
Organoruthenium Complexes with Benzo-Fused Pyrithiones Overcome Platinum Resistance in Ovarian Cancer Cells

 ,
,  , and
, and
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Physicochemical Characterization
2.3. Syntheses of Ligands and Complexes
2.4. Aqueous Stability
2.5. Human Cancer Cell Culture
2.6. Determination of Antiproliferative Activity
2.7. Induction of Apoptosis
2.8. Cell Cycle Analysis
2.9. Wound Healing Assay
2.10. Colony Formation Assay
2.11. Induction of Reactive Oxygen Species (ROS)
2.12. Evaluation of Mitochondrial Function
2.13. Thioredoxin Reductase Assay
2.14. Cellular Accumulation of Ruthenium
2.15. Statistical Analysis
3. Results and Discussion
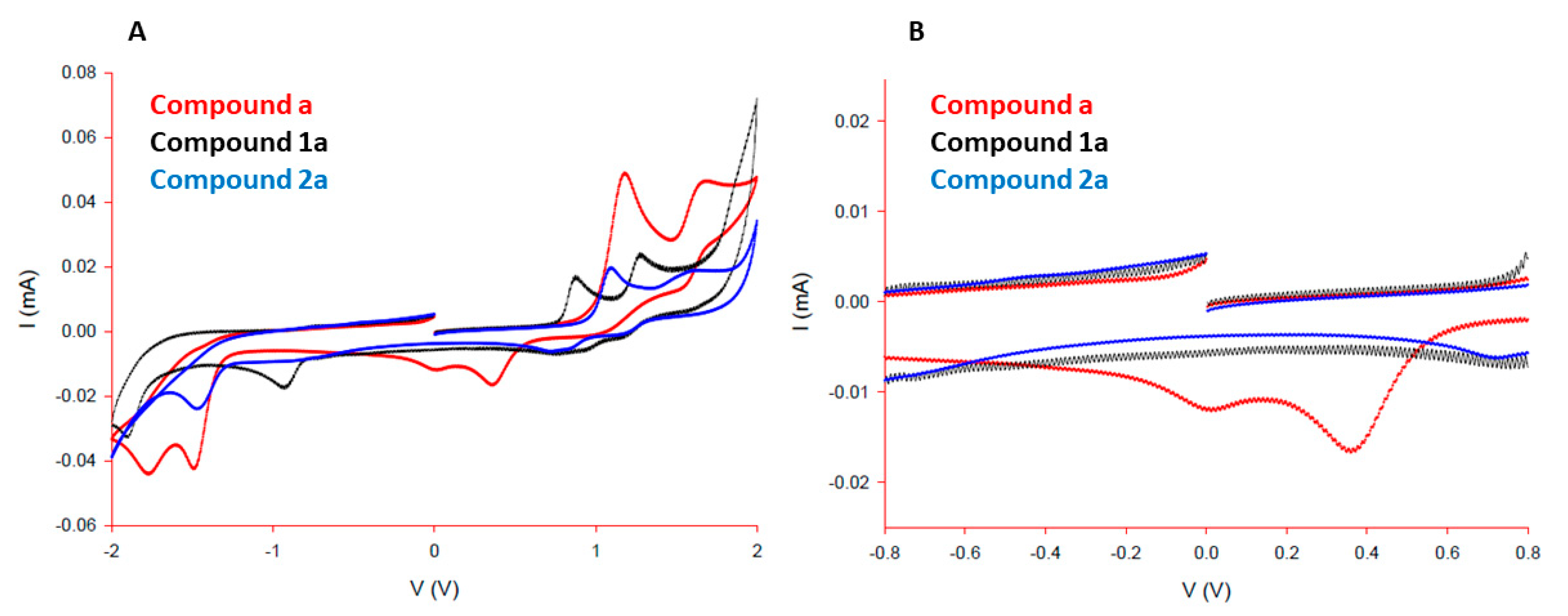
3.1. Syntheses and Crystal Structures of the Ligands and Complexes
3.2. NMR Stability in Solution
3.3. Antiproliferative Activity
3.4. Cellular Accumulation of Ruthenium
3.5. Interactions in Biological Media
3.6. Explorations of the Mechanism of Action
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Luo, G.; Li, M.; Guo, P.; Xiao, Y.; Ji, H.; Hao, Y. Global patterns and trends in ovarian cancer incidence: Age, period and birth cohort analysis. BMC Cancer 2019, 19, 984. [Google Scholar] [CrossRef]
- Momenimovahed, Z.; Tiznobaik, A.; Taheri, S.; Salehiniya, H. Ovarian cancer in the world: Epidemiology and risk factors. Int. J. Womens Health 2019, 11, 287–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaum, N.; Crosbie, E.J.; Edmondson, R.J.; Smith, M.J.; Evans, D.G. Epithelial ovarian cancer risk: A review of the current genetic landscape. Clin. Genet. 2020, 97, 54–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian cancer. Nat. Rev. Dis. Primers 2016, 2, 16061. [Google Scholar] [CrossRef]
- McLemore, M.R.; Miaskowski, C.; Aouizerat, B.E.; Chen, L.M.; Dodd, M.J. Epidemiological and genetic factors associated with ovarian cancer. Cancer Nurs. 2009, 32, 281–288. [Google Scholar] [CrossRef]
- Cortez, A.J.; Tudrej, P.; Kujawa, K.A.; Lisowska, K.M. Advances in ovarian cancer therapy. Cancer Chemother. Pharmacol. 2018, 81, 17–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, F.; Shen, J.; Shi, H.; Hornicek, F.J.; Kan, Q.; Duan, Z. Novel mechanisms and approaches to overcome multidrug resistance in the treatment of ovarian cancer. Biochim. Biophys. Acta Rev. Cancer 2016, 1866, 266–275. [Google Scholar] [CrossRef]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of multidrug resistance in cancer chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef] [PubMed]
- Gothe, Y.; Marzo, T.; Messori, L.; Metzler-Nolte, N. Iridium(I) compounds as prospective anticancer agents: Solution chemistry, antiproliferative profiles and protein interactions for a series of iridium(I) N-heterocyclic carbene complexes. Chem. Eur. J. 2016, 22, 12487–12494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coverdale, J.P.C.; Romero-Canelón, I.; Sanchez-Cano, C.; Clarkson, G.J.; Habtemariam, A.; Wills, M.; Sadler, P.J. Asymmetric transfer hydrogenation by synthetic catalysts in cancer cells. Nat. Chem. 2018, 10, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Hanif, M.; Babak, M.V.; Hartinger, C.G. Development of anticancer agents: Wizardry with osmium. Drug Discov. Today 2014, 19, 1640–1648. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Huang, H. Future potential of osmium complexes as anticancer drug candidates, photosensitizers and organelle-targeted probes. Dalton Trans. 2018, 47, 14841–14854. [Google Scholar] [CrossRef] [PubMed]
- Almodares, Z.; Lucas, S.J.; Crossley, B.D.; Basri, A.M.; Pask, C.M.; Hebden, A.J.; Phillips, R.M.; McGowan, P.C. Rhodium, iridium, and ruthenium half-sandwich picolinamide complexes as anticancer agents. Inorg. Chem. 2014, 53, 727–736. [Google Scholar] [CrossRef]
- Threatt, S.D.; Synold, T.W.; Wu, J.; Barton, J.K. In vivo anticancer activity of a rhodium metalloinsertor in the HCT116 tumor model. Proc. Natl. Acad. Sci. USA 2020, 117, 17535–17542. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Jakupec, M.A.; Zorbas-Seifried, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P.J.; Keppler, B.K. KP1019, a new redox-active anticancer agent—Preclinical development and results of a clinical phase I study in tumor patients. Chem. Biodivers. 2008, 5, 2140–2155. [Google Scholar] [CrossRef] [PubMed]
- Leijen, S.; Burgers, S.A.; Baas, P.; Pluim, D.; Tibben, M.; van Werkhoven, E.; Alessio, E.; Sava, G.; Beijnen, J.H.; Schellens, J.H.M. Phase I/II study with ruthenium compound NAMI-A and gemcitabine in patients with non-small cell lung cancer after first line therapy. Investig. New Drugs 2015, 33, 201–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessio, E.; Messori, L. NAMI-A and KP1019/1339, two iconic ruthenium anticancer drug candidates face-to-face: A case story in medicinal inorganic chemistry. Molecules 2019, 24, 1995. [Google Scholar] [CrossRef] [Green Version]
- Coverdale, J.P.C.; Laroiya-McCarron, T.; Romero-Canelón, I. Designing ruthenium anticancer drugs: What have we learnt from the key drug candidates? Inorganics 2019, 7, 31. [Google Scholar] [CrossRef] [Green Version]
- Soldevila-Barreda, J.J.; Romero-Canelón, I.; Habtemariam, A.; Sadler, P.J. Transfer hydrogenation catalysis in cells as a new approach to anticancer drug design. Nat. Commun. 2015, 6, 6582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wernitznig, D.; Kiakos, K.; Del Favero, G.; Harrer, N.; Machat, H.; Osswald, A.; Jakupec, M.A.; Wernitznig, A.; Sommergruber, W.; Keppler, B.K. First-in-class ruthenium anticancer drug (KP1339/IT-139) induces an immunogenic cell death signature in colorectal spheroids in vitro. Metallomics 2019, 11, 1044–1048. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Gupta, P.; Chen, Y.; Wang, E.; Ji, L.; Chao, H.; Chen, Z.S. The development of anticancer ruthenium(II) complexes: From single molecule compounds to nanomaterials. Chem. Soc. Rev. 2017, 46, 5771–5804. [Google Scholar] [CrossRef] [PubMed]
- Flocke, L.S.; Trondl, R.; Jakupec, M.A.; Keppler, B.K. Molecular mode of action of NKP-1339—A clinically investigated ruthenium-based drug—involves ER- and ROS-related effects in colon carcinoma cell lines. Investig. New Drugs 2016, 34, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Bergamo, A.; Masi, A.; Peacock, A.F.A.; Habtemariam, A.; Sadler, P.J.; Sava, G. In vivo tumour and metastasis reduction and in vitro effects on invasion assays of the ruthenium RM175 and osmium AFAP51 organometallics in the mammary cancer model. J. Inorg. Biochem. 2010, 104, 79–86. [Google Scholar] [CrossRef]
- Frühauf, S.; Zeller, W.J. In vitro evaluation of platinum, titanium and ruthenium metal complexes in cisplatin-sensitive and -resistant rat ovarian tumors. Cancer Chemother. Pharmacol. 1991, 27, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.E.; Aird, R.E.; del Socorro Murdoch, P.; Chen, H.; Cummings, J.; Hughes, N.D.; Parsons, S.; Parkin, A.; Boyd, G.; Jodrell, D.I.; et al. Inhibition of cancer cell growth by ruthenium(II) arene complexes. J. Med. Chem. 2001, 44, 3616–3621. [Google Scholar] [CrossRef]
- Aird, R.E.; Cummings, J.; Ritchie, A.A.; Muir, M.; Morris, R.E.; Chen, H.; Sadler, P.J.; Jodrell, D.I. In vitro and in vivo activity and cross resistance profiles of novel ruthenium (II) organometallic arene complexes in human ovarian cancer. Br. J. Cancer 2002, 86, 1652–1657. [Google Scholar] [CrossRef] [Green Version]
- Grozav, A.; Balacescu, O.; Balacescu, L.; Cheminel, T.; Berindan-Neagoe, I.; Therrien, B. Synthesis, anticancer activity, and genome profiling of thiazolo arene ruthenium complexes. J. Med. Chem. 2015, 58, 8475–8490. [Google Scholar] [CrossRef] [Green Version]
- Mühlgassner, G.; Bartel, C.; Schmid, W.F.; Jakupec, M.A.; Arion, V.B.; Keppler, B.K. Biological activity of ruthenium and osmium arene complexes with modified paullones in human cancer cells. J. Inorg. Biochem. 2012, 116, 180–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seršen, S.; Kljun, J.; Kryeziu, K.; Panchuk, R.; Alte, B.; Körner, W.; Heffeter, P.; Berger, W.; Turel, I. Structure-related mode-of-action differences of anticancer organoruthenium complexes with β-diketonates. J. Med. Chem. 2015, 58, 3984–3996. [Google Scholar] [CrossRef]
- Habtemariam, A.; Melchart, M.; Fernandez, R.; Parsons, S.; Oswald, I.D.H.; Parkin, A.; Fabbiani, F.P.A.; Davidson, J.E.; Dawson, A.; Aird, R.E.; et al. Structure-activity relationships for cytotoxic ruthenium(II) arene complexes containing N,N-, N,O-, and O,O-chelating ligands. J. Med. Chem. 2006, 49, 6858–6868. [Google Scholar] [CrossRef] [PubMed]
- Vock, C.A.; Renfrew, A.K.; Scopelliti, R.; Juillerat-Jeanneret, L.; Dyson, P.J. Influence of the diketonato ligand on the cytotoxicities of [Ru(η6-p-cymene)-(R2acac)(PTA)]+ complexes (PTA = 1,3,5-triaza-7-phosphaadamantane). Eur. J. Inorg. Chem. 2008, 1661–1671. [Google Scholar] [CrossRef]
- Pettinari, R.; Marchetti, F.; Condello, F.; Pettinari, C.; Lupidi, G.; Scopelliti, R.; Mukhopadhyay, S.; Riedel, T.; Dyson, P.J. Ruthenium(II)-arene RAPTA type complexes containing curcumin and bisdemethoxycurcumin display potent and selective anticancer activity. Organometallics 2014, 33, 3709–3715. [Google Scholar] [CrossRef]
- Caruso, F.; Rossi, M.; Benson, A.; Opazo, C.; Freedman, D.; Monti, E.; Gariboldi, M.B.; Shaulky, J.; Marchetti, F.; Pettinari, R.; et al. Ruthenium-arene complexes of curcumin: X-ray and density functional theory structure, synthesis, and spectroscopic characterization, in vitro antitumor activity, and DNA docking studied of (p-cymene)Ru(curcuminato)chloro. J. Med. Chem. 2012, 55, 1072–1081. [Google Scholar] [CrossRef]
- Gatti, A.; Habtemariam, A.; Romero-Canelόn, I.; Song, J.I.; Heer, B.; Clarkson, G.J.; Rogolino, D.; Sadler, P.J.; Carcelli, M. Half-sandwich arene ruthenium(II) and osmium(II) thiosemicarbazone complexes: Solution behavior and antiproliferative activity. Organometallics 2018, 37, 891–899. [Google Scholar] [CrossRef]
- Basto, A.P.; Anghel, N.; Rubbiani, R.; Müller, J.; Stibal, D.; Giannini, F.; Süss-Fink, G.; Balmer, V.; Gasser, G.; Furrer, J.; et al. Targeting of the mitochondrion by dinuclear thiolato-bridged arene ruthenium complexes in cancer cells and in the apicomplexan parasite Neospora caninum. Metallomics 2019, 11, 462–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.A.; Katritzky, A.R. N-oxides and related compounds. Part XVII. The tautomerism of mercapto- and acylamino-pyridine 1-oxides. J. Chem. Soc. 1960, 2937–2942. [Google Scholar] [CrossRef]
- Kljun, J.; Anko, M.; Traven, K.; Sinreih, M.; Pavlič, R.; Peršič, Š.; Ude, Ž.; Codina, E.E.; Stojan, J.; Lanišnik, R.T.; et al. Pyrithione-based ruthenium complexes as inhibitors of aldo-keto reductase 1C enzymes and anticancer agents. Dalton Trans. 2016, 45, 11791–11800. [Google Scholar] [CrossRef] [Green Version]
- Kladnik, J.; Kljun, J.; Burmeister, H.; Ott, I.; Romero-Canelón, I.; Turel, I. Towards identification of essential structural elements of organoruthenium(II)-pyrithionato complexes for anticancer activity. Chem. Eur. J. 2019, 25, 14169–14182. [Google Scholar] [CrossRef] [PubMed]
- Marković, K.; Milačič, R.; Marković, S.; Kladnik, J.; Turel, I.; Ščančar, J. Binding kinetics of ruthenium pyrithione chemotherapeutic candidates to human serum proteins studied by HPLC-ICP-MS. Molecules 2020, 25, 1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ristovski, S.; Uzelac, M.; Kljun, J.; Lipec, T.; Uršič, M.; Zemljič, J.Š.; Žužek, M.C.; Trobec, T.; Frangež, R.; Sepčić, K.; et al. Organoruthenium prodrugs as a new class of cholinesterase and glutathione-S-transferase inhibitors. ChemMedChem 2018, 13, 2166–2176. [Google Scholar] [CrossRef] [PubMed]
- Kladnik, J.; Ristovski, S.; Kljun, J.; Defant, A.; Mancini, I.; Sepčić, K.; Turel, I. Structural isomerism and enhanced lipophilicity of pyrithione ligands of organoruthenium(II) complexes increase inhibition on AChE and BuChE. Int. J. Mol. Sci. 2020, 21, 5628. [Google Scholar] [CrossRef]
- Daigle, D.J.; Decuir, T.J.; Robertson, J.B.; Darensbourg, D.J. 1,3,5‐Triaz‐7‐phosphatricyclo[3.3.1.13,7]decane and derivatives. In Inorganic Syntheses; Darensbourg, M.Y., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1998; Volume 32, pp. 40–45. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Blachly, P.G.; McCammon, J.A.; Cohen, S.M. Exploring the influence of the protein environment on metal-binding pharmacophores. J. Med. Chem. 2014, 57, 7126–7135. [Google Scholar] [CrossRef]
- Lam, N.Y.S.; Truong, D.; Burmeister, H.; Babak, M.V.; Holtkamp, H.U.; Movassaghi, S.; Ayine-Tora, D.M.; Zafar, A.; Kubanik, M.; Oehninger, L.; et al. From catalysis to cancer: Toward structure–activity relationships for benzimidazol-2-ylidene-derived N-heterocyclic-carbene complexes as anticancer agents. Inorg. Chem. 2018, 57, 14427–14434. [Google Scholar] [CrossRef]
- Oehninger, L.; Alborzinia, H.; Ludewig, S.; Baumann, K.; Wölfl, S.; Ott, I. From catalysts to bioactive organometallics: Do Grubbs catalysts trigger biological effects? ChemMedChem 2011, 6, 2142–2145. [Google Scholar] [CrossRef]
- Schatzschneider, U.; Niesel, J.; Ott, I.; Gust, R.; Alborzinia, H.; Wolfl, S. Cellular uptake, cytotoxicity, and metabolic profiling of human cancer cells treated with ruthenium(II) polypyridyl complexes [Ru(bpy)2(N-N)]Cl2 with N-N = bpy, phen, dpq, dppz, and dppn. ChemMedChem 2008, 3, 1104–1109. [Google Scholar] [CrossRef]
- Pizarro, A.M.; Habtemariam, A.; Sadler, P.J. Activation mechanisms for organometallic anticancer complexes. In Medicinal Organometallic Chemistry; Jaouen, G., Metzler-Nolte, N., Eds.; (Topics in Organometallic Chemistry); Springer: Berlin, Germany, 2010; Volume 32, pp. 21–56. [Google Scholar] [CrossRef]
- Briš, A.; Jašik, J.; Turel, I.; Roithova, J. Anti-cancer organoruthenium(II) complexes and their interactions with cysteine and its analogues. A mass-spectrometric study. Dalton Trans. 2019, 48, 2626–2634. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Hou, Z.; Cui, S.; Cao, K.; Yuan, S.; Sun, M.; Kljun, J.; Huang, G.; Turel, I.; Liu, Y. Covalent versus noncovalent binding of ruthenium η6-p-cymene complexes to zinc-finger protein NCp7. Chem. Eur. J. 2019, 25, 12789–12794. [Google Scholar] [CrossRef] [PubMed]
- Namiecińska, E.; Sadowska, B.; Więckowska-Szakiel, M.; Dołęga, A.; Pasternak, B.; Grazul, M.; Budzisz, E. Anticancer and antimicrobial properties of novel η6-p-cymene ruthenium(II) complexes containing a N,S-type ligand, their structural and theoretical characterization. RSC Adv. 2019, 9, 38629–38645. [Google Scholar] [CrossRef] [Green Version]
- Oehninger, L.; Stefanopoulou, M.; Alborzinia, H.; Schur, J.; Ludewig, S.; Namikawa, K.; Muñoz-Castro, A.; Köster, R.W.; Baumann, K.; Wölfl, S.; et al. Evaluation of arene ruthenium(II) N-heterocyclic carbene complexes as organometallics interacting with thiol and selenol containing biomolecules. Dalton Trans. 2013, 42, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Demoro, B.; de Almeida, R.F.M.; Marques, F.; Matos, C.P.; Otero, L.; Pessoa, J.C.; Santos, I.; Rodriguez, A.; Moreno, V.; Lorenzo, J.; et al. Screening organometallic binuclear thiosemicarbazone ruthenium complexes as potential anti-tumour agents: Cytotoxic activity and human serum albumin binding mechanism. Dalton Trans. 2013, 42, 7131–7146. [Google Scholar] [CrossRef]
- Mitra, R.; Das, S.; Shinde, S.V.; Sinha, S.; Somasundaram, K.; Samuelson, A.G. Anticancer activity of hydrogen-bond-stabilized half-sandwich RuII complexes with heterocycles. Chem. Eur. J. 2012, 18, 12278–12291. [Google Scholar] [CrossRef]
- Needham, R.J.; Sanchez-Cano, C.; Zhang, X.; Romero-Canelón, I.; Habtemariam, A.; Cooper, M.S.; Meszaros, L.; Clarkson, G.J.; Blower, P.J.; Sadler, P.J. In-cell activation of organo-osmium(II) anticancer complexes. Angew. Chem. Int. Ed. 2017, 56, 1017–1020. [Google Scholar] [CrossRef]
- Chow, M.J.; Ang, W.H. Organoruthenium(II)-arene complexes: Structural building blocks for anticancer drug discovery. In Inorganic and Organometallic Transition Metal Complexes with Biological Molecules and Living Cells; Lo, K.K.W., Ed.; Elsevier Academic Press Inc: San Diego, CA, USA, 2017; pp. 119–146. [Google Scholar]
- Boivin, M.; Lane, D.; Piché, A.; Rancourt, C. CA125 (MUC16) tumor antigen selectively modulates the sensitivity of ovarian cancer cells to genotoxic drug-induced apoptosis. Gynecol. Oncol. 2009, 115, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Furlong, M.T.; Hough, C.D.; Sherman-Baust, C.A.; Pizer, E.S.; Morin, P.J. Evidence for the colonic origin of ovarian cancer cell line SW626. J. Natl. Cancer Inst. 1999, 91, 1327–1328. [Google Scholar] [CrossRef] [Green Version]
- Puckett, C.A.; Ernst, R.J.; Barton, J.K. Exploring the cellular accumulation of metal complexes. Dalton Trans. 2010, 39, 1159–1170. [Google Scholar] [CrossRef] [Green Version]
- Renfrew, A.K. Transition metal complexes with bioactive ligands: Mechanisms for selective ligand release and applications for drug delivery. Metallomics 2014, 6, 1324–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Tao, L.; Fan, L.X.; Huang, K.; Luo, H.M.; Ge, H.; Wang, X.Y.; Wang, Q. Cx32 mediates cisplatin resistance in human ovarian cancer cells by affecting drug efflux transporter expression and activating the EGFR-Akt pathway. Mol. Med. Rep. 2019, 19, 2287–2296. [Google Scholar] [CrossRef] [PubMed]
- Bugarcic, T.; Novakova, O.; Halamikova, A.; Zerzankova, L.; Vrana, O.; Kasparkova, J.; Habtemariam, A.; Parsons, S.; Sadler, P.J.; Brabec, V. Cytotoxicity, cellular uptake, and DNA interactions of new monodentate ruthenium(II) complexes containing terphenyl arenes. J. Med. Chem. 2008, 51, 5310–5319. [Google Scholar] [CrossRef] [PubMed]
- Scolaro, C.; Chaplin, A.B.; Hartinger, C.G.; Bergamo, A.; Cocchietto, M.; Keppler, B.K.; Sava, G.; Dyson, P.J. Tuning the hydrophobicity of ruthenium(II)-arene (RAPTA) drugs to modify uptake, biomolecular interactions and efficacy. Dalton Trans. 2007, 43, 5065–5072. [Google Scholar] [CrossRef]
- Flagg, E.W.; Coates, R.J.; Jones, D.P.; Eley, J.W.; Gunter, E.W.; Jackson, B.; Greenberg, R.S. Plasma total glutathione in humans and its association with demographic and health-related factors. Br. J. Nutr. 1993, 70, 797–808. [Google Scholar] [CrossRef]
- Arner, E.S.J.; Holmgren, A. The thioredoxin system in cancer. Semin. Cancer Biol. 2006, 16, 420–426. [Google Scholar] [CrossRef]
- Nguyen, P.; Awwad, R.T.; Smart, D.D.K.; Spitz, D.R.; Gius, D. Thioredoxin reductase as a novel molecular target for cancer therapy. Cancer Lett. 2006, 236, 164–174. [Google Scholar] [CrossRef]
- Sasada, T.; Iwata, S.; Sato, N.; Kitaoka, Y.; Hirota, K.; Nakamura, K.; Nishiyama, A.; Taniguchi, Y.; Takabayashi, A.; Yodoi, J. Redox control of resistance to cis-diamminedichloroplatinum (II) (CDDP)—Protective effect of human thioredoxin against CDDP-induced cytotoxicity. J. Clin. Investig. 1996, 97, 2268–2276. [Google Scholar] [CrossRef] [Green Version]
- Marzano, C.; Gandin, V.; Folda, A.; Scutari, G.; Bindoli, A.; Rigobello, M.P. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free Radic. Biol. Med. 2007, 42, 872–881. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Ryter, S.W.; Kim, H.P.; Hoetzel, A.; Park, J.W.; Nakahira, K.; Wang, X.; Choi, A.M.K. Mechanisms of cell death in oxidative stress. Antioxid. Redox Signal. 2007, 9, 49–89. [Google Scholar] [CrossRef]
- Jungwirth, U.; Kowol, C.R.; Keppler, B.K.; Hartinger, C.G.; Berger, W.; Heffeter, P. Anticancer activity of metal complexes: Involvement of redox processes. Antioxid. Redox Signal. 2011, 15, 1085–1127. [Google Scholar] [CrossRef] [Green Version]
- Coverdale, J.P.C.; Bridgewater, H.E.; Song, J.I.; Smith, N.A.; Barry, N.P.E.; Bagley, I.; Sadler, P.J.; Romero-Canelón, I. In vivo selectivity and localization of reactive oxygen species (ROS) induction by osmium anticancer complexes that circumvent platinum resistance. J. Med. Chem. 2018, 61, 9246–9255. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.C.; Zhang, Y.; Jie, X.M.; She, J.; Dongye, G.Z.; Zhong, Y.; Deng, Y.Y.; Wang, J.; Guo, B.Y.; Chen, L.M. Ruthenium(II) salicylate complexes inducing ROS-mediated apoptosis by targeting thioredoxin reductase. J. Inorg. Biochem. 2019, 193, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jiang, G.B.; Yao, J.H.; Wang, X.Z.; Wang, J.; Han, B.J.; Xie, Y.Y.; Lin, G.J.; Huang, H.L.; Liu, Y.J. Ruthenium(II) complexes: DNA-binding, cytotoxicity, apoptosis, cellular localization, cell cycle arrest, reactive oxygen species, mitochondrial membrane potential and western blot analysis. J. Photochem. Photobiol. B 2014, 140, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Bolitho, E.M.; Coverdale, J.P.C.; Bridgewater, H.E.; Clarkson, G.J.; Quinn, P.D.; Sanchez-Cano, C.; Sadler, P.J. Tracking reactions of asymmetric organo-osmium transfer hydrogenation catalysts in cancer cells. Angew. Chem. Int. Ed. 2021, 60, 6462–6472. [Google Scholar] [CrossRef] [PubMed]
- Bragado, P.; Armesilla, A.; Silva, A.; Porras, A. Apoptosis by cisplatin requires p53 mediated p38 alpha MAPK activation through ROS generation. Apoptosis 2007, 12, 1733–1742. [Google Scholar] [CrossRef]
- Rosell, R.; Mendez, P.; Isla, D.; Taron, M. Platinum resistance related to a functional NER pathway. J. Thorac. Oncol. 2007, 2, 1063–1066. [Google Scholar] [CrossRef] [Green Version]
- de Carvalho, N.C.; Neves, S.P.; Dias, R.B.; Valverde, L.D.F.; Sales, C.B.S.; Rocha, C.A.G.; Soares, M.B.P.; dos Santos, E.R.; Oliveira, R.M.M.; Carlos, R.M.; et al. A novel ruthenium complex with xanthoxylin induces S-phase arrest and causes ERK1/2-mediated apoptosis in HepG2 cells through a p53-independent pathway. Cell Death Dis. 2018, 9, 79. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) [a] | |||||
|---|---|---|---|---|---|---|
| A2780 | A549 | HCT116 | OE19 | HEPG2 | PC3 | |
| a | 8.6 ± 0.2 | 11.6 ± 0.8 | 17.5 ± 0.4 | 12.1 ± 0.4 | 32.5 ± 0.6 | 16.5 ± 0.3 |
| 1a | 1.0 ± 0.08 | 4.5 ± 0.3 | 14.3 ± 0.9 | 10.9 ± 0.2 | 29.5 ± 0.3 | 5.1 ± 0.2 |
| 2a | >50 [b] | >50 [b] | >50 [b] | >50 [b] | >50 [b] | >50 [b] |
| b | 12.4 ± 0.6 | 10.4 ± 0.2 | 15.1 ± 0.4 | 16.7 ± 0.5 | 29.1 ± 0.9 | 13.1 ± 0.2 |
| 1b | 2.2 ± 0.3 | 5.8 ± 0.5 | 8.4 ± 0.3 | 11.4 ± 0.6 | 25.9 ± 0.4 | 3.9 ± 0.6 |
| 2b | >50 [b] | >50 [b] | >50 [b] | >50 [b] | >50 [b] | >50 [b] |
| Cisplatin | 1.2 ± 0.3 | 3.2 ± 0.1 | 5.2 ± 0.3 | 8.7 + 0.9 | 5.7 ± 0.9 | 4.1 ± 0.5 |
| Compound | IC50 (µM) [a] | ||||
|---|---|---|---|---|---|
| A2780 | SKOV3 | SW626 | A2780Cis | A2780ADR | |
| a | 8.6 ± 0.2 | 22.5 ± 0.4 | 8.4 ± 0.6 | n.d. [c] | n.d. [c] |
| 1a | 1.0 ± 0.08 | 6.4 ± 0.2 | 3.8 ± 0.4 | 1.1 ± 0.05 | 1.6 ± 0.2 |
| 2a | >50 [b] | >50 [b] | >50 [b] | n.d. [c] | n.d. [c] |
| b | 12.4 ± 0.6 | 20.3 ± 0.6 | 6.4 ± 0.5 | n.d. [c] | n.d. [c] |
| 1b | 2.2 ± 0.3 | 5.1 ± 0.2 | 2.8 ± 0.4 | 2.5 ± 0.1 | 2.8 ± 0.4 |
| 2b | >50 [b] | >50 [b] | >50 [b] | n.d. [c] | n.d. [c] |
| Cisplatin | 1.2 ± 0.3 | 16.8 ± 0.8 | 15.7 ±0.8 | 13.4 ± 0.3 | 8.9 ± 0.5 |
| Complex | nmol Ru/mg Protein [a] |
|---|---|
| 1a | 1.30 ± 0.22 |
| 2a | 1.02 ± 0.15 |
| Untreated | 0.03 ± 0.09 |
| Complex | G1 | G2/M | S |
|---|---|---|---|
| 1a | 72.5 ± 0.5 | 12.6 ± 0.7 | 14.9 ± 0.6 |
| Untreated | 62.3 ± 0.3 | 17.8 ± 0.4 | 19.9 ± 0.8 |
| Complex | PI−/ANN− | PI+/ANN− | PI−/ANN+ | PI+/ANN+ |
|---|---|---|---|---|
| 1a | 76.5 ± 0.9 ** | 11.2 ± 0.8 ** | 4.1 ± 0.4 ** | 8.4 ± 0.7 ** |
| Untreated | 95.6 ± 0.7 | 2 ± 1 | 2 ± 1 | 0.3 ± 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kladnik, J.; Coverdale, J.P.C.; Kljun, J.; Burmeister, H.; Lippman, P.; Ellis, F.G.; Jones, A.M.; Ott, I.; Romero-Canelón, I.; Turel, I. Organoruthenium Complexes with Benzo-Fused Pyrithiones Overcome Platinum Resistance in Ovarian Cancer Cells. Cancers 2021, 13, 2493. https://doi.org/10.3390/cancers13102493
Kladnik J, Coverdale JPC, Kljun J, Burmeister H, Lippman P, Ellis FG, Jones AM, Ott I, Romero-Canelón I, Turel I. Organoruthenium Complexes with Benzo-Fused Pyrithiones Overcome Platinum Resistance in Ovarian Cancer Cells. Cancers. 2021; 13(10):2493. https://doi.org/10.3390/cancers13102493
Chicago/Turabian StyleKladnik, Jerneja, James P. C. Coverdale, Jakob Kljun, Hilke Burmeister, Petra Lippman, Francesca G. Ellis, Alan M. Jones, Ingo Ott, Isolda Romero-Canelón, and Iztok Turel. 2021. "Organoruthenium Complexes with Benzo-Fused Pyrithiones Overcome Platinum Resistance in Ovarian Cancer Cells" Cancers 13, no. 10: 2493. https://doi.org/10.3390/cancers13102493
APA StyleKladnik, J., Coverdale, J. P. C., Kljun, J., Burmeister, H., Lippman, P., Ellis, F. G., Jones, A. M., Ott, I., Romero-Canelón, I., & Turel, I. (2021). Organoruthenium Complexes with Benzo-Fused Pyrithiones Overcome Platinum Resistance in Ovarian Cancer Cells. Cancers, 13(10), 2493. https://doi.org/10.3390/cancers13102493