
IR-Surviving NSCLC Cells Exhibit Different Patterns of Molecular and Cellular Reactions Relating to the Multifraction Irradiation Regimen and p53-Family Proteins Expression

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Irradiation
2.3. Clonogenic Assay and Soft Agar Assay for Anchorage-Independent Growth
2.4. Cell Proliferation Assay
2.5. MTT Test
2.6. Migration Assay (Scratch Wound Test)
2.7. Flow Cytometry Analysis of FRA1
2.8. Western Blotting
2.9. Immunofluorescence Analysis of N-Cadherin
2.10. Flow Cytometry Analysis of Cellular Polyploidy
2.11. Statistical Analysis
3. Results
3.1. MFR-Surviving Cell Phenotype and Radiosensitivity after Treatment by Two Different Irradiation Regimens
3.2. Effect of MFR Regimen on Anchorage-Independent Transfomation Efficiency and Radiosensitivity of NSCLC Sublines
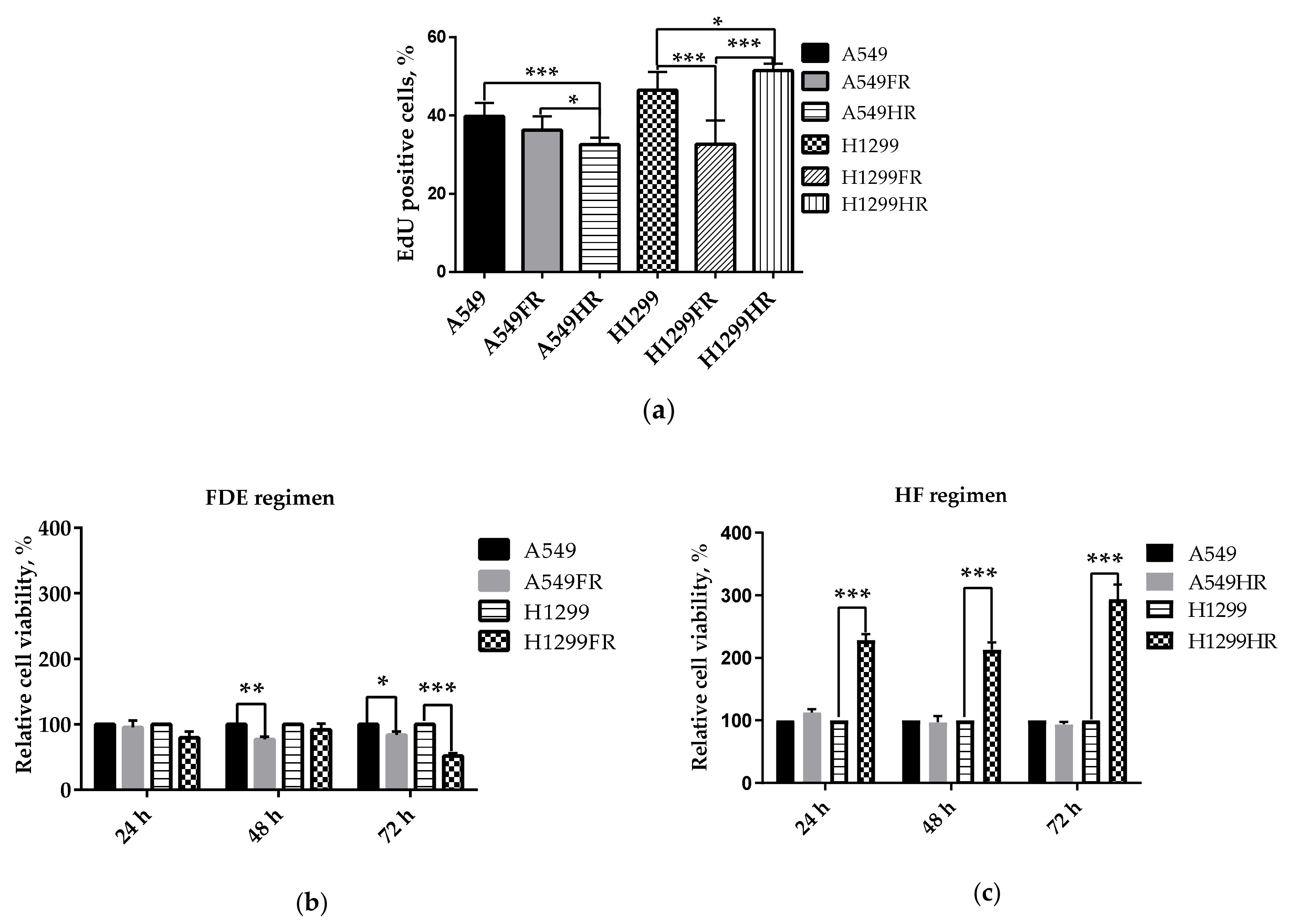
3.3. The Impact of Radiation Exposure Regimens on Proliferative and Metabolical Activity of MFR-Surivived NSCLC Cells
3.4. MFR Exposure Caused Partial EMT-Program Activation and 1D Migration of Surviving Sublines
3.5. FRA1 Expressed in NSCLC Cells in p53 Status-Related Manner
3.6. p63 and p73 Expression Changed Subsequent to MFR
3.7. Polyploidy May Be Involved in Decreased Radiosensitivity of IR-Surviving H1299 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Perez, C.A.; Pajak, T.F.; Rubin, P.; Simpson, J.R.; Mohiuddin, M.; Brady, L.W.; Perez-Tamayo, R.; Rotman, M. Long-term observations of the patterns of failure in patients with unresectable non-oat cell carcinoma of the lung treated with definitive radiotherapy. Report by the Radiation Therapy Oncology Group. Cancer 1987, 59, 1874–1881. [Google Scholar] [CrossRef]
- Brianzoni, E.; Rossi, G.; Ancidei, S.; Berbellini, A.; Capoccetti, F.; Cidda, C.; D’Avenia, P.; Fattori, S.; Montini, G.C.; Valentini, G.; et al. Radiotherapy planning: PET/CT scanner performances in the definition of gross tumour volume and clinical target volume. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.; Lang, S.; Rowbottom, C.; Guckenberger, M.; Faivre-Finn, C.; Committee, I.A.R.T. Intensity-modulated radiotherapy for lung cancer: Current status and future developments. J. Thorac. Oncol. 2014, 9, 1598–1608. [Google Scholar] [CrossRef] [Green Version]
- Bradley, J.D.; Naqa, I.E.; Drzymala, R.E.; Trovo, M.; Jones, G.; Denning, M.D. Stereotactic body radiation therapy for early-stage non-small-cell lung cancer: The pattern of failure is distant. Int. J. Radiat. Oncol. Biol. Phys. 2010, 77, 1146–1150. [Google Scholar] [CrossRef]
- Stephans, K.L.; Woody, N.M.; Reddy, C.A.; Varley, M.; Magnelli, A.; Zhuang, T.; Qi, P.; Videtic, G.M.M. Tumor control and toxicity for common stereotactic body radiation therapy dose-fractionation regimens in stage I non-small cell lung cancer. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Shibamoto, Y.; Hashizume, C.; Kondo, T.; Iwata, H.; Tomita, N.; Ogino, H. Repeat stereotactic body radiotherapy (SBRT) for local recurrence of non-small cell lung cancer and lung metastasis after first SBRT. Radiat. Oncol. 2018, 13, 136. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, W.R.; Gabani, P.; Nikitas, J.; Robinson, C.G.; Bradley, J.D.; Roach, M.C. Repeat stereotactic body radiation therapy (SBRT) for salvage of isolated local recurrence after definitive lung SBRT. Radiother. Oncol. 2020, 142, 230–235. [Google Scholar] [CrossRef]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047. [Google Scholar] [CrossRef]
- Park, B.J.; Lee, S.J.; Kim, J.I.; Lee, S.J.; Lee, C.H.; Chang, S.G.; Park, J.H.; Chi, S.G. Frequent alteration of p63 expression in human primary bladder carcinomas. Cancer Res. 2000, 60, 3370–3374. [Google Scholar]
- Koga, F.; Kawakami, S.; Fujii, Y.; Saito, K.; Ohtsuka, Y.; Iwai, A.; Ando, N.; Takizawa, T.; Kageyama, Y.; Kihara, K. Impaired p63 expression associates with poor prognosis and uroplakin III expression in invasive urothelial carcinoma of the bladder. Clin. Cancer Res. 2003, 9, 5501–5507. [Google Scholar]
- Tucci, P.; Agostini, M.; Grespi, F.; Markert, E.K.; Terrinoni, A.; Vousden, K.H.; Muller, P.A.; Dotsch, V.; Kehrloesser, S.; Sayan, B.S.; et al. Loss of p63 and its microRNA-205 target results in enhanced cell migration and metastasis in prostate cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 15312–15317. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Mercado-Uribe, I.; Hanash, S.; Liu, J. iTRAQ-based proteomic analysis of polyploid giant cancer cells and budding progeny cells reveals several distinct pathways for ovarian cancer development. PLoS ONE 2013, 8, e80120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, J.; Luo, S.; Lechler, T.; Zhang, J.Y. FRA1 promotes squamous cell carcinoma growth and metastasis through distinct AKT and c-Jun dependent mechanisms. Oncotarget 2016, 7, 34371–34383. [Google Scholar] [CrossRef] [PubMed]
- Diesch, J.; Sanij, E.; Gilan, O.; Love, C.; Tran, H.; Fleming, N.I.; Ellul, J.; Amalia, M.; Haviv, I.; Pearson, R.B.; et al. Widespread FRA1-dependent control of mesenchymal transdifferentiation programs in colorectal cancer cells. PLoS ONE 2014, 9, e88950. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Liu, H.; Mu, X.; Cui, J.; Peng, Z. Dysregulation of Fra1 expression by Wnt/beta-catenin signalling promotes glioma aggressiveness through epithelial-mesenchymal transition. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- Risolino, M.; Mandia, N.; Iavarone, F.; Dardaei, L.; Longobardi, E.; Fernandez, S.; Talotta, F.; Bianchi, F.; Pisati, F.; Spaggiari, L.; et al. Transcription factor PREP1 induces EMT and metastasis by controlling the TGF-beta-SMAD3 pathway in non-small cell lung adenocarcinoma. Proc. Natl. Acad. Sci. USA 2014, 111, E3775–E3784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usui, A.; Hoshino, I.; Akutsu, Y.; Sakata, H.; Nishimori, T.; Murakami, K.; Kano, M.; Shuto, K.; Matsubara, H. The molecular role of Fra-1 and its prognostic significance in human esophageal squamous cell carcinoma. Cancer 2012, 118, 3387–3396. [Google Scholar] [CrossRef] [PubMed]
- Sayan, A.E.; Stanford, R.; Vickery, R.; Grigorenko, E.; Diesch, J.; Kulbicki, K.; Edwards, R.; Pal, R.; Greaves, P.; Jariel-Encontre, I.; et al. Fra-1 controls motility of bladder cancer cells via transcriptional upregulation of the receptor tyrosine kinase AXL. Oncogene 2012, 31, 1493–1503. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Chen, S.; Tan, X.; Li, N.; Liu, C.; Li, Z.; Liu, Z.; Stupack, D.G.; Reisfeld, R.A.; Xiang, R. Fra-1 promotes breast cancer chemosensitivity by driving cancer stem cells from dormancy. Cancer Res. 2012, 72, 3451–3456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, G.; Chen, X.; Fang, X.; Wang, D.; Xie, M.; Chen, Q. Fra-1 is upregulated in lung cancer tissues and inhibits the apoptosis of lung cancer cells by the P53 signaling pathway. Oncology Rep. 2016, 35, 447–453. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, G.; Choy, H.; Bradley, J.; Rosenzweig, K.E.; Bogart, J.; Curran, W.J., Jr.; Gore, E.; Langer, C.; Louie, A.V.; Lutz, S.; et al. Definitive radiation therapy in locally advanced non-small cell lung cancer: Executive summary of an American Society for Radiation Oncology (ASTRO) evidence-based clinical practice guideline. Pract. Radiat. Oncol. 2015, 5, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Guadamillas, M.C.; Cerezo, A.; Pozo, M.A.D. Overcoming anoikis-pathways to anchorage-independent growth in cancer. J. Cell Sci. 2011, 124, 3189–3197. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.I.; Freedman, V.H.; Risser, R.; Pollack, R. Tumorigenicity of virus-transformed cells in nude mice is correlated specifically with anchorage independent growth in vitro. Proc. Natl. Acad. Sci. USA 1975, 72, 4435–4439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Pan, D.; Du, Y.; Jia, R.; Li, X.; Hu, B. Induction of cancer stem-like cells in A549 cells after exposure to carbon ions and X-rays. J. Int. J. Radiat. Res. 2019, 17, 25–35. [Google Scholar]
- Morata-Tarifa, C.; Jimenez, G.; Garcia, M.A.; Entrena, J.M.; Grinan-Lison, C.; Aguilera, M.; Picon-Ruiz, M.; Marchal, J.A. Low adherent cancer cell subpopulations are enriched in tumorigenic and metastatic epithelial-to-mesenchymal transition-induced cancer stem-like cells. Sci. Rep. 2016, 6, 18772. [Google Scholar] [CrossRef] [Green Version]
- Castillo, V.; Valenzuela, R.; Huidobro, C.; Contreras, H.R.; Castellon, E.A. Functional characteristics of cancer stem cells and their role in drug resistance of prostate cancer. Int. J. Oncol. 2014, 45, 985–994. [Google Scholar] [CrossRef]
- Puzio-Kuter, A.M. The role of p53 in metabolic regulation. Genes Cancer 2011, 2, 385–391. [Google Scholar] [CrossRef]
- Buch, K.; Peters, T.; Nawroth, T.; Sanger, M.; Schmidberger, H.; Langguth, P. Determination of cell survival after irradiation via clonogenic assay versus multiple MTT Assay—A comparative study. Radiat. Oncol. 2012, 7, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, M.V.; Herst, P.M.; Tan, A.S. Tetrazolium dyes as tools in cell biology: New insights into their cellular reduction. Biotechnol. Annu. Rev. 2005, 11, 127–152. [Google Scholar] [CrossRef]
- Adiseshaiah, P.; Vaz, M.; Machireddy, N.; Kalvakolanu, D.V.; Reddy, S.P. A Fra-1-dependent, matrix metalloproteinase driven EGFR activation promotes human lung epithelial cell motility and invasion. J. Cell Physiol. 2008, 216, 405–412. [Google Scholar] [CrossRef] [Green Version]
- Endo, S.; Fujita, M.; Yamada, S.; Imadome, K.; Nakayama, F.; Isozaki, T.; Yasuda, T.; Imai, T.; Matsubara, H. Fra1 enhances the radioresistance of colon cancer cells to X-ray or C-ion radiation. Oncol. Rep. 2018, 39, 1112–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajanne, R.; Miettinen, P.; Tenhunen, M.; Leppa, S. Transcription factor AP-1 promotes growth and radioresistance in prostate cancer cells. Int. J. Oncol. 2009, 35, 1175–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiewe, T.; Putzer, B.M. Role of p73 in malignancy: Tumor suppressor or oncogene? Cell Death Differ. 2002, 9, 237–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuddihy, A.R.; Bristow, R.G. The p53 protein family and radiation sensitivity: Yes or no? Cancer Metastasis Rev. 2004, 23, 237–257. [Google Scholar] [CrossRef]
- Mirzayans, R.; Andrais, B.; Murray, D. Roles of polyploid/multinucleated giant cancer cells in metastasis and disease relapse following anticancer treatment. Cancers 2018, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Herbein, G.; Nehme, Z. Polyploid giant cancer cells, a hallmark of oncoviruses and a new therapeutic challenge. Front. Oncol. 2020, 10, 567116. [Google Scholar] [CrossRef]
- Boudreau, H.E.; Casterline, B.W.; Burke, D.J.; Leto, T.L. Wild-type and mutant p53 differentially regulate NADPH oxidase 4 in TGF-beta-mediated migration of human lung and breast epithelial cells. Br. J. Cancer 2014, 110, 2569–2582. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Vousden, K.H. p53: New roles in metabolism. Trends Cell Biol. 2007, 17, 286–291. [Google Scholar] [CrossRef]
- Kondoh, H.; Lleonart, M.E.; Gil, J.; Wang, J.; Degan, P.; Peters, G.; Martinez, D.; Carnero, A.; Beach, D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005, 65, 177–185. [Google Scholar]
- Hardie, D.G. The AMP-activated protein kinase pathway—New players upstream and downstream. J. Cell Sci. 2004, 117, 5479–5487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [Green Version]
- Ellisen, L.W.; Ramsayer, K.D.; Johannessen, C.M.; Yang, A.; Beppu, H.; Minda, K.; Oliner, J.D.; McKeon, F.; Haber, D.A. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol. Cell 2002, 10, 995–1005. [Google Scholar] [CrossRef]
- Dewhurst, S.M.; McGranahan, N.; Burrell, R.A.; Rowan, A.J.; Gronroos, E.; Endesfelder, D.; Joshi, T.; Mouradov, D.; Gibbs, P.; Ward, R.L.; et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 2014, 4, 175–185. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.; Ivanov, A.; Wheatley, S.P.; Kosmacek, E.A.; Ianzini, F.; Anisimov, A.P.; Mackey, M.; Davis, P.J.; Plakhins, G.; Illidge, T.M. Endopolyploidy in irradiated p53-deficient tumour cell lines: Persistence of cell division activity in giant cells expressing Aurora-B kinase. Cell Biol. Int. 2008, 32, 1044–1056. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, Y.; Igarashi, A.; Kikuchi, I.; Obata, Y.; Fukumoto, Y.; Yamaguchi, N. Bleomycin-induced over-replication involves sustained inhibition of mitotic entry through the ATM/ATR pathway. Exp. Cell Res. 2009, 315, 2515–2528. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; Denchi, E.L.; Lange, T.D. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell 2010, 141, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.L.; Sengupta, S.; Gurdziel, K.; Bell, G.W.; Jacks, T.; Flores, E.R. p63 and p73 transcriptionally regulate genes involved in DNA repair. PLoS Genet. 2009, 5, e1000680. [Google Scholar] [CrossRef]
- Crum, C.P.; McKeon, F.D. p63 in epithelial survival, germ cell surveillance, and neoplasia. Annu. Rev. Pathol. 2010, 5, 349–371. [Google Scholar] [CrossRef] [PubMed]
- Su, P.; Yuan, Y.; Huang, Y.; Wang, W.; Zhang, Z. Anorectal malformation associated with a mutation in the P63 gene in a family with split hand-foot malformation. Int. J. Colorectal. Dis. 2013, 28, 1621–1627. [Google Scholar] [CrossRef] [Green Version]
- Talos, F.; Nemajerova, A.; Flores, E.R.; Petrenko, O.; Moll, U.M. p73 suppresses polyploidy and aneuploidy in the absence of functional p53. Mol. Cell 2007, 27, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Oren, M. p53: Guardian of ploidy. Mol. Oncol. 2011, 5, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; Lange, T.D. Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer Cell 2012, 21, 765–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erenpreisa, J.A.; Cragg, M.S.; Fringes, B.; Sharakhov, I.; Illidge, T.M. Release of mitotic descendants by giant cells from irradiated Burkitt’s lymphoma cell line. Cell Biol. Int. 2000, 24, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, M.; Guernsey, D.L.; Rajaraman, M.M.; Rajaraman, R. Neosis: A novel type of cell division in cancer. Cancer Biol. Ther. 2004, 3, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Marrazzo, E.; Marchini, S.; Tavecchio, M.; Alberio, T.; Previdi, S.; Erba, E.; Rotter, V.; Broggini, M. The expression of the ΔNp73β isoform of p73 leads to tetraploidy. Eur. J. Cancer 2009, 45, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Yeung, K.T.; Yang, J. Epithelial-mesenchymal transition in tumor metastasis. Mol. Oncol. 2017, 11, 28–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaeffer, D.; Somarelli, J.A.; Hanna, G.; Palmer, G.M.; Garcia-Blanco, M.A. Cellular migration and invasion uncoupled: Increased migration is not an inexorable consequence of epithelial-to-mesenchymal transition. Mol. Cell Biol. 2014, 34, 3486–3499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollestelle, A.; Peeters, J.K.; Smid, M.; Timmermans, M.; Verhoog, L.C.; Westenend, P.J.; Heine, A.A.; Chan, A.; Sieuwerts, A.M.; Wiemer, E.A.; et al. Loss of E-cadherin is not a necessity for epithelial to mesenchymal transition in human breast cancer. Breast Cancer Res. Treat. 2013, 138, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pustovalova, M.; Alhaddad, L.; Smetanina, N.; Chigasova, A.; Blokhina, T.; Chuprov-Netochin, R.; Osipov, A.N.; Leonov, S. The p53-53BP1-Related Survival of A549 and H1299 human lung cancer cells after multifractionated radiotherapy demonstrated different response to additional acute X-ray exposure. Int. J. Mol. Sci. 2020, 21, 3342. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, D.; Bunjobpol, W.; Sabapathy, K. Interplay between TAp73 protein and selected activator protein-1 (AP-1) family members promotes AP-1 target gene activation and cellular growth. J. Biol. Chem. 2015, 290, 18636–18649. [Google Scholar] [CrossRef] [Green Version]
- Newbold, K.; Partridge, M.; Cook, G.; Sohaib, S.A.; Charles-Edwards, E.; Rhys-Evans, P.; Harrington, K.; Nutting, C. Advanced imaging applied to radiotherapy planning in head and neck cancer: A clinical review. Br. J. Radiol. 2006, 79, 554–561. [Google Scholar] [CrossRef]
- Thwaites, D.I.; Tuohy, J.B. Back to the future: The history and development of the clinical linear accelerator. Phys. Med. Biol. 2006, 51, R343–R362. [Google Scholar] [CrossRef] [PubMed]
- Mohan, R.; Wu, Q.; Manning, M.; Schmidt-Ullrich, R. Radiobiological considerations in the design of fractionation strategies for intensity-modulated radiation therapy of head and neck cancers. Int. J. Radiat. Oncol. Biol. Phys. 2000, 46, 619–630. [Google Scholar] [CrossRef]
- Prasanna, A.; Ahmed, M.M.; Mohiuddin, M.; Coleman, C.N. Exploiting sensitization windows of opportunity in hyper and hypo-fractionated radiation therapy. J. Thorac. Dis. 2014, 6, 287–302. [Google Scholar] [CrossRef]
- Baumann, P.; Nyman, J.; Hoyer, M.; Wennberg, B.; Gagliardi, G.; Lax, I.; Drugge, N.; Ekberg, L.; Friesland, S.; Johansson, K.A.; et al. Outcome in a prospective phase II trial of medically inoperable stage I non-small-cell lung cancer patients treated with stereotactic body radiotherapy. J. Clin. Oncol. 2009, 27, 3290–3296. [Google Scholar] [CrossRef]
- Fakiris, A.J.; McGarry, R.C.; Yiannoutsos, C.T.; Papiez, L.; Williams, M.; Henderson, M.A.; Timmerman, R. Stereotactic body radiation therapy for early-stage non-small-cell lung carcinoma: Four-year results of a prospective phase II study. Int. J. Radiat. Oncol. Biol. Phys. 2009, 75, 677–682. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhaddad, L.; Pustovalova, M.; Blokhina, T.; Chuprov-Netochin, R.; Osipov, A.N.; Leonov, S. IR-Surviving NSCLC Cells Exhibit Different Patterns of Molecular and Cellular Reactions Relating to the Multifraction Irradiation Regimen and p53-Family Proteins Expression. Cancers 2021, 13, 2669. https://doi.org/10.3390/cancers13112669
Alhaddad L, Pustovalova M, Blokhina T, Chuprov-Netochin R, Osipov AN, Leonov S. IR-Surviving NSCLC Cells Exhibit Different Patterns of Molecular and Cellular Reactions Relating to the Multifraction Irradiation Regimen and p53-Family Proteins Expression. Cancers. 2021; 13(11):2669. https://doi.org/10.3390/cancers13112669
Chicago/Turabian StyleAlhaddad, Lina, Margarita Pustovalova, Taisia Blokhina, Roman Chuprov-Netochin, Andreyan N. Osipov, and Sergey Leonov. 2021. "IR-Surviving NSCLC Cells Exhibit Different Patterns of Molecular and Cellular Reactions Relating to the Multifraction Irradiation Regimen and p53-Family Proteins Expression" Cancers 13, no. 11: 2669. https://doi.org/10.3390/cancers13112669
APA StyleAlhaddad, L., Pustovalova, M., Blokhina, T., Chuprov-Netochin, R., Osipov, A. N., & Leonov, S. (2021). IR-Surviving NSCLC Cells Exhibit Different Patterns of Molecular and Cellular Reactions Relating to the Multifraction Irradiation Regimen and p53-Family Proteins Expression. Cancers, 13(11), 2669. https://doi.org/10.3390/cancers13112669