Circumventing Drug Treatment? Intrinsic Lethal Effects of Polyethyleneimine (PEI)-Functionalized Nanoparticles on Glioblastoma Cells Cultured in Stem Cell Conditions

 ,
,  , , , ,
, , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
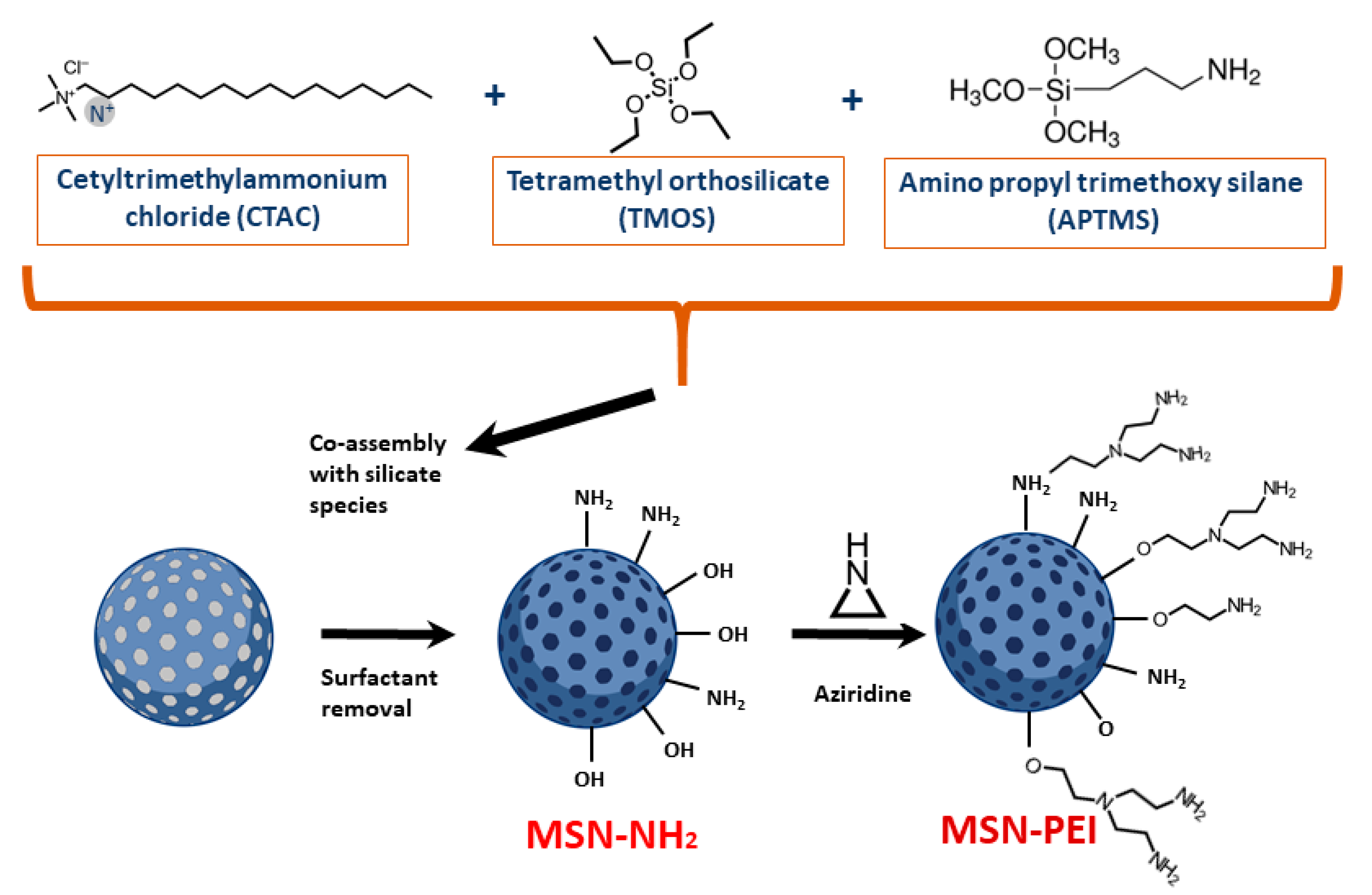
2.1. Preparation and Characterization of Hyperbranched PEI-Functionalized Mesoporous Silica Nanoparticles (PEI-MSNs)
2.2. Cell Culture
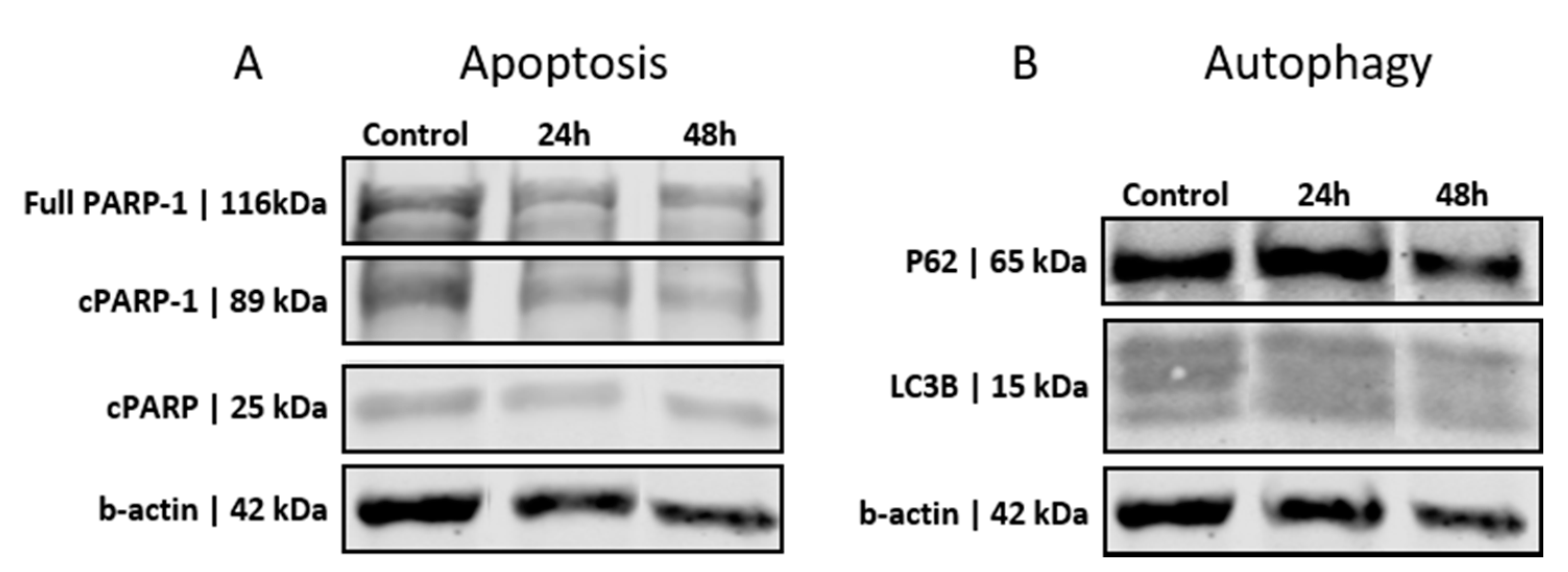
2.3. Western Blotting and Antibodies
2.4. Flow Cytometry
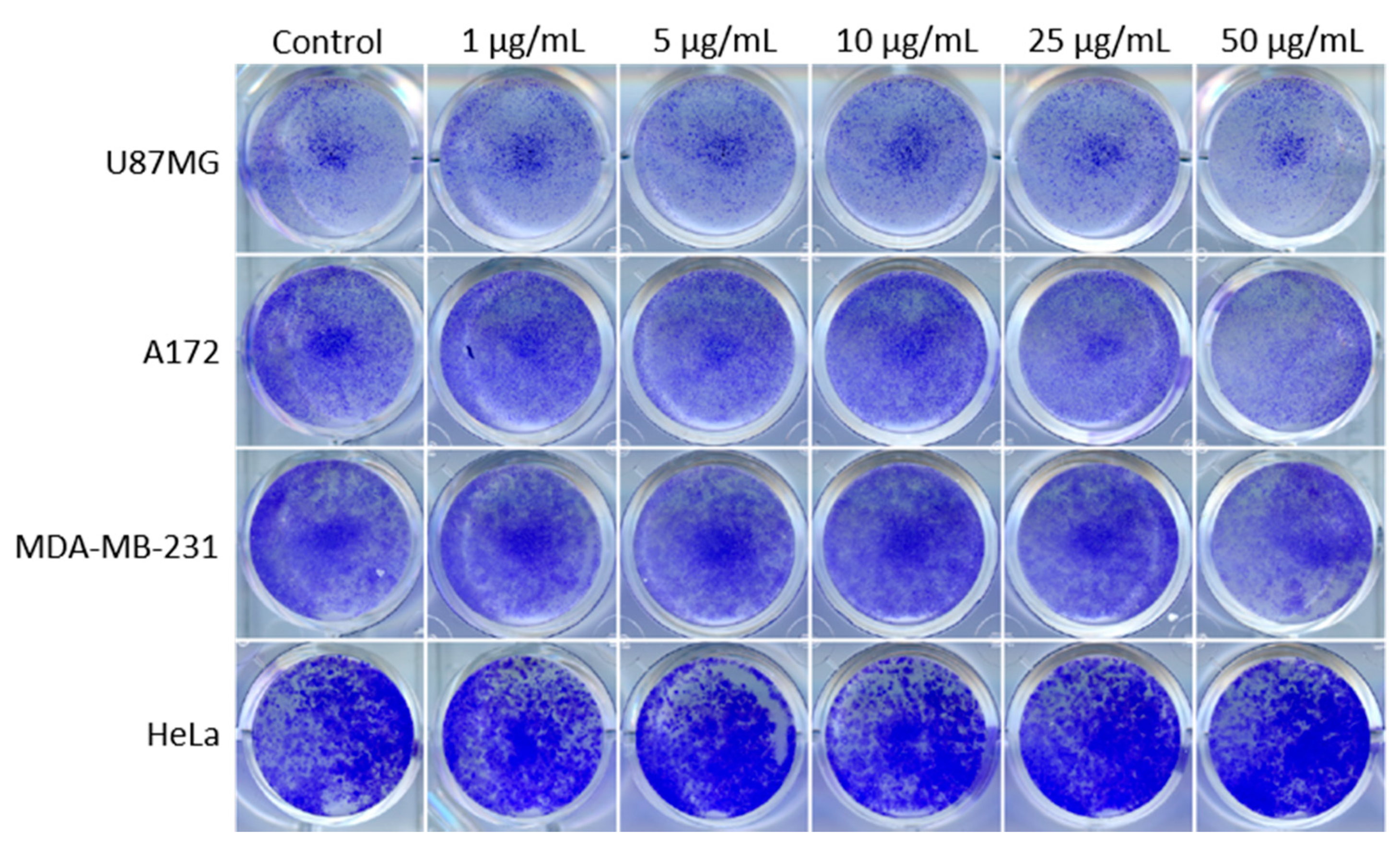
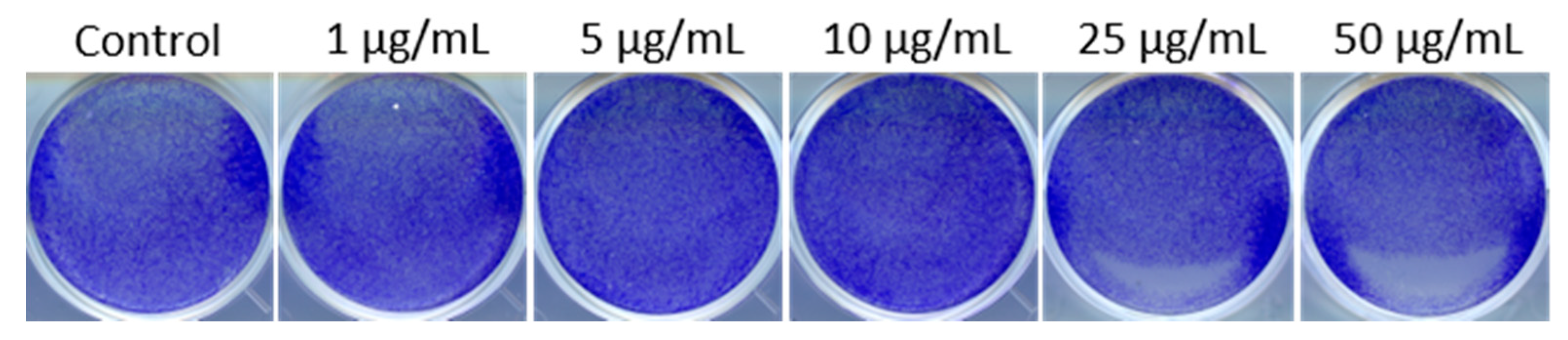
2.5. Colony Formation Assay
2.6. Light Microscopy
2.6.1. Immunofluorescence (Early Endosomes and Lysosomes)
2.6.2. Mitochondrial Staining
2.7. Transmission Electron Microscopy (TEM)
2.8. In Vitro Blood–Brain Tumor Barrier
2.9. In Vivo Procedures
3. Results
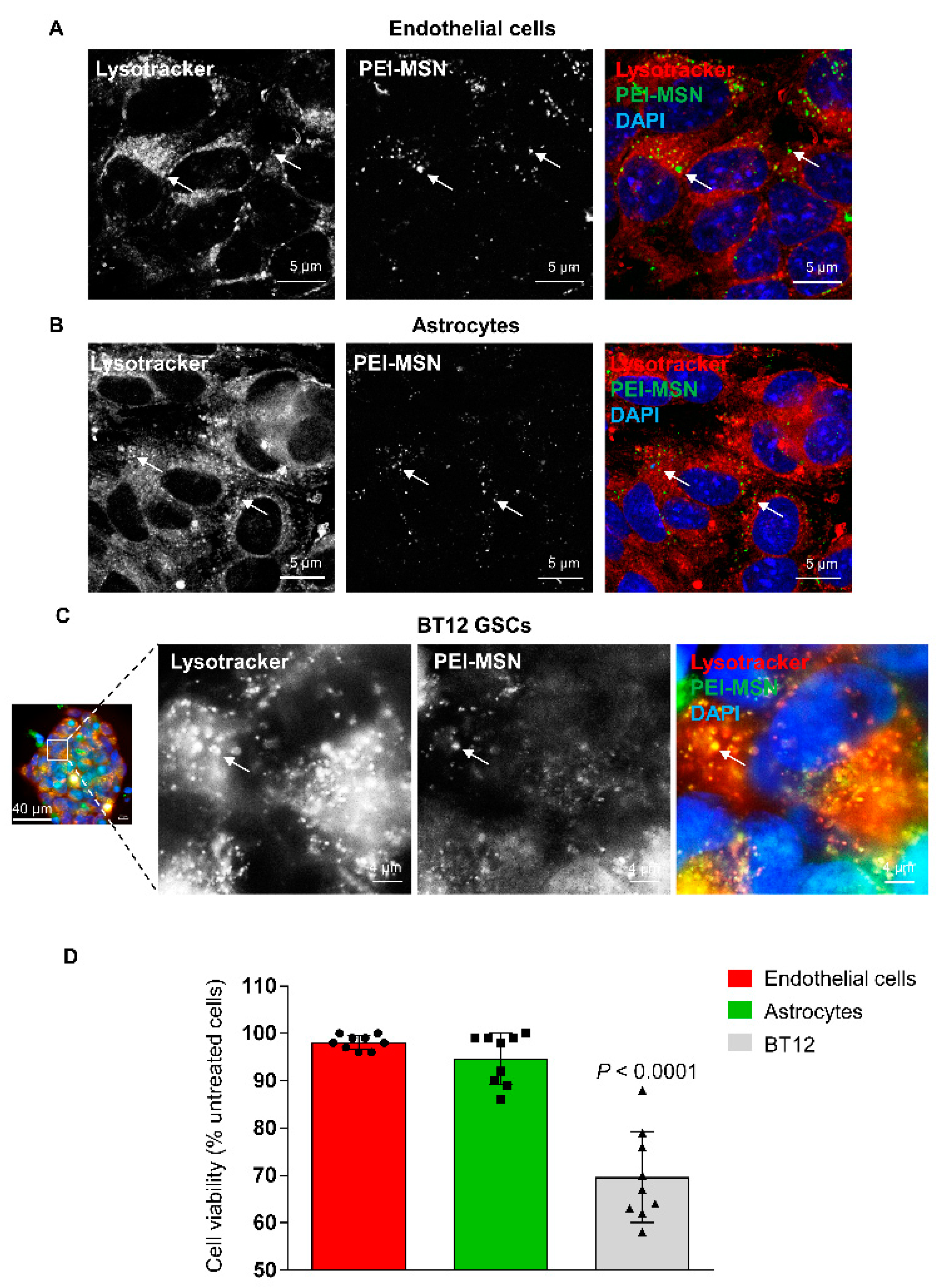
3.1. PEI-MSNs Exhibit Specific Toxicity towards GSCs Cultured under Stem Cell Conditions
3.2. GSCs Show No Induction of Apoptosis or Autophagy after PEI-MSN Treatment
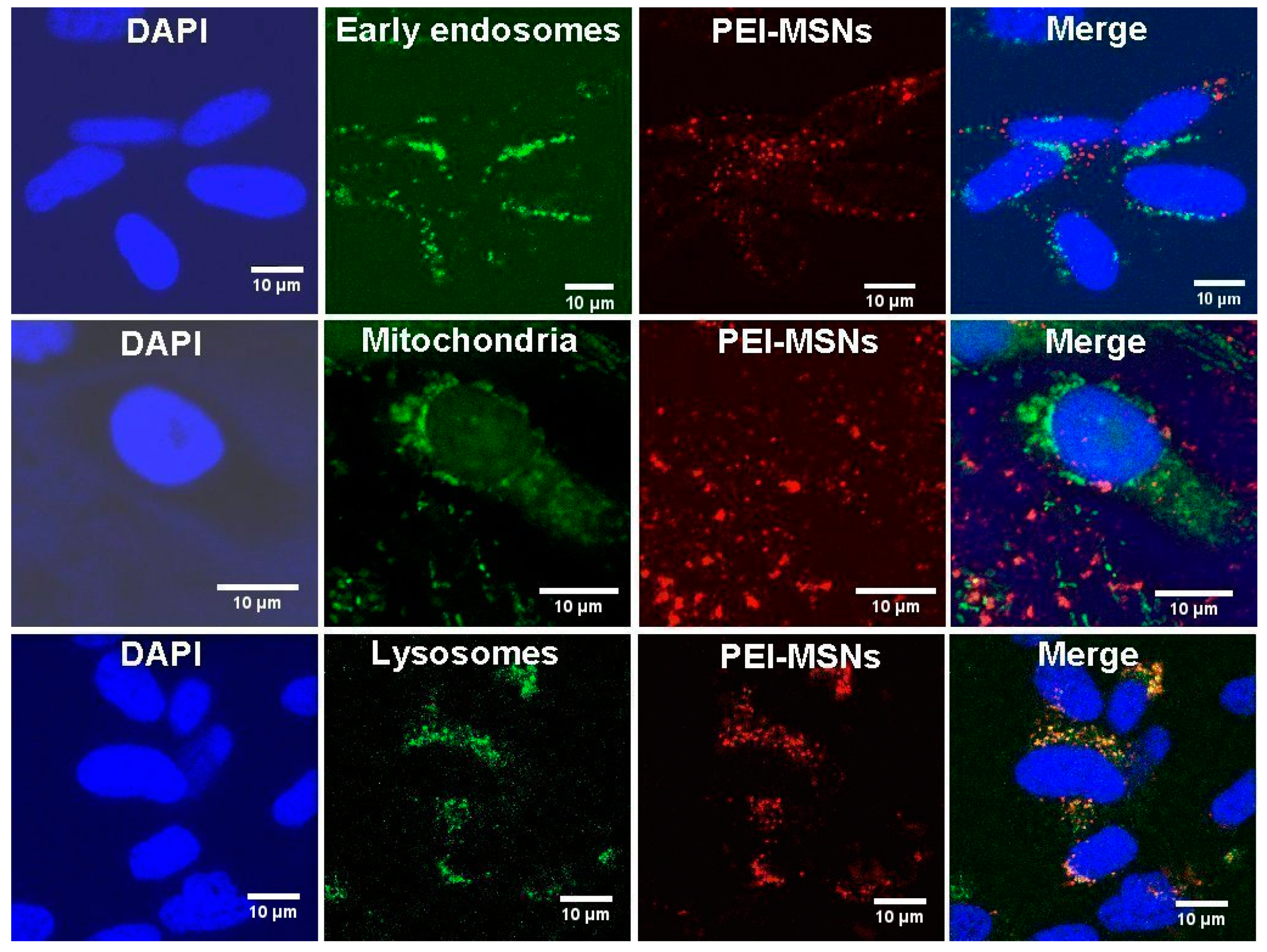
3.3. PEI-MSNs Localize within the Cytoplasmic Space and Lysosomes
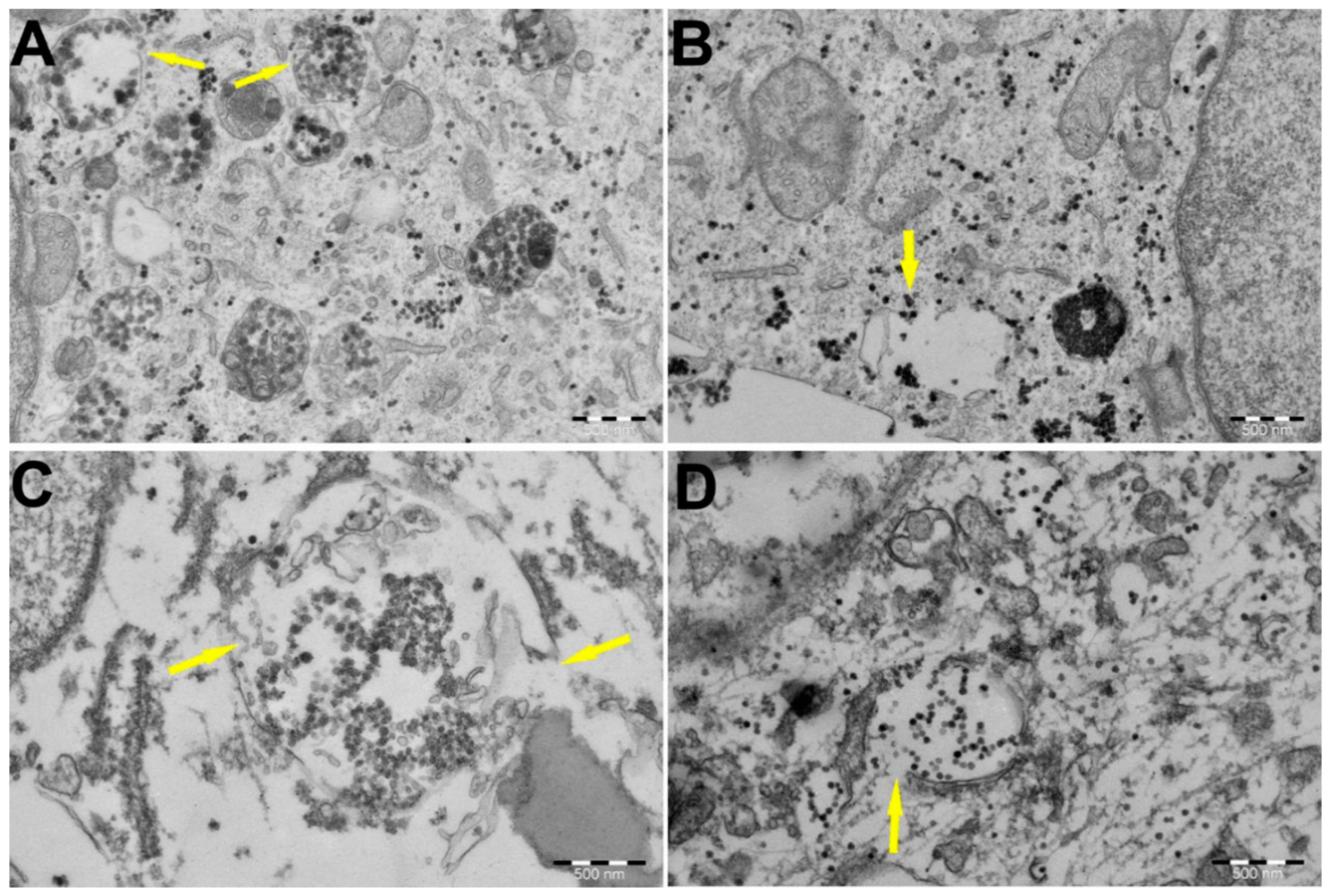
3.4. PEI-MSNs Cause Lysosomal Membrane Rupture in GSCs, Leading to Cell Death
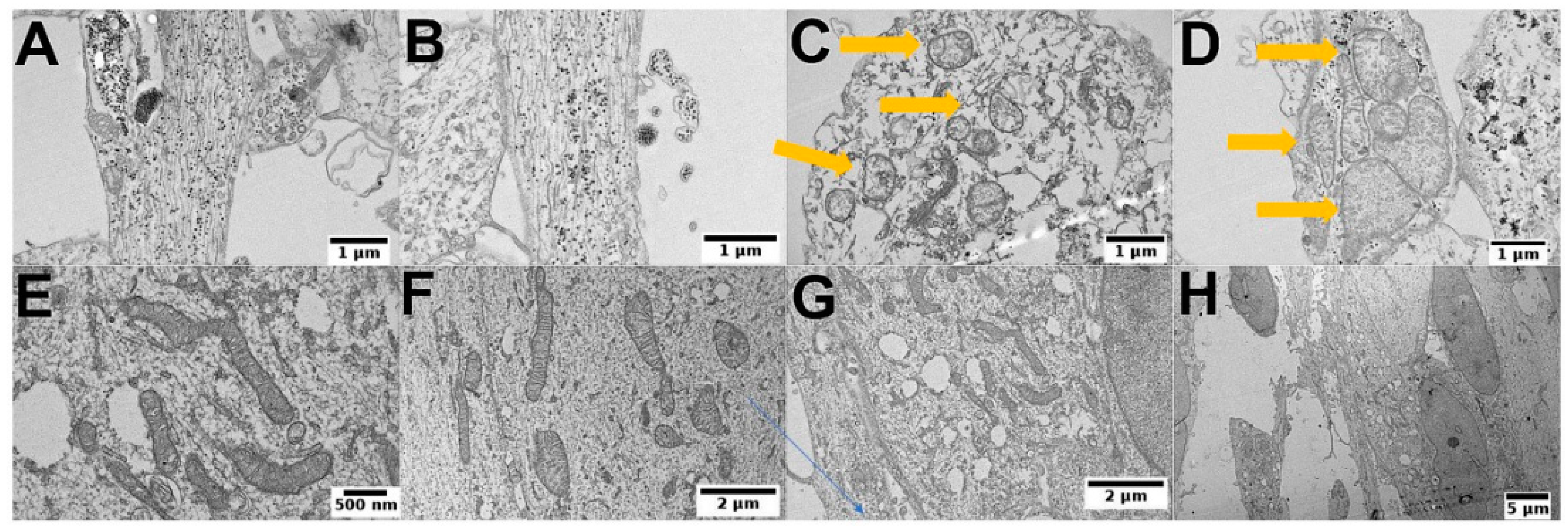
3.5. PEI-MSNs Cause Morphological Abnormalities in GSCs
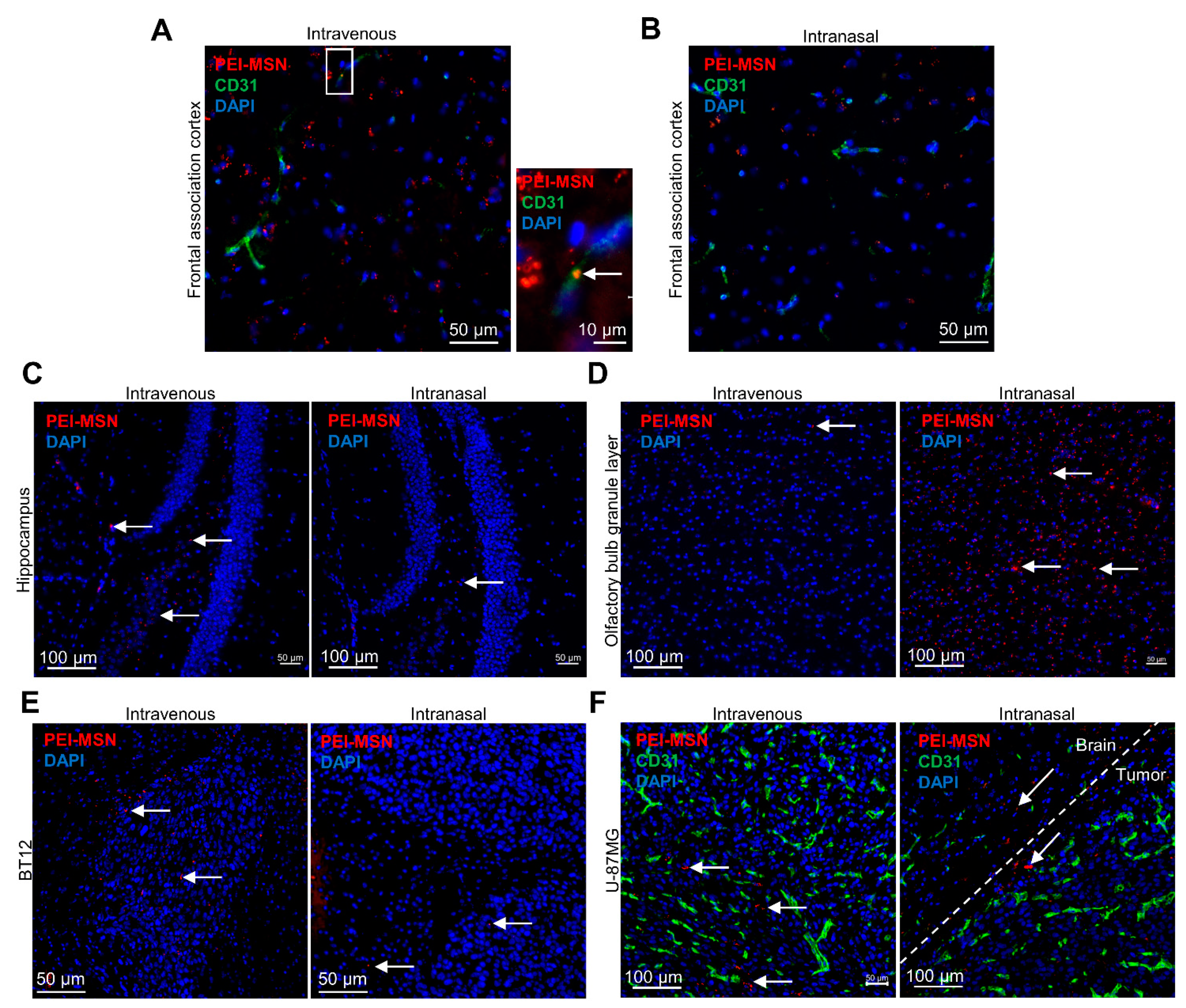
3.6. PEI-MSNs cross the Neurovascular Unit In Vitro and In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Holland, E.C. Glioblastoma multiforme: The terminator. Proc. Natl. Acad. Sci. USA 2000, 97, 6242–6244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar]
- Eramo, A.; Vitiani, L.R.; Zeuner, A.; Pallini, R.; Lotti, F.; Sette, G.; Pilozzi, E.; LaRocca, L.M.; Peschle, C.; De Maria, R. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006, 13, 1238–1241. [Google Scholar] [CrossRef] [Green Version]
- Haar, C.P.; Hebbar, P.; Wallace, G.C.; Das, A.; Vandergrift, W.A.; Smith, J.A.; Giglio, P.; Patel, S.J.; Ray, S.K.; Banik, N.L. Drug Resistance in Glioblastoma: A Mini Review. Neurochem. Res. 2012, 37, 1192–1200. [Google Scholar] [CrossRef]
- Syed, M.; Liermann, J.; Verma, V.; Bernhardt, D.; Bougatf, N.; Paul, A.; Rieken, S.; Debus, J.; Adeberg, S. Survival and recurrence patterns of multifocal glioblastoma after radiation therapy. Cancer Manag. Res. 2018, 10, 4229–4235. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karaman, D.Ş.; Sarparanta, M.P.; Rosenholm, J.M.; Airaksinen, A.J. Multimodality Imaging of Silica and Silicon Materials In Vivo. Adv. Mater. 2018, 30, e1703651. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Kim, G.R.; Yoon, J.; Kim, S.E.; Yoo, J.S.; Piao, Y. In vivo delineation of glioblastoma by targeting tumor-associated macrophages with near-infrared fluorescent silica coated iron oxide nanoparticles in orthotopic xenografts for surgical guidance. Sci. Rep. 2018, 8, 11122. [Google Scholar] [CrossRef]
- Baghirov, H.; Karaman, D.; Viitala, T.; Duchanoy, A.; Lou, Y.-R.; Mamaeva, V.; Pryazhnikov, E.; Khiroug, L.; Davies, C.D.L.; Sahlgren, C.; et al. Feasibility Study of the Permeability and Uptake of Mesoporous Silica Nanoparticles across the Blood-Brain Barrier. PLoS ONE 2016, 11, e0160705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulin-Sarfraz, T.; Pryazhnikov, E.; Zhang, J.; Khiroug, L.; Rosenholm, J.M. Chemical and photonic interactions in vitro and in vivo between fluorescent tracer and nanoparticle-based scavenger for enhanced molecular imaging. Mater. Today Bio 2019, 2, 100010. [Google Scholar] [CrossRef] [PubMed]
- Shevtsov, M.; Parr, M.A.; Ryzhov, V.A.; Zemtsova, E.G.; Arbenin, A.Y.; Ponomareva, A.N.; Smirnov, V.M.; Multhoff, G. Zero-valent Fe confined mesoporous silica nanocarriers (Fe(0) @ MCM-41) for targeting experimental orthotopic glioma in rats. Sci. Rep. 2016, 6, 29247. [Google Scholar] [CrossRef] [PubMed]
- Shahein, S.A.; Aboul-Enein, A.M.; Higazy, I.M.; Abou-Elella, F.; Lojkowski, W.; Ahmed, E.R.; A Mousa, S.; AbouAitah, K. Targeted anticancer potential against glioma cells of thymoquinone delivered by mesoporous silica core-shell nanoformulations with pH-dependent release. Int. J. Nanomed. 2019, 14, 5503–5526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turan, O.; Bielecki, P.A.; Perera, V.; Lorkowski, M.; Covarrubias, G.; Tong, K.; Yun, A.; Loutrianakis, G.; Raghunathan, S.; Park, Y.; et al. Treatment of Glioblastoma Using Multicomponent Silica Nanoparticles. Adv. Ther. 2019, 2, 1900118. [Google Scholar] [CrossRef] [PubMed]
- Dogra, P.; Adolphi, N.L.; Wang, Z.; Lin, Y.-S.; Butler, K.S.; Durfee, P.N.; Croissant, J.G.; Noureddine, A.; Coker, E.N.; Bearer, E.L.; et al. Establishing the effects of mesoporous silica nanoparticle properties on in vivo disposition using imaging-based pharmacokinetics. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Saroj, S.; Rajput, S.J. Composite smart mesoporous silica nanoparticles as promising therapeutic and diagnostic candidates: Recent trends and applications. J. Drug Deliv. Sci. Technol. 2018, 44, 349–365. [Google Scholar] [CrossRef]
- von Baeckmann, C.; Guillet-Nicolas, R.; Renfer, D.; Kählig, H.; Kleitz, F. A Toolbox for the Synthesis of Multifunctionalized Mesoporous Silica Nanoparticles for Biomedical Applications. ACS Omega 2018, 3, 17496–17510. [Google Scholar] [CrossRef]
- Shi, Y.; Miller, M.L.; Di Pasqua, A.J. Biocompatibility of Mesoporous Silica Nanoparticles? Comments Inorg. Chem. 2015, 36, 61–80. [Google Scholar] [CrossRef]
- Liu, J.; Liu, T.; Pan, J.; Liu, S.; Lu, G. (Max) Advances in Multicompartment Mesoporous Silica Micro/Nanoparticles for Theranostic Applications. Annu. Rev. Chem. Biomol. Eng. 2018, 9, 389–411. [Google Scholar] [CrossRef]
- Gisbert-Garzarán, M.; Vallet-Regí, M. Influence of the Surface Functionalization on the Fate and Performance of Mesoporous Silica Nanoparticles. Nanomaterials 2020, 10, 916. [Google Scholar] [CrossRef]
- Sábio, R.M.; Meneguin, A.B.; Ribeiro, T.C.; Silva, R.R.; Chorilli, M. New insights towards mesoporous silica nanoparticles as a technological platform for chemotherapeutic drugs delivery. Int. J. Pharm. 2019, 564, 379–409. [Google Scholar] [CrossRef]
- Rosenholm, J.M.; Penninkangas, A.; Lindén, M. Amino-functionalization of large-pore mesoscopically ordered silica by a one-step hyperbranching polymerization of a surface-grown polyethyleneimine. Chem. Commun. 2006, 3909–3911. [Google Scholar] [CrossRef]
- Desai, D.; Karaman, D.Ş.; Prabhakar, N.; Tadayon, S.; Duchanoy, A.; Toivola, D.M.; Rajput, S.; Näreoja, T.; Rosenholm, J.M. Design considerations for mesoporous silica nanoparticulate systems in facilitating biomedical applications. Open Mater. Sci. 2014, 1, 16–43. [Google Scholar] [CrossRef] [Green Version]
- Prabhakar, N.; Zhang, J.; Desai, D.; Casals, E.; Gulin-Sarfraz, T.; Näreoja, T.; Westermarck, J.; Rosenholm, J.M. Stimuli-responsive hybrid nanocarriers developed by controllable integration of hyperbranched PEI with mesoporous silica nanoparticles for sustained intracellular siRNA delivery. Int. J. Nanomed. 2016, 11, 6591–6608. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Chen, H.; Wang, L.; Zhao, L.; Cheng, Y.; Wang, A.; Wang, F.; Zhang, X. A Dual Receptor Targeting- and BBB Penetrating- Peptide Functionalized Polyethyleneimine Nanocomplex for Secretory Endostatin Gene Delivery to Malignant Glioma. Int. J. Nanomed. 2020, 15, 8875–8892. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.V.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano–bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Boussif, O.; Lezoualc’H, F.; Zanta, M.A.; Mergny, M.D.; Scherman, D.; Demeneix, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar] [CrossRef] [Green Version]
- Godbey, W.T.; Barry, M.A.; Saggau, P.; Wu, K.K.; Mikos, A.G. Poly(ethylenimine)-mediated transfection: A new paradigm for gene delivery. J. Biomed. Mater. Res. 2000, 51, 321–328. [Google Scholar] [CrossRef]
- Akinc, A.; Thomas, M.; Klibanov, A.M.; Langer, R. Exploring polyethylenimine-mediated DNA transfection and the proton sponge hypothesis. J. Gene Med. 2004, 7, 657–663. [Google Scholar] [CrossRef]
- Zhang, J.; Niemelä, M.; Westermarck, J.; Rosenholm, J. Mesoporous silica nanoparticles with redox-responsive surface linkers for charge-reversible loading and release of short oligonucleotides. Dalton Trans. 2014, 43, 4115–4126. [Google Scholar] [CrossRef]
- Le Joncour, V.; Filppu, P.; Hyvönen, M.; Holopainen, M.; Turunen, S.P.; Sihto, H.; Burghardt, I.; Joensuu, H.; Tynninen, O.; Jääskeläinen, J.; et al. Vulnerability of invasive glioblastoma cells to lysosomal membrane destabilization. EMBO Mol. Med. 2019, 11, e9034. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Guzmán, C.; Bagga, M.; Kaur, A.; Westermarck, J.; Abankwa, D. ColonyArea: An ImageJ Plugin to Automatically Quantify Colony Formation in Clonogenic Assays. PLoS ONE 2014, 9, e92444. [Google Scholar] [CrossRef]
- Prabhakar, N.; Khan, M.H.; Peurla, M.; Chang, H.-C.; Hänninen, P.E.; Rosenholm, J.M. Intracellular Trafficking of Fluorescent Nanodiamonds and Regulation of Their Cellular Toxicity. ACS Omega 2017, 2, 2689–2693. [Google Scholar] [CrossRef] [Green Version]
- Vancha, A.R.; Govindaraju, S.; Parsa, K.V.L.; Jasti, M.; González-García, M.; Ballestero, R.P. Use of polyethyleneimine polymer in cell culture as attachment factor and lipofection enhancer. BMC Biotechnol. 2004, 4, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, A.C. Molecular hurdles in polyfectin design and mechanistic background to polycation induced cytotoxicity. Adv. Drug Deliv. Rev. 2006, 58, 1523–1531. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Symonds, P.; Murray, J.C.; Hunter, A.C.; Debska, G.; Szewczyk, A. A two-stage poly(ethylenimine)-mediated cytotoxicity: Implications for gene transfer/therapy. Mol. Ther. 2005, 11, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Florea, B.I.; Meaney, C.; Junginger, H.E.; Borchard, G. Transfection efficiency and toxicity of polyethylenimine in differentiated Calu-3 and nondifferentiated COS-1 cell cultures. AAPS PharmSci 2002, 4, 1–11. [Google Scholar] [CrossRef]
- Kafil, V.; Omidi, Y. Cytotoxic Impacts of Linear and Branched Polyethylenimine Nanostructures in A431 Cells. BioImpacts 2011, 1, 23–30. [Google Scholar]
- Chaitanya, G.V.; Alexander, J.S.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.-W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of Poly(ADP-Ribose) Polymerase-1-Dependent Cell Death by Apoptosis-Inducing Factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Mahfouz, R.Z.; Sharma, R.K.; Poenicke, K.; Jha, R.; Paasch, U.; Grunewald, S.; Agarwal, A. Evaluation of poly(ADP-ribose) polymerase cleavage (cPARP) in ejaculated human sperm fractions after induction of apoptosis. Fertil. Steril. 2009, 91, 2210–2220. [Google Scholar] [CrossRef] [PubMed]
- Rusten, T.E.; Stenmark, H. p62, an autophagy hero or culprit? Nat. Cell Biol. 2010, 12, 207–209. [Google Scholar] [CrossRef]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Koukourakis, M.I.; Kalamida, D.; Giatromanolaki, A.; Zois, C.E.; Sivridis, E.; Pouliliou, S.; Mitrakas, A.; Gatter, K.C.; Harris, A.L. Autophagosome Proteins LC3A, LC3B and LC3C Have Distinct Subcellular Distribution Kinetics and Expression in Cancer Cell Lines. PLoS ONE 2015, 10, e0137675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. In Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2008; Volume 445, pp. 77–88. [Google Scholar]
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780–782. [Google Scholar] [CrossRef]
- Oh, N.; Park, J.-H. Endocytosis and exocytosis of nanoparticles in mammalian cells. Int. J. Nanomed. 2014, 9, 51–63. [Google Scholar]
- Zhang, S.; Gao, H.; Bao, G. Physical Principles of Nanoparticle Cellular Endocytosis. ACS Nano 2015, 9, 8655–8671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klumperman, J.; Raposo, G. The Complex Ultrastructure of the Endolysosomal System. Cold Spring Harb. Perspect. Biol. 2014, 6, a016857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonker, C.T.; De Heus, C.; Faber, L.; Brink, C.T.; Potze, L.; Fermie, J.; Liv, N.; Klumperman, J. An adapted protocol to overcome endosomal damage caused by polyethylenimine (PEI) mediated transfections. Matters 2017, 3, 201711000012. [Google Scholar] [CrossRef] [Green Version]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed]
- Bieber, T.; Meissner, W.; Kostin, S.; Niemann, A.; Elsasser, H.-P. Intracellular route and transcriptional competence of polyethylenimine–DNA complexes. J. Control. Release 2002, 82, 441–454. [Google Scholar] [CrossRef]
- Wang, F.; Salvati, A.; Boya, P. Lysosome-dependent cell death and deregulated autophagy induced by amine-modified polystyrene nanoparticles. Open Biol. 2018, 8, 170271. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Puebla, A.; Boya, P. Lysosomal membrane permeabilization in cell death: New evidence and implications for health and disease. Ann. N. Y. Acad. Sci. 2016, 1371, 30–44. [Google Scholar] [CrossRef]
- Wang, F.; Gómez-Sintes, R.; Boya, P. Lysosomal membrane permeabilization and cell death. Traffic 2018, 19, 918–931. [Google Scholar] [CrossRef]
- Le Joncour, V.; Karaman, S.; Laakkonen, P.M. Predicting In Vivo Payloads Delivery using a Blood-brain Tumor-barrier in a Dish. J. Vis. Exp. 2019, e59384. [Google Scholar] [CrossRef] [Green Version]
- Bruinsmann, F.A.; Vaz, G.R.; Alves, A.D.C.S.; Aguirre, T.; Pohlmann, A.R.; Guterres, S.S.; Sonvico, F. Nasal Drug Delivery of Anticancer Drugs for the Treatment of Glioblastoma: Preclinical and Clinical Trials. Molecules 2019, 24, 4312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdő, F.; Bors, L.A.; Farkas, D.; Bajza, Á.; Gizurarson, S. Evaluation of intranasal delivery route of drug administration for brain targeting. Brain Res. Bull. 2018, 143, 155–170. [Google Scholar] [CrossRef]
- Mamaeva, V.; Rosenholm, J.M.; Bate-Eya, L.T.; Bergman, L.; Peuhu, E.; Duchanoy, A.; E Fortelius, L.; Landor, S.; Toivola, D.M.; Lindén, M.; et al. Mesoporous Silica Nanoparticles as Drug Delivery Systems for Targeted Inhibition of Notch Signaling in Cancer. Mol. Ther. 2011, 19, 1538–1546. [Google Scholar] [CrossRef] [Green Version]
- Vermeulen, L.M.; De Smedt, S.C.; Remaut, K.; Braeckmans, K. The proton sponge hypothesis: Fable or fact? Eur. J. Pharm. Biopharm. 2018, 129, 184–190. [Google Scholar] [CrossRef]
- Wojnilowicz, M.; Glab, A.; Bertucci, A.; Caruso, F.; Cavalieri, F. Super-resolution Imaging of Proton Sponge-Triggered Rupture of Endosomes and Cytosolic Release of Small Interfering RNA. ACS Nano 2019, 13, 187–202. [Google Scholar] [CrossRef]
- Jacobs, K.A.; André-Grégoire, G.; Maghe, C.; Thys, A.; Li, Y.; Harford-Wright, E.; Trillet, K.; Douanne, T.; Nicolau, C.; Frénel, J.; et al. Paracaspase MALT1 regulates glioma cell survival by controlling endo-lysosome homeostasis. EMBO J. 2020, 39, e102030. [Google Scholar] [CrossRef] [PubMed]
- Pellosi, D.S.; De Paula, L.B.; De Melo, M.T.; Tedesco, A.C. Targeted and Synergic Glioblastoma Treatment: Multifunctional Nanoparticles Delivering Verteporfin as Adjuvant Therapy for Temozolomide Chemotherapy. Mol. Pharm. 2019, 16, 1009–1024. [Google Scholar] [CrossRef] [PubMed]
- Pucci, C.; De Pasquale, D.; Marino, A.; Martinelli, C.; Lauciello, S.; Ciofani, G. Hybrid Magnetic Nanovectors Promote Selective Glioblastoma Cell Death through a Combined Effect of Lysosomal Membrane Permeabilization and Chemotherapy. ACS Appl. Mater. Interfaces 2020, 12, 29037–29055. [Google Scholar] [CrossRef]
- De Pasquale, D.; Marino, A.; Tapeinos, C.; Pucci, C.; Rocchiccioli, S.; Michelucci, E.; Finamore, F.; McDonnell, L.; Scarpellini, A.; Lauciello, S.; et al. Homotypic targeting and drug delivery in glioblastoma cells through cell membrane-coated boron nitride nanotubes. Mater. Des. 2020, 192, 108742. [Google Scholar] [CrossRef] [PubMed]
- Norouzi, M.; Yathindranath, V.; Thliveris, J.A.; Kopec, B.M.; Siahaan, T.J.; Miller, D.W. Doxorubicin-loaded iron oxide nanoparticles for glioblastoma therapy: A combinational approach for enhanced delivery of nanoparticles. Sci. Rep. 2020, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prabhakar, N.; Merisaari, J.; Le Joncour, V.; Peurla, M.; Karaman, D.Ş.; Casals, E.; Laakkonen, P.; Westermarck, J.; Rosenholm, J.M. Circumventing Drug Treatment? Intrinsic Lethal Effects of Polyethyleneimine (PEI)-Functionalized Nanoparticles on Glioblastoma Cells Cultured in Stem Cell Conditions. Cancers 2021, 13, 2631. https://doi.org/10.3390/cancers13112631
Prabhakar N, Merisaari J, Le Joncour V, Peurla M, Karaman DŞ, Casals E, Laakkonen P, Westermarck J, Rosenholm JM. Circumventing Drug Treatment? Intrinsic Lethal Effects of Polyethyleneimine (PEI)-Functionalized Nanoparticles on Glioblastoma Cells Cultured in Stem Cell Conditions. Cancers. 2021; 13(11):2631. https://doi.org/10.3390/cancers13112631
Chicago/Turabian StylePrabhakar, Neeraj, Joni Merisaari, Vadim Le Joncour, Markus Peurla, Didem Şen Karaman, Eudald Casals, Pirjo Laakkonen, Jukka Westermarck, and Jessica M. Rosenholm. 2021. "Circumventing Drug Treatment? Intrinsic Lethal Effects of Polyethyleneimine (PEI)-Functionalized Nanoparticles on Glioblastoma Cells Cultured in Stem Cell Conditions" Cancers 13, no. 11: 2631. https://doi.org/10.3390/cancers13112631
APA StylePrabhakar, N., Merisaari, J., Le Joncour, V., Peurla, M., Karaman, D. Ş., Casals, E., Laakkonen, P., Westermarck, J., & Rosenholm, J. M. (2021). Circumventing Drug Treatment? Intrinsic Lethal Effects of Polyethyleneimine (PEI)-Functionalized Nanoparticles on Glioblastoma Cells Cultured in Stem Cell Conditions. Cancers, 13(11), 2631. https://doi.org/10.3390/cancers13112631