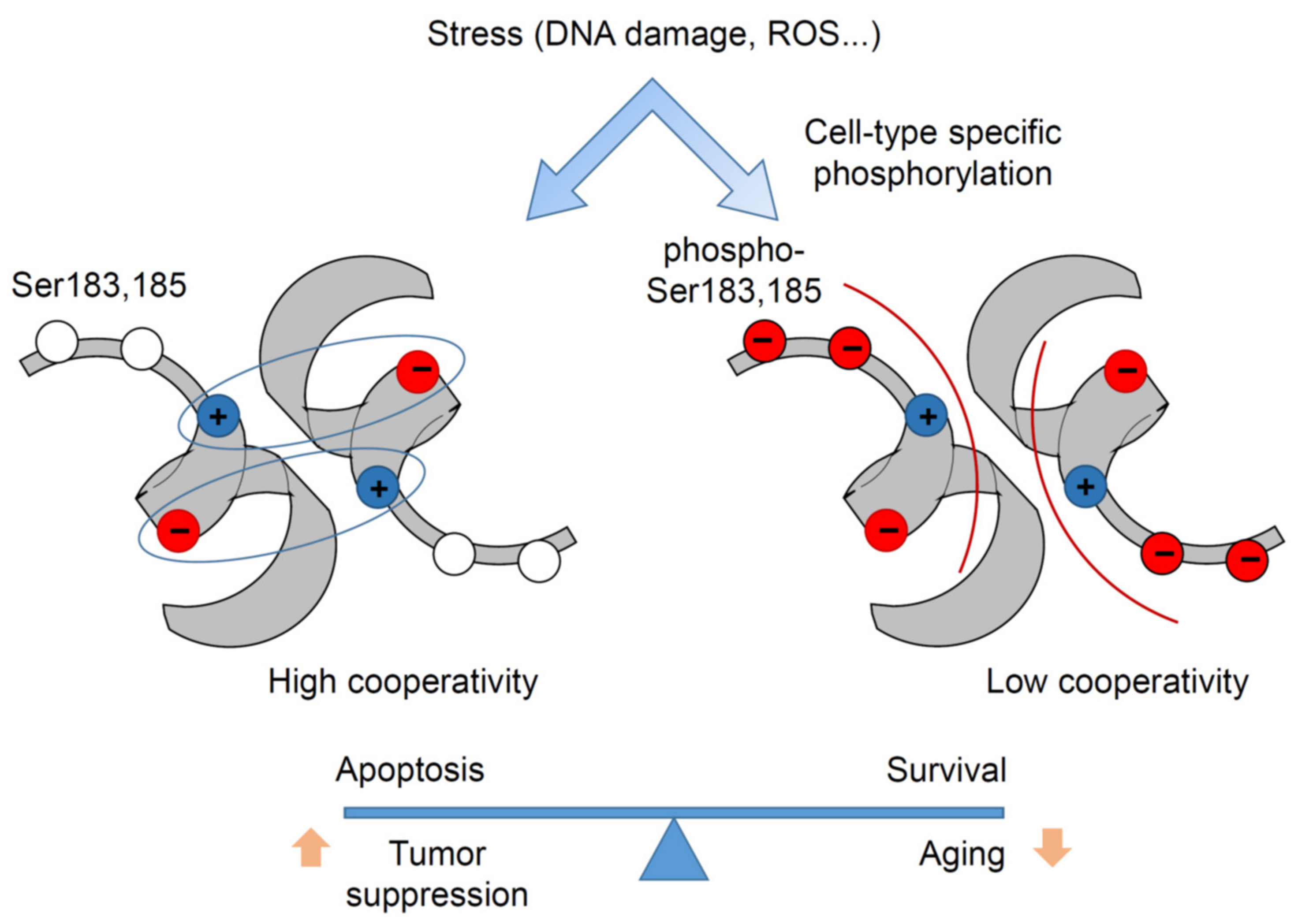{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Cooperative DNA Binding by p53
3. Cancer-Associated Mutations of the DBD—Structural Implications
3.1. Stuctural Mutations
3.2. Contact Mutations
3.3. Cooperativity Mutations
4. Functional Consequences of Cooperativity Mutations
4.1. Effects of Cooperativity Mutations on p53 DNA Binding
4.2. Cooperativity Mutations Affect Transcriptional Activity of p53
5. Cooperativity and Tumor Suppression
5.1. Trp53E177R Mouse
5.2. Trp53R178C Mouse
5.3. Trp53R178E Mouse
6. Mechanisms of Regulation of DNA Binding Cooperativity
6.1. H1 Helix Phosphorylation
6.2. Contribution of CTD to Cooperativity
7. Cooperativity Mutations and Therapy
7.1. Boosting Mitochondrial Apoptosis Using BH3-Mimetics
7.2. ROS-Mediated Therapy
7.3. Targeting Senescence
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Belyi, V.A.; Ak, P.; Markert, E.; Wang, H.; Hu, W.; Puzio-Kuter, A.; Levine, A.J. The origins and evolution of the p53 family of genes. Cold Spring Harb. Perspect. Biol. 2010, 2, a001198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, A.J. The many faces of p53: Something for everyone. J. Mol. Cell Biol. 2019, 11, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Khoury, M.P.; Bourdon, J.C. The isoforms of the p53 protein. Cold Spring Harb. Perspect. Biol. 2010, 2, a000927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieler, M.; Sanyal, S. P53 isoforms and their implications in cancer. Cancers 2018, 10, 288. [Google Scholar] [CrossRef] [Green Version]
- Joerger, A.C.; Fersht, A.R. The tumor suppressor p53: From structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2010, 2, a000919. [Google Scholar] [CrossRef]
- Kruse, J.P.; Gu, W. Modes of p53 Regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.; Gu, W. P53 post-translational modification: Deregulated in tumorigenesis. Trends Mol. Med. 2010, 16, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [Green Version]
- Marchenko, N.D.; Moll, U.M. Mitochondrial death functions of p53. Mol. Cell. Oncol. 2014, 1, e955995. [Google Scholar] [CrossRef]
- Vaseva, A.V.; Marchenko, N.D.; Ji, K.; Tsirka, S.E.; Holzmann, S.; Moll, U.M. P53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012, 149, 1536–1548. [Google Scholar] [CrossRef] [Green Version]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. P53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Chaiswing, L.; Velez, J.M.; Batinic-Haberle, I.; Colburn, N.H.; Oberley, T.D.; St. Clair, D.K. p53 translocation to mitochondria precedes its nuclear translocation and targets mitochondrial oxidative defense protein-manganese superoxide dismutase. Cancer Res. 2005, 65, 3745–3750. [Google Scholar] [CrossRef] [Green Version]
- Hampp, S.; Kiessling, T.; Buechle, K.; Mansilla, S.F.; Thomale, J.; Rall, M.; Ahn, J.; Pospiech, H.; Gottifredi, V.; Wiesmüller, L. DNA damage tolerance pathway involving DNA polymerase ι and the tumor suppressor p53 regulates DNA replication fork progression. Proc. Natl. Acad. Sci. USA 2016, 113, E4311–E4319. [Google Scholar] [CrossRef] [Green Version]
- Linke, S.P.; Sengupta, S.; Khabie, N.; Jeffries, B.A.; Buchhop, S.; Miska, S.; Henning, W.; Pedeux, R.; Wang, X.W.; Hofseth, L.J.; et al. p53 interacts with hRAD51 and hRAD54, and directly modulates homologous recombination. Cancer Res. 2003, 63, 2596–2605. [Google Scholar]
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 1370–1384. [Google Scholar] [CrossRef] [Green Version]
- Campbell, P.J.; Getz, G.; Korbel, J.O.; Stuart, J.M.; Jennings, J.L.; Stein, L.D.; Perry, M.D.; Nahal-Bose, H.K.; Ouellette, B.F.F.; Li, C.H.; et al. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Gerstung, M.; Jolly, C.; Leshchiner, I.; Dentro, S.C.; Gonzalez, S.; Rosebrock, D.; Mitchell, T.J.; Rubanova, Y.; Anur, P.; Yu, K.; et al. The evolutionary history of 2,658 cancers. Nature 2020, 578, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Leroy, B.; Anderson, M.; Soussi, T. TP53 mutations in human cancer: Database reassessment and prospects for the next decade. Hum. Mutat. 2014, 35, 672–688. [Google Scholar] [CrossRef]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef]
- Stiewe, T.; Haran, T.E. How mutations shape p53 interactions with the genome to promote tumorigenesis and drug resistance. Drug Resist. Updates 2018, 38, 27–43. [Google Scholar] [CrossRef]
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu. Rev. Biochem. 2016, 85, 375–404. [Google Scholar] [CrossRef]
- Kato, S.; Han, S.-Y.; Liu, W.; Otsuka, K.; Shibata, H.; Kanamaru, R.; Ishioka, C. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 8424–8429. [Google Scholar] [CrossRef] [Green Version]
- Kotler, E.; Shani, O.; Goldfeld, G.; Lotan-Pompan, M.; Tarcic, O.; Gershoni, A.; Hopf, T.A.; Marks, D.S.; Oren, M.; Segal, E. A Systematic p53 Mutation Library Links Differential Functional Impact to Cancer Mutation Pattern and Evolutionary Conservation. Mol. Cell 2018, 71, 178–190. [Google Scholar] [CrossRef] [Green Version]
- Boettcher, S.; Miller, P.G.; Sharma, R.; McConkey, M.; Leventhal, M.; Krivtsov, A.V.; Giacomelli, A.O.; Wong, W.; Kim, J.; Chao, S.; et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 2019, 365, 599–604. [Google Scholar] [CrossRef]
- Pavlakis, E.; Stiewe, T. p53′s extended reach: The mutant p53 secretome. Biomolecules 2020, 10, 307. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. Structural Biology of the Tumor Suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582. [Google Scholar] [CrossRef]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, C.D.; McLure, K.G.; Shields, M.A.; Lee, P.W.K. Biogenesis of p53 involves cotranslational dimerization of monomers and posttranslational dimerization of dimers. Implications on the dominant negative effect. J. Biol. Chem. 2002, 277, 12937–12945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopalan, S.; Huang, F.; Fersht, A.R. Single-molecule characterization of oligomerization kinetics and equilibria of the tumor suppressor p53. Nucleic Acids Res. 2011, 39, 2294–2303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, R.L.; Veprintsev, D.B.; Fersht, A.R. Cooperative binding of tetrameric p53 to DNA. J. Mol. Biol. 2004, 341, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, K.; Sakamoto, H.; Lewis, M.S.; Anderson, C.W.; Erickson, J.W.; Appella, E.; Xie, D. Phosphorylation of serine 392 stabilizes the tetramer formation of tumor suppressor protein p53. Biochemistry 1997, 36, 10117–10124. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Jaulent, A.M.; Wells, M.; Veprintsev, D.B.; Fersht, A.R. 14-3-3 activation of DNA binding of p53 by enhancing its association into tetramers. Nucleic Acids Res. 2008, 36, 5983–5991. [Google Scholar] [CrossRef] [Green Version]
- Gaglia, G.; Guan, Y.; Shah, J.V.; Lahav, G. Activation and control of p53 tetramerization in individual living cells. Proc. Natl. Acad. Sci. USA 2013, 110, 15497–15501. [Google Scholar] [CrossRef] [Green Version]
- Shakked, Z. Quaternary structure of p53: The light at the end of the tunnel. Proc. Natl. Acad. Sci. USA 2007, 104, 12231–12232. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.L.; Wu, Q.; Vega, V.B.; Chiu, K.P.; Ng, P.; Zhang, T.; Shahab, A.; Yong, H.C.; Fu, Y.T.; Weng, Z.; et al. A global map of p53 transcription-factor binding sites in the human genome. Cell 2006, 124, 207–219. [Google Scholar] [CrossRef] [Green Version]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef]
- McLure, K.G.; Lee, P.W.K. How p53 binds DNA as a tetramer. EMBO J. 1998, 17, 3342–3350. [Google Scholar] [CrossRef]
- Kitayner, M.; Rozenberg, H.; Kessler, N.; Rabinovich, D.; Shaulov, L.; Haran, T.E.; Shakked, Z. Structural Basis of DNA Recognition by p53 Tetramers. Mol. Cell 2006, 22, 741–753. [Google Scholar] [CrossRef]
- Tidow, H.; Melero, R.; Mylonas, E.; Freund, S.M.V.; Grossmann, J.G.; Carazo, J.M.; Svergun, D.I.; Valle, M.; Fersht, A.R. Quaternary structures of tumor suppressor p53 and a specific p53-DNA complex. Proc. Natl. Acad. Sci. USA 2007, 104, 12324–12329. [Google Scholar] [CrossRef] [Green Version]
- Klein, C.; Planker, E.; Diercks, T.; Kessler, H.; Künkele, K.P.; Lang, K.; Hansen, S.; Schwaiger, M. NMR Spectroscopy Reveals the Solution Dimerization Interface of p53 Core Domains Bound to Their Consensus DNA. J. Biol. Chem. 2001, 276, 49020–49027. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Schwedes, J.F.; Parks, D.; Mann, K.; Tegtmeyer, P. Interaction of p53 with its consensus DNA-binding site. Mol. Cell. Biol. 1995, 15, 2157–2165. [Google Scholar] [CrossRef] [Green Version]
- Malecka, K.A.; Ho, W.C.; Ho, W.C. Crystal structure of a p53 core tetramer bound to DNA. Oncogene 2009, 28, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Stefan, M.I.; Le Novère, N. Cooperative Binding. PLoS Comput. Biol. 2013, 9, e1003106. [Google Scholar] [CrossRef] [Green Version]
- Pauling, L. The Oxygen Equilibrium of Hemoglobin and Its Structural Interpretation. Proc. Natl. Acad. Sci. USA 1935, 21, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Jeffrey, P.D.; Gorina, S.; Pavletich, N.P. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science 1995, 267, 1498–1502. [Google Scholar] [CrossRef]
- Balagurumoorthy, P.; Sakamoto, H.; Lewis, M.S.; Zambrano, N.; Clore, G.M.; Gronenborn, A.M.; Appella, E.; Harrington, R.E. Four p53 DNA-binding domain peptides bind natural p53-response elements and bend the DNA. Proc. Natl. Acad. Sci. USA 1995, 92, 8591–8595. [Google Scholar] [CrossRef] [Green Version]
- Nagaich, A.K.; Zhurkin, V.B.; Sakamoto, H.; Gorin, A.A.; Clore, G.M.; Gronenborn, A.M.; Appella, E.; Harrington, R.E. Architectural accommodation in the complex of four p53 DNA binding domain peptides with the p21/waf1/cip1 DNA response element. J. Biol. Chem. 1997, 272, 14830–14841. [Google Scholar] [CrossRef] [Green Version]
- Schlereth, K.; Beinoraviciute-Kellner, R.; Zeitlinger, M.K.; Bretz, A.C.; Sauer, M.; Charles, J.P.; Vogiatzi, F.; Leich, E.; Samans, B.; Eilers, M.; et al. DNA Binding Cooperativity of p53 Modulates the Decision between Cell-Cycle Arrest and Apoptosis. Mol. Cell 2010, 38, 356–368. [Google Scholar] [CrossRef]
- Schlereth, K.; Heyl, C.; Krampitz, A.M.; Mernberger, M.; Finkernagel, F.; Scharfe, M.; Jarek, M.; Leich, E.; Rosenwald, A.; Stiewe, T. Characterization of the p53 Cistrome - DNA Binding Cooperativity Dissects p53′s Tumor Suppressor Functions. PLoS Genet. 2013, 9, e1003726. [Google Scholar] [CrossRef]
- Rippin, T.M.; Freund, S.M.V.; Veprintsev, D.B.; Fersht, A.R. Recognition of DNA by p53 core domain and location of intermolecular contacts of cooperative binding. J. Mol. Biol. 2002, 319, 351–358. [Google Scholar] [CrossRef]
- Dehner, A.; Klein, C.; Hansen, S.; Müller, L.; Buchner, J.; Schwaiger, M.; Kessler, H. Cooperative binding of p53 to DNA: Regulation by protein-protein interactions through a double salt bridge. Angew. Chem. Int. Ed. 2005, 44, 5247–5251. [Google Scholar] [CrossRef]
- Veprintsev, D.B.; Freund, S.M.V.; Andreeva, A.; Rutledge, S.E.; Tidow, H.; Pérez Cañadillas, J.M.; Blair, C.M.; Fersht, A.R. Core domain interactions in full-length p53 in solution. Proc. Natl. Acad. Sci. USA 2006, 103, 2115–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, W.C.; Fitzgerald, M.X.; Marmorstein, R. Structure of the p53 core domain dimer bound to DNA. J. Biol. Chem. 2006, 281, 20494–20502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitayner, M.; Rozenberg, H.; Rohs, R.; Suad, O.; Rabinovich, D.; Honig, B.; Shakked, Z. Diversity in DNA recognition by p53 revealed by crystal structures with Hoogsteen base pairs. Nat. Struct. Mol. Biol. 2010, 17, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Laptenko, O.; Shiff, I.; Freed-Pastor, W.; Zupnick, A.; Mattia, M.; Freulich, E.; Shamir, I.; Kadouri, N.; Kahan, T.; Manfredi, J.; et al. The p53 C Terminus Controls Site-Specific DNA Binding and Promotes Structural Changes within the Central DNA Binding Domain. Mol. Cell 2015, 57, 1034–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enthart, A.; Klein, C.; Dehner, A.; Coles, M.; Gemmecker, G.; Kessler, H.; Hagn, F. Solution structure and binding specificity of the p63 DNA binding domain. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Ethayathulla, A.S.; Tse, P.W.; Monti, P.; Nguyen, S.; Inga, A.; Fronza, G.; Viadiu, H. Structure of p73 DNA-binding domain tetramer modulates p73 transactivation. Proc. Natl. Acad. Sci. USA 2012, 109, 6066–6071. [Google Scholar] [CrossRef] [Green Version]
- Beno, I.; Rosenthal, K.; Levitine, M.; Shaulov, L.; Haran, T.E. Sequence-dependent cooperative binding of p53 to DNA targets and its relationship to the structural properties of the DNA targets. Nucleic Acids Res. 2011, 39, 1919–1932. [Google Scholar] [CrossRef] [Green Version]
- Nagaich, A.K.; Zhurkin, V.B.; Durell, S.R.; Jernigan, R.L.; Appella, E.; Harrington, R.E. p53-Induced DNA bending and twisting: p53 Tetramer binds on the outer side of a DNA loop and increases DNA twisting. Proc. Natl. Acad. Sci. USA 1999, 96, 1875–1880. [Google Scholar] [CrossRef] [Green Version]
- Sabapathy, K.; Lane, D.P. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 2017, 15, 13–30. [Google Scholar] [CrossRef]
- Friedler, A.; Veprintsev, D.B.; Hansson, L.O.; Fersht, A.R. Kinetic instability of p53 core domain mutants. Implications for rescue by small molecules. J. Biol. Chem. 2003, 278, 24108–24112. [Google Scholar] [CrossRef] [Green Version]
- Ang, H.C.; Joerger, A.C.; Mayer, S.; Fersht, A.R. Effects of common cancer mutations on stability and DNA binding of full-length p53 compared with isolated core domains. J. Biol. Chem. 2006, 281, 21934–21941. [Google Scholar] [CrossRef] [Green Version]
- Bullock, A.N.; Henckel, J.; Fersht, A.R. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: Definition of mutant states for rescue in cancer therapy. Oncogene 2000, 19, 1245–1256. [Google Scholar] [CrossRef] [Green Version]
- Joerger, A.C.; Ang, H.C.; Fersht, A.R. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl. Acad. Sci. USA 2006, 103, 15056–15061. [Google Scholar] [CrossRef] [Green Version]
- Shiraishi, K.; Kato, S.; Han, S.Y.; Liu, W.; Otsuka, K.; Sakayori, M.; Ishida, T.; Takeda, M.; Kanamaru, R.; Ohuchi, N.; et al. Isolation of Temperature-sensitive p53 Mutations from a Comprehensive Missense Mutation Library. J. Biol. Chem. 2004, 279, 348–355. [Google Scholar] [CrossRef] [Green Version]
- Dearth, L.R.; Qian, H.; Wang, T.; Baroni, T.E.; Zeng, J.; Chen, S.W.; Yi, S.Y.; Brachmann, R.K. Inactive full-length p53 mutants lacking dominant wild-type p53 inhibition highlight loss of heterozygosity as an important aspect of p53 status in human cancers. Carcinogenesis 2007, 28, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Paleček, E.; Ostatná, V.; Černocká, H.; Joerger, A.C.; Fersht, A.R. Electrocatalytic monitoring of metal binding and mutation-induced conformational changes in p53 at picomole level. J. Am. Chem. Soc. 2011, 133, 7190–7196. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.M.; Vousden, K.H. Characterization of Structural p53 Mutants Which Show Selective Defects in Apoptosis but Not Cell Cycle Arrest. Mol. Cell. Biol. 1998, 18, 3692–3698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, R.L.; Bates, S.; Vousden, K.H. Differential activation of target cellular promoters by p53 mutants with impaired apoptotic function. Mol. Cell. Biol. 1996, 16, 4952–4960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timofeev, O.; Klimovich, B.; Schneikert, J.; Wanzel, M.; Pavlakis, E.; Noll, J.; Mutlu, S.; Elmshäuser, S.; Nist, A.; Mernberger, M.; et al. Residual apoptotic activity of a tumorigenic p53 mutant improves cancer therapy responses. EMBO J. 2019, 38, e102096. [Google Scholar] [CrossRef]
- Qian, H.; Wang, T.; Naumovski, L.; Lopez, C.D.; Brachmann, R.K. Groups of p53 target genes involved in specific p53 downstream effects cluster into different classes of DNA binding sites. Oncogene 2002, 21, 7901–7911. [Google Scholar] [CrossRef] [Green Version]
- Szak, S.T.; Mays, D.; Pietenpol, J.A. Kinetics of p53 Binding to Promoter Sites In Vivo. Mol. Cell. Biol. 2001, 21, 3375–3386. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, R.L.; Veprintsev, D.B.; Bycroft, M.; Fersht, A.R. Comparative binding of p53 to its promoter and DNA recognition elements. J. Mol. Biol. 2005, 348, 589–596. [Google Scholar] [CrossRef]
- Schlereth, K.; Charles, J.P.; Bretz, A.C.; Stiewe, T. Life or death: p53-induced apoptosis requires DNA binding cooperativity. Cell Cycle 2010, 9, 4068–4076. [Google Scholar] [CrossRef] [Green Version]
- Timofeev, O.; Schlereth, K.; Wanzel, M.; Braun, A.; Nieswandt, B.; Pagenstecher, A.; Rosenwald, A.; Elsasser, H.P.; Stiewe, T. p53 DNA binding cooperativity is essential for apoptosis and tumor suppression in vivo. Cell Rep. 2013, 3, 1512–1525. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.G.; Lago, C.U.; Lee, J.E.; Park, J.H.; Donnelly, M.P.; Starost, M.F.; Liu, C.; Kwon, J.; Noguchi, A.C.; Ge, K.; et al. A Mouse Homolog of a Human TP53 Germline Mutation Reveals a Lipolytic Activity of p53. Cell Rep. 2020, 30, 783–792. [Google Scholar] [CrossRef] [Green Version]
- Zhao, K.; Chai, X.; Johnston, K.; Clements, A.; Marmorstein, R. Crystal Structure of the Mouse p53 Core DNA-binding Domain at 2.7 Å Resolution. J. Biol. Chem. 2001, 276, 12120–12127. [Google Scholar] [CrossRef] [Green Version]
- Garcia, P.B.; Attardi, L.D. Illuminating p53 function in cancer with genetically engineered mouse models. Semin. Cell Dev. Biol. 2014, 27, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Klimovich, B.; Stiewe, T.; Timofeev, O. Inactivation of Mdm2 restores apoptosis proficiency of cooperativity mutant p53 in vivo. Cell Cycle 2020, 19, 109–123. [Google Scholar] [CrossRef]
- Liu, G.; Parant, J.M.; Lang, G.; Chau, P.; Chavez-Reyes, A.; El-Naggar, A.K.; Multani, A.; Chang, S.; Lozano, G. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat. Genet. 2004, 36, 63–68. [Google Scholar] [CrossRef]
- Morton, J.P.; Timpson, P.; Karim, S.A.; Ridgway, R.A.; Athineos, D.; Doyle, B.; Jamieson, N.B.; Oien, K.A.; Lowy, A.M.; Brunton, V.G.; et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 246–251. [Google Scholar] [CrossRef] [Green Version]
- Butler, J.S.; Loh, S.N. Structure, function, and aggregation of the zinc-free form of the p53 DNA binding domain. Biochemistry 2003, 42, 2396–2403. [Google Scholar] [CrossRef]
- Sidransky, D.; Tokino, T.; Helzlsouer, K.; Zehnbauer, B.; Shelton, B.; Prestigiacomo, L.; Vogelstein, B.; Davidson, N.; Helzlsouer, K.; Rausch, G. Inherited p53 Gene Mutations in Breast Cancer. Cancer Res. 1992, 52, 2984–2986. [Google Scholar]
- Comel, A.; Sorrentino, G.; Capaci, V.; Del Sal, G. The cytoplasmic side of p53′s oncosuppressive activities. FEBS Lett. 2014, 588, 2600–2609. [Google Scholar] [CrossRef] [Green Version]
- Klimovich, B.; Mutlu, S.; Schneikert, J.; Elmshäuser, S.; Klimovich, M.; Nist, A.; Mernberger, M.; Timofeev, O.; Stiewe, T. Loss of p53 function at late stages of tumorigenesis confers ARF-dependent vulnerability to p53 reactivation therapy. Proc. Natl. Acad. Sci. USA 2019, 116, 22288–22293. [Google Scholar] [CrossRef] [Green Version]
- Gu, B.; Zhu, W.G. Surf the Post-translational Modification Network of p53 Regulation. Int. J. Biol. Sci. 2012, 8, 672–684. [Google Scholar] [CrossRef] [Green Version]
- Ho, T.; Tan, B.X.; Lane, D. How the other half lives: What p53 does when it is not being a transcription factor. Int. J. Mol. Sci. 2020, 21, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeHart, C.J.; Chahal, J.S.; Flint, S.J.; Perlman, D.H. Extensive Post-translational Modification of Active and Inactivated Forms of Endogenous p53. Mol. Cell. Proteom. 2014, 13, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeHart, C.J.; Perlman, D.H.; Flint, S.J. Impact of the Adenoviral E4 Orf3 Protein on the Activity and Posttranslational Modification of p53. J. Virol. 2015, 89, 3209–3220. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Ma, C.A.; Zhao, Y.; Jain, A. Aurora B interacts with NIR-p53, leading to p53 phosphorylation in its DNA-binding domain and subsequent functional suppression. J. Biol. Chem. 2011, 286, 2236–2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gully, C.P.; Velazquez-Torres, G.; Shin, J.-H.; Fuentes-Mattei, E.; Wang, E.; Carlock, C.; Chen, J.; Rothenberg, D.; Adams, H.P.; Choi, H.H.; et al. Aurora B kinase phosphorylates and instigates degradation of p53. Proc. Natl. Acad. Sci. USA 2012, 9, E1513–E1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Muzio, L. Lo The crucial role of protein phosphorylation in cell signalingand its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skinner, J.J.; Wang, S.; Lee, J.; Ong, C.; Sommese, R.; Sivaramakrishnan, S.; Koelmel, W.; Hirschbeck, M.; Schindelin, H.; Kisker, C.; et al. Conserved salt-bridge competition triggered by phosphorylation regulates the protein interactome. Proc. Natl. Acad. Sci. USA 2017, 114, 13453–13458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timofeev, O.; Koch, L.; Niederau, C.; Tscherne, A.; Schneikert, J.; Klimovich, M.; Elmshauser, S.; Zeitlinger, M.; Mernberger, M.; Nist, A.; et al. Phosphorylation control of P53 dna-binding cooperativity balances tumorigenesis and aging. Cancer Res. 2020, 80, 5231–5244. [Google Scholar] [CrossRef]
- Bell, S.; Klein, C.; Müller, L.; Hansen, S.; Buchner, J. p53 contains large unstructured regions in its native state. J. Mol. Biol. 2002, 14, 535–562. [Google Scholar] [CrossRef]
- Rustandi, R.R.; Baldisseri, D.M.; Weber, D.J. Structure of the negative regulatory domain of p53 bound to S100B(ββ). Nat. Struct. Biol. 2000, 7, 570–574. [Google Scholar] [CrossRef]
- Huart, A.-S.; Hupp, T. Evolution of Conformational Disorder & Diversity of the P53 Interactome. Biodiscovery 2013. Available online: https://biodiscovery.pensoft.net/article/8952/ (accessed on 16 May 2021). [CrossRef]
- Liu, Y.; Tavana, O.; Gu, W. P53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019, 11, 564–577. [Google Scholar] [CrossRef] [Green Version]
- McKinney, K.; Mattia, M.; Gottifredi, V.; Prives, C. p53 linear diffusion along DNA requires its C terminus. Mol. Cell 2004, 16, 413–424. [Google Scholar] [CrossRef]
- Hamard, P.J.; Lukin, D.J.; Manfredi, J.J. p53 basic C terminus regulates p53 functions through DNA binding modulation of subset of target genes. J. Biol. Chem. 2012, 287, 22397–22407. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kim, K.; Choi, J.; Heo, K.; Baek, H.J.; Roeder, R.G.; An, W. P53 requires an intact C-terminal domain for DNA binding and transactivation. J. Mol. Biol. 2012, 415, 843–854. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, R.L.; Freund, S.M.V.; Veprintsev, D.B.; Bycroft, M.; Fersht, A.R. Regulation of DNA binding of p53 by its C-terminal domain. J. Mol. Biol. 2004, 342, 801–811. [Google Scholar] [CrossRef]
- Tafvizi, A.; Huang, F.; Fersht, A.R.; Mirny, L.A.; van Oijen, A.M. A single-molecule characterization of p53 search on DNA. Proc. Natl. Acad. Sci. USA 2011, 108, 563–568. [Google Scholar] [CrossRef] [Green Version]
- Tafvizi, A.; Huang, F.; Leith, J.S.; Fersht, A.R.; Mirny, L.A.; Van Oijen, A.M. Tumor suppressor p53 slides on DNA with low friction and high stability. Biophys. J. 2008, 95, L01–L03. [Google Scholar] [CrossRef] [Green Version]
- Friedler, A.; Veprintsev, D.B.; Freund, S.M.V.; Von Glos, K.I.; Fersht, A.R. Modulation of binding of DNA to the C-terminal domain of p53 by acetylation. Structure 2005, 13, 629–636. [Google Scholar] [CrossRef] [Green Version]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Mujtaba, S.; He, Y.; Zeng, L.; Yan, S.; Plotnikova, O.; Sachchidanand; Sanchez, R.; Zeleznik-Le, N.J.; Ronai, Z.; Zhou, M.M. Structural Mechanism of the Bromodomain of the Coactivator CBP in p53 Transcriptional Activation. Mol. Cell 2004, 13, 251–263. [Google Scholar] [CrossRef]
- Poyurovsky, M.V.; Katz, C.; Laptenko, O.; Beckerman, R.; Lokshin, M.; Ahn, J.; Byeon, I.-J.L.; Gabizon, R.; Mattia, M.; Zupnick, A.; et al. The C terminus of p53 binds the N-terminal domain of MDM2. Nat. Struct. Mol. Biol. 2010, 17, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Kon, N.; Lasso, G.; Jiang, L.; Leng, W.; Zhu, W.G.; Qin, J.; Honig, B.; Gu, W. Acetylation-regulated interaction between p53 and SET reveals a widespread regulatory mode. Nature 2016, 538, 118–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlev, N.A.; Liu, L.; Chehab, N.H.; Mansfield, K.; Harris, K.G.; Halazonetis, T.D.; Berger, S.L. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell 2001, 8, 1243–1254. [Google Scholar] [CrossRef]
- Li, A.G.; Piluso, L.G.; Cai, X.; Gadd, B.J.; Ladurner, A.G.; Liu, X. An Acetylation Switch in p53 Mediates Holo-TFIID Recruitment. Mol. Cell 2007, 28, 408–421. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.E.; Woelker, B.; Reed, M.; Wang, P.; Tegtmeyer, P. Reciprocal interference between the sequence-specific core and nonspecific C-terminal DNA binding domains of p53: Implications for regulation. Mol. Cell. Biol. 1997, 17, 6255–6264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinosa, J.M.; Emerson, B.M. Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol. Cell 2001, 8, 57–69. [Google Scholar] [CrossRef]
- Luo, J.; Li, M.; Tang, Y.; Laszkowska, M.; Roeder, R.G.; Gu, W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 2259–2264. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Blay, J.Y.; Italiano, A.; Le Cesne, A.; Penel, N.; Zhi, J.; Heil, F.; Rueger, R.; Graves, B.; Ding, M.; et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: An exploratory proof-of-mechanism study. Lancet Oncol. 2012, 13, 1133–1140. [Google Scholar] [CrossRef]
- Andreeff, M.; Kelly, K.R.; Yee, K.; Assouline, S.; Strair, R.; Popplewell, L.; Bowen, D.; Martinelli, G.; Drummond, M.W.; Vyas, P.; et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin. Cancer Res. 2016, 22, 868–876. [Google Scholar] [CrossRef] [Green Version]
- Merino, D.; Kelly, G.L.; Lessene, G.; Wei, A.H.; Roberts, A.W.; Strasser, A. BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 2018, 34, 879–891. [Google Scholar] [CrossRef] [Green Version]
- Le Pen, J.; Laurent, M.; Sarosiek, K.; Vuillier, C.; Gautier, F.; Montessuit, S.; Martinou, J.C.; Letaï, A.; Braun, F.; Juin, P.P. Constitutive p53 heightens mitochondrial apoptotic priming and favors cell death induction by BH3 mimetic inhibitors of BCL-xL. Cell Death Dis. 2016, 7, e2083. [Google Scholar] [CrossRef]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Bève, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt Plus Azacitidine in TP53 -Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia: A Phase II Study by the Groupe Francophone des Myélodysplasies (GFM). J. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53 -Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef]
- Lambert, J.M.R.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. PRIMA-1 Reactivates Mutant p53 by Covalent Binding to the Core Domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018, 9, 439. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.J.N.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Lambert, J.M.R.; Moshfegh, A.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. Mutant p53 reactivation by PRIMA-1MET induces multiple signaling pathways converging on apoptosis. Oncogene 2010, 29, 1329–1338. [Google Scholar] [CrossRef] [Green Version]
- Zandi, R.; Selivanova, G.; Christensen, C.L.; Gerds, T.A.; Willumsen, B.M.; Poulsen, H.S. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin. Cancer Res. 2011, 17, 2830–2841. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.J.N.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. PRIMA-1MET synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005, 9, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fransson, Å.; Glaessgen, D.; Alfredsson, J.; Wiman, K.G.; Bajalica-Lagercrantz, S.; Mohell, N. Strong synergy with APR-246 and DNA-damaging drugs in primary cancer cells from patients with TP53 mutant High-Grade Serous ovarian cancer. J. Ovarian Res. 2016, 9, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krayem, M.; Journe, F.; Wiedig, M.; Morandini, R.; Najem, A.; Salès, F.; Van Kempen, L.C.; Sibille, C.; Awada, A.; Marine, J.C.; et al. P53 Reactivation by PRIMA-1Met (APR-246) sensitises V600E/KBRAF melanoma to vemurafenib. Eur. J. Cancer 2016, 55, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Zhang, M.Q.Z.; Conserva, F.; Hosny, G.; Selivanova, G.; Bykov, V.J.N.; Arnér, E.S.J.; Wiman, K.G. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013, 4, e881. [Google Scholar] [CrossRef]
- Tessoulin, B.; Descamps, G.; Moreau, P.; Maïga, S.; Lodé, L.; Godon, C.; Marionneau-Lambot, S.; Oullier, T.; Le Gouill, S.; Amiot, M.; et al. PRIMA-1 Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood 2015, 124, 1626–1637. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, N.; Kajiyama, H.; Nakamura, K.; Utsumi, F.; Niimi, K.; Mitsui, H.; Sekiya, R.; Suzuki, S.; Shibata, K.; Callen, D.; et al. PRIMA-1 MET induces apoptosis through accumulation of intracellular reactive oxygen species irrespective of p53 status and chemo-sensitivity in epithelial ovarian cancer cells. Oncol. Rep. 2016, 35, 2543–2552. [Google Scholar] [CrossRef] [Green Version]
- Birsen, R.; Larrue, C.; Decroocq, J.; Johnson, N.; Guiraud, N.; Gotanegre, M.; Cantero-Aguilar, L.; Grignano, E.; Huynh, T.; Fontenay, M.; et al. APR-246 induces early cell death by ferroptosis in acute myeloid leukemia. Haematologica 2021. [Google Scholar] [CrossRef]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC−/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef] [Green Version]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef]
- Jackson, J.G.; Pant, V.; Li, Q.; Chang, L.L.; Quint??s-Cardama, A.; Garza, D.; Tavana, O.; Yang, P.; Manshouri, T.; Li, Y.; et al. P53-Mediated Senescence Impairs the Apoptotic Response to Chemotherapy and Clinical Outcome in Breast Cancer. Cancer Cell 2012, 21, 793–806. [Google Scholar] [CrossRef] [Green Version]
- Bertheau, P.; Plassa, F.; Espié, M.; Turpin, E.; De Roquancourt, A.; Marty, M.; Lerebours, F.; Beuzard, Y.; Janin, A.; De Thé, H. Effect of mutated TP53 on response of advanced breast cancers to high-dose chemotherapy. Lancet 2002, 360, 852–854. [Google Scholar] [CrossRef]
- Ungerleider, N.A.; Rao, S.G.; Shahbandi, A.; Yee, D.; Niu, T.; Frey, W.D.; Jackson, J.G. Breast cancer survival predicted by TP53 mutation status differs markedly depending on treatment. Breast Cancer Res. 2018, 20, 115. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Shahbandi, A.; Rao, S.G.; Anderson, A.Y.; Frey, W.D.; Olayiwola, J.O.; Ungerleider, N.A.; Jackson, J.G. BH3 mimetics selectively eliminate chemotherapy-induced senescent cells and improve response in TP53 wild-type breast cancer. Cell Death Differ. 2020, 27, 3097–3116. [Google Scholar] [CrossRef]
- Myrianthopoulos, V.; Evangelou, K.; Vasileiou, P.V.S.; Cooks, T.; Vassilakopoulos, T.P.; Pangalis, G.A.; Kouloukoussa, M.; Kittas, C.; Georgakilas, A.G.; Gorgoulis, V.G. Senescence and senotherapeutics: A new field in cancer therapy. Pharmacol. Ther. 2019, 193, 31–49. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timofeev, O.; Stiewe, T. Rely on Each Other: DNA Binding Cooperativity Shapes p53 Functions in Tumor Suppression and Cancer Therapy. Cancers 2021, 13, 2422. https://doi.org/10.3390/cancers13102422
Timofeev O, Stiewe T. Rely on Each Other: DNA Binding Cooperativity Shapes p53 Functions in Tumor Suppression and Cancer Therapy. Cancers. 2021; 13(10):2422. https://doi.org/10.3390/cancers13102422
Chicago/Turabian StyleTimofeev, Oleg, and Thorsten Stiewe. 2021. "Rely on Each Other: DNA Binding Cooperativity Shapes p53 Functions in Tumor Suppression and Cancer Therapy" Cancers 13, no. 10: 2422. https://doi.org/10.3390/cancers13102422
APA StyleTimofeev, O., & Stiewe, T. (2021). Rely on Each Other: DNA Binding Cooperativity Shapes p53 Functions in Tumor Suppression and Cancer Therapy. Cancers, 13(10), 2422. https://doi.org/10.3390/cancers13102422