
An Overview of the Recent Development of Anticancer Agents Targeting the HIF-1 Transcription Factor

 , , , , , , and
, , , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
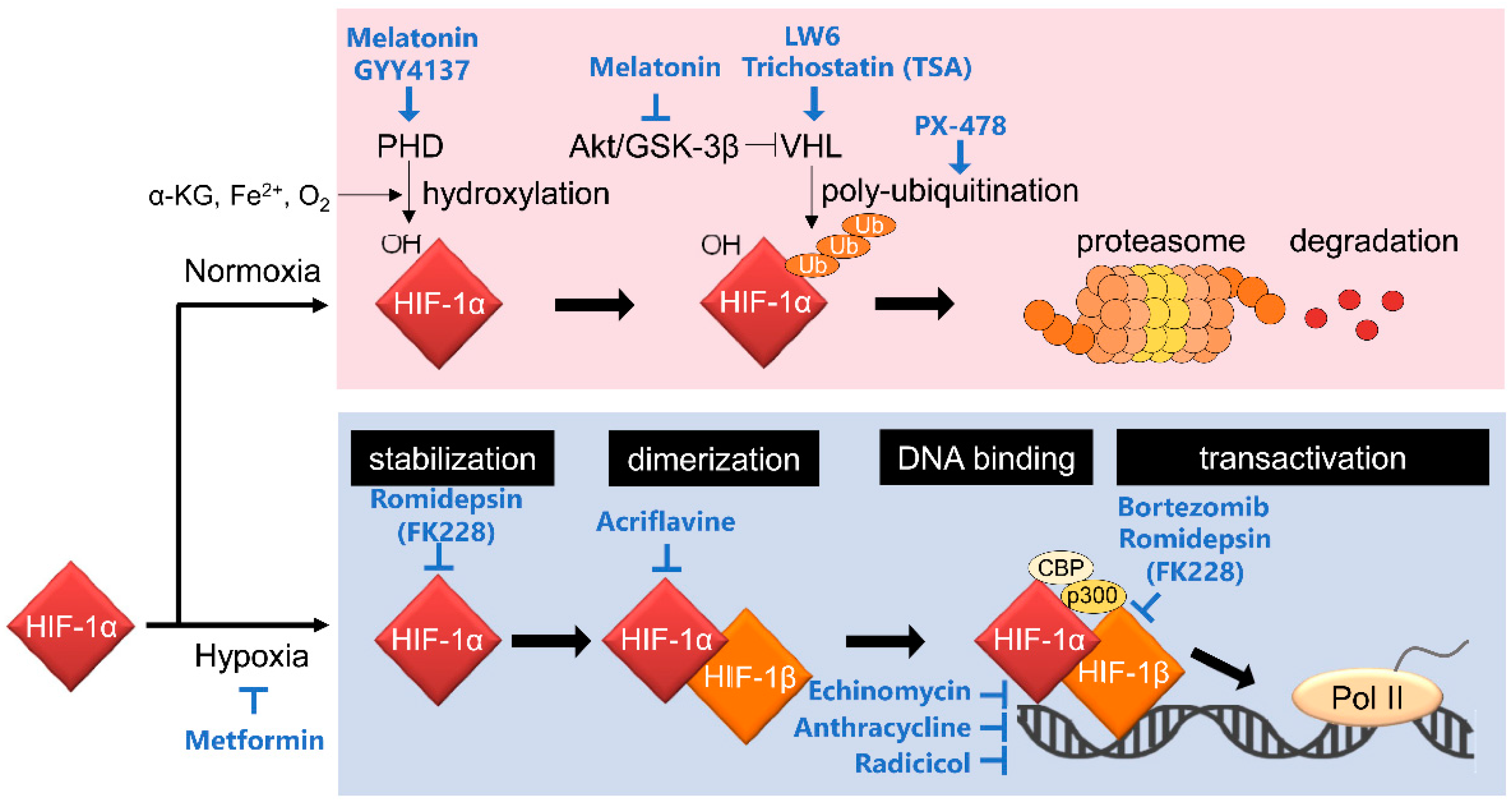
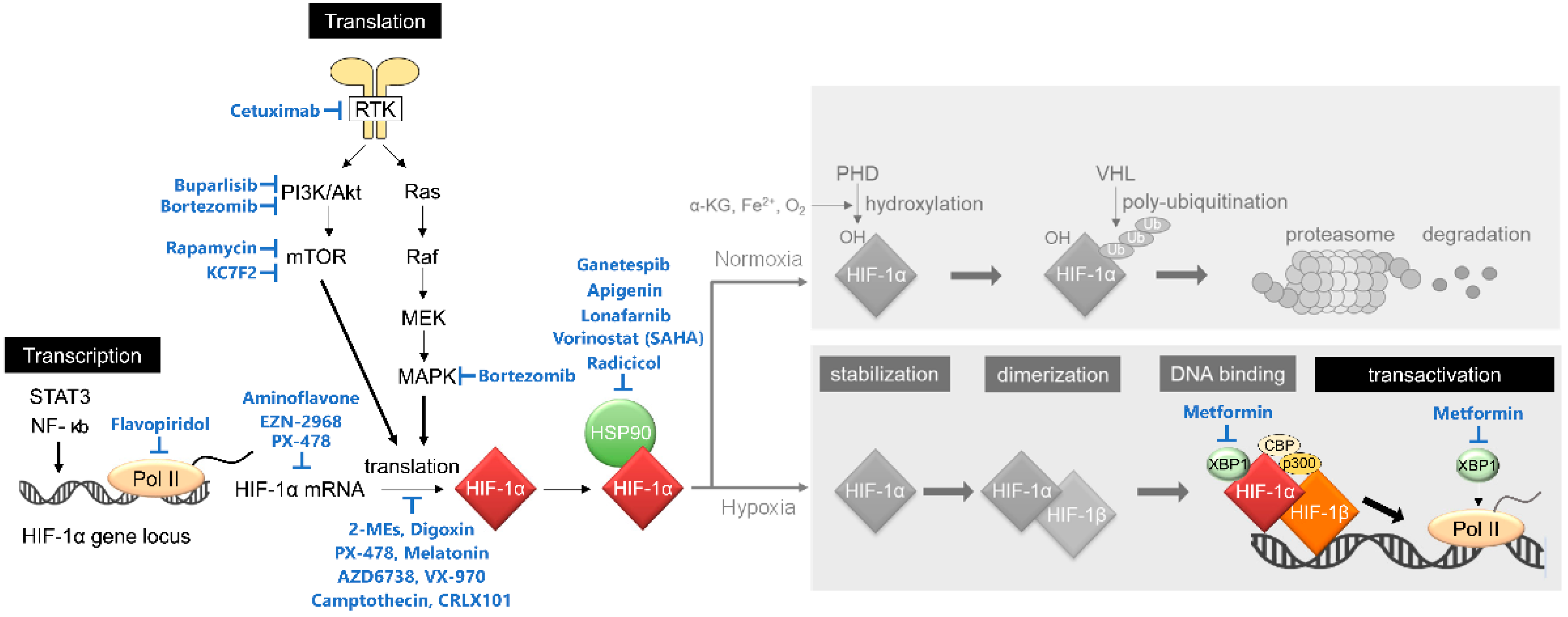
2. Mechanisms of Action of HIF-1 Inhibitors
2.1. Inhibitors of Transcriptional Initiation of HIF-1α
- Flavopiridol
- Aminoflavone (AF)
2.2. Inhibitor of HIF-1α mRNA Stabilization
- EZN-2968
2.3. Inhibitors of Translational Initiation of HIF-1α
2.3.1. Targeting the RTK/PI3K/Akt/mTOR Pathway
- Rapamycin
- Cetuximab
- Buparlisib
- KC7F2
2.3.2. Inhibitors of Microtubule Dynamics and Na+/K+ ATPase
- 2-MEs
- Digoxin
2.3.3. Translational Regulation of HIF-1α by ATR
- AZD6738 and VX-970
2.3.4. Antitumor Effect of This Class of Drugs Inhibiting Translational Initiation of HIF-1α
2.4. Inhibitors of Stabilization of the HIF-1α Protein
2.4.1. Drugs Increasing PHD2/VHL Activity
- Melatonin and its derivative, NB-5-MT
- LW6
- GYY4137
2.4.2. Inhibitors Mediating HIF-1α Stability through HSP90
- Ganetespib
- Apigenin
- Lonafarnib
2.4.3. Inhibitors Mediating HIF-1α Stability through Histone Deacetylases (HDACs)
- Vorinostat (SAHA)
- Romidepsin (FK228)
- Trichostatin (TSA)
2.4.4. Antitumor Effect of This Class of Drugs Inhibiting Stabilization of the HIF-1α Protein
2.5. Inhibitor of HIF-1 Dimerization
- Acriflavine
2.6. Inhibitors of HIF-1 DNA Binding
- Echinomycin
- Anthracycline
- Radicicol
2.7. Inhibitors of HIF-1α Transactivation Activity
- Bortezomib
2.8. Inhibitors of HIF-1α at Multiple Levels
- PX-478
- Camptothecin and its analogues
- CRLX101
- Metformin
3. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Kizaka-Kondoh, S.; Inoue, M.; Harada, H.; Hiraoka, M. Tumor hypoxia: A target for selective cancer therapy. Cancer Sci. 2003, 94, 1021–1028. [Google Scholar] [CrossRef]
- Vaupel, P.; Mayer, A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Yeom, C.J.; Goto, Y.; Zhu, Y.; Hiraoka, M.; Harada, H. Microenvironments and cellular characteristics in the micro tumor cords of malignant solid tumors. Int. J. Mol. Sci. 2012, 13, 13949–13965. [Google Scholar] [CrossRef] [Green Version]
- Harada, H.; Kizaka-Kondoh, S.; Li, G.; Itasaka, S.; Shibuya, K.; Inoue, M.; Hiraoka, M. Significance of HIF-1-active cells in angiogenesis and radioresistance. Oncogene 2007, 26, 7508–7516. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef] [Green Version]
- Berchner-Pfannschmidt, U.; Frede, S.; Wotzlaw, C.; Fandrey, J. Imaging of the hypoxia-inducible factor pathway: Insights into oxygen sensing. Eur. Respir. J. 2008, 32, 210–217. [Google Scholar] [CrossRef]
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Ohh, M.; Park, C.W.; Ivan, M.; Hoffman, M.A.; Kim, T.Y.; Huang, L.E.; Pavletich, N.; Chau, V.; Kaelin, W.G. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat. Cell Biol. 2000, 2, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, K.; Makino, Y.; Pereira, T.; Poellinger, L. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000, 19, 4298–4309. [Google Scholar] [CrossRef] [Green Version]
- Hewitson, K.S.; McNeill, L.A.; Riordan, M.V.; Tian, Y.M.; Bullock, A.N.; Welford, R.W.; Elkins, J.M.; Oldham, N.J.; Bhattacharya, S.; Gleadle, J.M.; et al. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J. Biol. Chem. 2002, 277, 26351–26355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 2002, 295, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef] [Green Version]
- Isaacs, J.S.; Jung, Y.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.L.; Merino, M.; Trepel, J.; Zbar, B.; Toro, J.; et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 2005, 8, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burr, S.P.; Costa, A.S.; Grice, G.L.; Timms, R.T.; Lobb, I.T.; Freisinger, P.; Dodd, R.B.; Dougan, G.; Lehner, P.J.; Frezza, C.; et al. Mitochondrial Protein Lipoylation and the 2-Oxoglutarate Dehydrogenase Complex Controls HIF1alpha Stability in Aerobic Conditions. Cell Metab 2016, 24, 740–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef]
- Koyasu, S.; Kobayashi, M.; Goto, Y.; Hiraoka, M.; Harada, H. Regulatory mechanisms of hypoxia-inducible factor 1 activity: Two decades of knowledge. Cancer Sci 2018, 109, 560–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- von Sonntag, C. Advanced oxidation processes: Mechanistic aspects. Water Sci. Technol. 2008, 58, 1015–1021. [Google Scholar] [CrossRef]
- Zepeda, A.B.; Pessoa, A., Jr.; Castillo, R.L.; Figueroa, C.A.; Pulgar, V.M.; Farias, J.G. Cellular and molecular mechanisms in the hypoxic tissue: Role of HIF-1 and ROS. Cell Biochem. Funct. 2013, 31, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Dang, E.V.; Barbi, J.; Yang, H.Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.R.; et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 2011, 146, 772–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, S.A.; Pober, J.S. IFN-alpha induces transcription of hypoxia-inducible factor-1alpha to inhibit proliferation of human endothelial cells. J. Immunol. 2008, 181, 1052–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [Green Version]
- Koshikawa, N.; Hayashi, J.; Nakagawara, A.; Takenaga, K. Reactive oxygen species-generating mitochondrial DNA mutation up-regulates hypoxia-inducible factor-1alpha gene transcription via phosphatidylinositol 3-kinase-Akt/protein kinase C/histone deacetylase pathway. J. Biol. Chem. 2009, 284, 33185–33194. [Google Scholar] [CrossRef] [Green Version]
- Yeom, C.J.; Zeng, L.; Goto, Y.; Morinibu, A.; Zhu, Y.; Shinomiya, K.; Kobayashi, M.; Itasaka, S.; Yoshimura, M.; Hur, C.G.; et al. LY6E: A conductor of malignant tumor growth through modulation of the PTEN/PI3K/Akt/HIF-1 axis. Oncotarget 2016, 7, 65837–65848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, H.; Itasaka, S.; Kizaka-Kondoh, S.; Shibuya, K.; Morinibu, A.; Shinomiya, K.; Hiraoka, M. The Akt/mTOR pathway assures the synthesis of HIF-1alpha protein in a glucose- and reoxygenation-dependent manner in irradiated tumors. J. Biol. Chem. 2009, 284, 5332–5342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laughner, E.; Taghavi, P.; Chiles, K.; Mahon, P.C.; Semenza, G.L. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: Novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol. Cell Biol. 2001, 21, 3995–4004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Callapina, M.; Goodall, G.J.; Brune, B. Functional integrity of nuclear factor kappaB, phosphatidylinositol 3’-kinase, and mitogen-activated protein kinase signaling allows tumor necrosis factor alpha-evoked Bcl-2 expression to provoke internal ribosome entry site-dependent translation of hypoxia-inducible factor 1alpha. Cancer Res. 2004, 64, 9041–9048. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.V.; Baek, J.H.; Zhang, H.; Diez, R.; Cole, R.N.; Semenza, G.L. RACK1 competes with HSP90 for binding to HIF-1alpha and is required for O(2)-independent and HSP90 inhibitor-induced degradation of HIF-1alpha. Mol. Cell 2007, 25, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Goto, Y.; Zeng, L.; Yeom, C.J.; Zhu, Y.; Morinibu, A.; Shinomiya, K.; Kobayashi, M.; Hirota, K.; Itasaka, S.; Yoshimura, M.; et al. UCHL1 provides diagnostic and antimetastatic strategies due to its deubiquitinating effect on HIF-1alpha. Nat. Commun. 2015, 6, 6153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gojo, I.; Zhang, B.; Fenton, R.G. The cyclin-dependent kinase inhibitor flavopiridol induces apoptosis in multiple myeloma cells through transcriptional repression and down-regulation of Mcl-1. Clin. Cancer Res. 2002, 8, 3527–3538. [Google Scholar]
- Lam, L.T.; Pickeral, O.K.; Peng, A.C.; Rosenwald, A.; Hurt, E.M.; Giltnane, J.M.; Averett, L.M.; Zhao, H.; Davis, R.E.; Sathyamoorthy, M.; et al. Genomic-scale measurement of mRNA turnover and the mechanisms of action of the anti-cancer drug flavopiridol. Genome Biol. 2001, 2, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newcomb, E.W.; Ali, M.A.; Schnee, T.; Lan, L.; Lukyanov, Y.; Fowkes, M.; Miller, D.C.; Zagzag, D. Flavopiridol downregulates hypoxia-mediated hypoxia-inducible factor-1alpha expression in human glioma cells by a proteasome-independent pathway: Implications for in vivo therapy. Neuro-Oncol. 2005, 7, 225–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, M.A.; Kortmansky, J.; Motwani, M.; Drobnjak, M.; Gonen, M.; Yi, S.; Weyerbacher, A.; Cordon-Cardo, C.; Lefkowitz, R.; Brenner, B.; et al. A phase I clinical trial of the sequential combination of irinotecan followed by flavopiridol. Clin. Cancer Res. 2005, 11, 3836–3845. [Google Scholar] [CrossRef] [Green Version]
- Van Veldhuizen, P.J.; Faulkner, J.R.; Lara, P.N., Jr.; Gumerlock, P.H.; Goodwin, J.W.; Dakhil, S.R.; Gross, H.M.; Flanigan, R.C.; Crawford, E.D. A phase II study of flavopiridol in patients with advanced renal cell carcinoma: Results of Southwest Oncology Group Trial 0109. Cancer Chemother. Pharmacol. 2005, 56, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Terzuoli, E.; Puppo, M.; Rapisarda, A.; Uranchimeg, B.; Cao, L.; Burger, A.M.; Ziche, M.; Melillo, G. Aminoflavone, a ligand of the aryl hydrocarbon receptor, inhibits HIF-1alpha expression in an AhR-independent fashion. Cancer Res. 2010, 70, 6837–6848. [Google Scholar] [CrossRef] [Green Version]
- Callero, M.A.; Suarez, G.V.; Luzzani, G.; Itkin, B.; Nguyen, B.; Loaiza-Perez, A.I. Aryl hydrocarbon receptor activation by aminoflavone: New molecular target for renal cancer treatment. Int. J. Oncol. 2012, 41, 125–134. [Google Scholar] [CrossRef]
- Kuffel, M.J.; Schroeder, J.C.; Pobst, L.J.; Naylor, S.; Reid, J.M.; Kaufmann, S.H.; Ames, M.M. Activation of the antitumor agent aminoflavone (NSC 686288) is mediated by induction of tumor cell cytochrome P450 1A1/1A2. Mol. Pharmacol. 2002, 62, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Loaiza-Perez, A.I.; Kenney, S.; Boswell, J.; Hollingshead, M.; Alley, M.C.; Hose, C.; Ciolino, H.P.; Yeh, G.C.; Trepel, J.B.; Vistica, D.T.; et al. Aryl hydrocarbon receptor activation of an antitumor aminoflavone: Basis of selective toxicity for MCF-7 breast tumor cells. Mol. Cancer Ther. 2004, 3, 715–725. [Google Scholar] [PubMed]
- Greenberger, L.M.; Horak, I.D.; Filpula, D.; Sapra, P.; Westergaard, M.; Frydenlund, H.F.; Albaek, C.; Schroder, H.; Orum, H. A RNA antagonist of hypoxia-inducible factor-1alpha, EZN-2968, inhibits tumor cell growth. Mol. Cancer Ther. 2008, 7, 3598–3608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, W.; Rapisarda, A.; Park, S.R.; Kinders, R.J.; Chen, A.; Melillo, G.; Turkbey, B.; Steinberg, S.M.; Choyke, P.; Doroshow, J.H.; et al. Pilot trial of EZN-2968, an antisense oligonucleotide inhibitor of hypoxia-inducible factor-1 alpha (HIF-1alpha), in patients with refractory solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 343–348. [Google Scholar] [CrossRef]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shackelford, D.B.; Vasquez, D.S.; Corbeil, J.; Wu, S.; Leblanc, M.; Wu, C.L.; Vera, D.R.; Shaw, R.J. mTOR and HIF-1alpha-mediated tumor metabolism in an LKB1 mouse model of Peutz-Jeghers syndrome. Proc. Natl. Acad. Sci. USA 2009, 106, 11137–11142. [Google Scholar] [CrossRef] [Green Version]
- Thomas, G.V.; Tran, C.; Mellinghoff, I.K.; Welsbie, D.S.; Chan, E.; Fueger, B.; Czernin, J.; Sawyers, C.L. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat. Med. 2006, 12, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Liang, K.; Lu, Y.; Fan, Z. The anti-EGFR antibody cetuximab sensitizes human head and neck squamous cell carcinoma cells to radiation in part through inhibiting radiation-induced upregulation of HIF-1alpha. Cancer Lett. 2012, 322, 78–85. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.M.; Zhao, Z.L.; Ma, S.R.; Yu, G.T.; Liu, B.; Zhang, L.; Zhang, W.F.; Kulkarni, A.B.; Sun, Z.J.; Zhao, Y.F. Epidermal growth factor receptor inhibition reduces angiogenesis via hypoxia-inducible factor-1alpha and Notch1 in head neck squamous cell carcinoma. PLoS ONE 2015, 10, e0119723. [Google Scholar] [CrossRef] [Green Version]
- Baselga, J.; Im, S.A.; Iwata, H.; Cortes, J.; De Laurentiis, M.; Jiang, Z.; Arteaga, C.L.; Jonat, W.; Clemons, M.; Ito, Y.; et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 904–916. [Google Scholar] [CrossRef]
- Narita, T.; Yin, S.; Gelin, C.F.; Moreno, C.S.; Yepes, M.; Nicolaou, K.C.; Van Meir, E.G. Identification of a novel small molecule HIF-1alpha translation inhibitor. Clin. Cancer Res. 2009, 15, 6128–6136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbonaro, M.; O’Brate, A.; Giannakakou, P. Microtubule disruption targets HIF-1alpha mRNA to cytoplasmic P-bodies for translational repression. J. Cell Biol. 2011, 192, 83–99. [Google Scholar] [CrossRef] [Green Version]
- LaVallee, T.M.; Burke, P.A.; Swartz, G.M.; Hamel, E.; Agoston, G.E.; Shah, J.; Suwandi, L.; Hanson, A.D.; Fogler, W.E.; Sidor, C.F.; et al. Significant antitumor activity in vivo following treatment with the microtubule agent ENMD-1198. Mol. Cancer Ther. 2008, 7, 1472–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabjeesh, N.J.; Escuin, D.; LaVallee, T.M.; Pribluda, V.S.; Swartz, G.M.; Johnson, M.S.; Willard, M.T.; Zhong, H.; Simons, J.W.; Giannakakou, P. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell 2003, 3, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Matei, D.; Schilder, J.; Sutton, G.; Perkins, S.; Breen, T.; Quon, C.; Sidor, C. Activity of 2 methoxyestradiol (Panzem NCD) in advanced, platinum-resistant ovarian cancer and primary peritoneal carcinomatosis: A Hoosier Oncology Group trial. Gynecol. Oncol. 2009, 115, 90–96. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Richardson, P.G.; Lacy, M.Q.; Dispenzieri, A.; Greipp, P.R.; Witzig, T.E.; Schlossman, R.; Sidor, C.F.; Anderson, K.C.; Gertz, M.A. Novel therapy with 2-methoxyestradiol for the treatment of relapsed and plateau phase multiple myeloma. Clin. Cancer Res. 2007, 13, 6162–6167. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Qian, D.Z.; Tan, Y.S.; Lee, K.; Gao, P.; Ren, Y.R.; Rey, S.; Hammers, H.; Chang, D.; Pili, R.; et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc. Natl. Acad. Sci. USA 2008, 105, 19579–19586. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Zhang, H.; Iwase, T.; Shen, J.; Semenza, G.L.; Campochiaro, P.A. Digoxin inhibits retinal ischemia-induced HIF-1alpha expression and ocular neovascularization. FASEB J. 2010, 24, 1759–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leszczynska, K.B.; Dobrynin, G.; Leslie, R.E.; Ient, J.; Boumelha, A.J.; Senra, J.M.; Hawkins, M.A.; Maughan, T.; Mukherjee, S.; Hammond, E.M. Preclinical testing of an Atr inhibitor demonstrates improved response to standard therapies for esophageal cancer. Radiother. Oncol. 2016, 121, 232–238. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.J.; Jung, Y.; Song, Y.S.; Park, S.; Park, Y.; Lee, H.J. Enhanced anti-angiogenic activity of novel melatonin-like agents. J. Pineal. Res. 2021, e12739. [Google Scholar] [CrossRef]
- Kim, K.J.; Choi, J.S.; Kang, I.; Kim, K.W.; Jeong, C.H.; Jeong, J.W. Melatonin suppresses tumor progression by reducing angiogenesis stimulated by HIF-1 in a mouse tumor model. J. Pineal Res. 2013, 54, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Lee, H.J.; Jeong, S.J.; Lee, H.J.; Kim, H.S.; Chen, C.Y.; Lee, E.O.; Kim, S.H. Sphingosine kinase 1 pathway is involved in melatonin-induced HIF-1alpha inactivation in hypoxic PC-3 prostate cancer cells. J. Pineal Res. 2011, 51, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Hwang, M.S.; Suh, S.I.; Baek, W.K. Melatonin down-regulates HIF-1 alpha expression through inhibition of protein translation in prostate cancer cells. J. Pineal Res. 2009, 46, 415–421. [Google Scholar] [CrossRef]
- Maleki Dana, P.; Reiter, R.J.; Hallajzadeh, J.; Asemi, Z.; Mansournia, M.A.; Yousefi, B. Melatonin as a potential inhibitor of kidney cancer: A survey of the molecular processes. IUBMB Life 2020, 72, 2355–2365. [Google Scholar] [CrossRef] [PubMed]
- Jardim-Perassi, B.V.; Lourenco, M.R.; Doho, G.M.; Grigolo, I.H.; Gelaleti, G.B.; Ferreira, L.C.; Borin, T.F.; Moschetta, M.G.; Pires de Campos Zuccari, D.A. Melatonin Regulates Angiogenic Factors under Hypoxia in Breast Cancer Cell Lines. Anticancer Agents Med. Chem. 2016, 16, 347–358. [Google Scholar] [CrossRef]
- Lee, K.; Kang, J.E.; Park, S.K.; Jin, Y.; Chung, K.S.; Kim, H.M.; Kang, M.R.; Lee, M.K.; Song, K.B.; Yang, E.G.; et al. LW6, a novel HIF-1 inhibitor, promotes proteasomal degradation of HIF-1alpha via upregulation of VHL in a colon cancer cell line. Biochem. Pharmacol. 2010, 80, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Prabhudesai, S.; Zhang, Y.; Rao, G.; Thirugnanam, K.; Hossen, M.N.; Dwivedi, S.K.D.; Ramchandran, R.; Mukherjee, P.; Bhattacharya, R. Cystathione beta-synthase regulates HIF-1alpha stability through persulfidation of PHD2. Sci. Adv. 2020, 6, eaaz8534. [Google Scholar] [CrossRef]
- Lee, Z.W.; Zhou, J.; Chen, C.S.; Zhao, Y.; Tan, C.H.; Li, L.; Moore, P.K.; Deng, L.W. The slow-releasing hydrogen sulfide donor, GYY4137, exhibits novel anti-cancer effects in vitro and in vivo. PLoS ONE 2011, 6, e21077. [Google Scholar] [CrossRef] [Green Version]
- Ying, W.; Du, Z.; Sun, L.; Foley, K.P.; Proia, D.A.; Blackman, R.K.; Zhou, D.; Inoue, T.; Tatsuta, N.; Sang, J.; et al. Ganetespib, a unique triazolone-containing Hsp90 inhibitor, exhibits potent antitumor activity and a superior safety profile for cancer therapy. Mol. Cancer Ther. 2012, 11, 475–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagaraju, G.P.; Park, W.; Wen, J.; Mahaseth, H.; Landry, J.; Farris, A.B.; Willingham, F.; Sullivan, P.S.; Proia, D.A.; El-Hariry, I.; et al. Antiangiogenic effects of ganetespib in colorectal cancer mediated through inhibition of HIF-1alpha and STAT-3. Angiogenesis 2013, 16, 903–917. [Google Scholar] [CrossRef]
- Xiang, L.; Gilkes, D.M.; Chaturvedi, P.; Luo, W.; Hu, H.; Takano, N.; Liang, H.; Semenza, G.L. Ganetespib blocks HIF-1 activity and inhibits tumor growth, vascularization, stem cell maintenance, invasion, and metastasis in orthotopic mouse models of triple-negative breast cancer. J. Mol. Med. 2014, 92, 151–164. [Google Scholar] [CrossRef] [Green Version]
- Socinski, M.A.; Goldman, J.; El-Hariry, I.; Koczywas, M.; Vukovic, V.; Horn, L.; Paschold, E.; Salgia, R.; West, H.; Sequist, L.V.; et al. A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clin. Cancer Res. 2013, 19, 3068–3077. [Google Scholar] [CrossRef] [Green Version]
- Madunic, J.; Madunic, I.V.; Gajski, G.; Popic, J.; Garaj-Vrhovac, V. Apigenin: A dietary flavonoid with diverse anticancer properties. Cancer Lett. 2018, 413, 11–22. [Google Scholar] [CrossRef]
- Zhao, M.; Ma, J.; Zhu, H.Y.; Zhang, X.H.; Du, Z.Y.; Xu, Y.J.; Yu, X.D. Apigenin inhibits proliferation and induces apoptosis in human multiple myeloma cells through targeting the trinity of CK2, Cdc37 and Hsp90. Mol. Cancer 2011, 10, 104. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Zhou, Q.; Liu, L.Z.; Xia, C.; Hu, X.; Shi, X.; Jiang, B.H. Apigenin inhibits tumor angiogenesis through decreasing HIF-1alpha and VEGF expression. Carcinogenesis 2007, 28, 858–864. [Google Scholar] [CrossRef] [Green Version]
- Melstrom, L.G.; Salabat, M.R.; Ding, X.Z.; Strouch, M.J.; Grippo, P.J.; Mirzoeva, S.; Pelling, J.C.; Bentrem, D.J. Apigenin down-regulates the hypoxia response genes: HIF-1alpha, GLUT-1, and VEGF in human pancreatic cancer cells. J. Surg. Res. 2011, 167, 173–181. [Google Scholar] [CrossRef]
- Osada, M.; Imaoka, S.; Funae, Y. Apigenin suppresses the expression of VEGF, an important factor for angiogenesis, in endothelial cells via degradation of HIF-1alpha protein. FEBS Lett. 2004, 575, 59–63. [Google Scholar] [CrossRef] [Green Version]
- Chaponis, D.; Barnes, J.W.; Dellagatta, J.L.; Kesari, S.; Fast, E.; Sauvageot, C.; Panagrahy, D.; Greene, E.R.; Ramakrishna, N.; Wen, P.Y.; et al. Lonafarnib (SCH66336) improves the activity of temozolomide and radiation for orthotopic malignant gliomas. J. Neurooncol. 2011, 104, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Khuri, F.R.; Glisson, B.S.; Kim, E.S.; Statkevich, P.; Thall, P.F.; Meyers, M.L.; Herbst, R.S.; Munden, R.F.; Tendler, C.; Zhu, Y.; et al. Phase I study of the farnesyltransferase inhibitor lonafarnib with paclitaxel in solid tumors. Clin. Cancer Res. 2004, 10, 2968–2976. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.S.; Kies, M.S.; Fossella, F.V.; Glisson, B.S.; Zaknoen, S.; Statkevich, P.; Munden, R.F.; Summey, C.; Pisters, K.M.; Papadimitrakopoulou, V.; et al. Phase II study of the farnesyltransferase inhibitor lonafarnib with paclitaxel in patients with taxane-refractory/resistant nonsmall cell lung carcinoma. Cancer 2005, 104, 561–569. [Google Scholar] [CrossRef]
- Milojkovic Kerklaan, B.; Dieras, V.; Le Tourneau, C.; Mergui-Roelvink, M.; Huitema, A.D.; Rosing, H.; Beijnen, J.H.; Marreaud, S.; Govaerts, A.S.; Piccart-Gebhart, M.J.; et al. Phase I study of lonafarnib (SCH66336) in combination with trastuzumab plus paclitaxel in Her2/neu overexpressing breast cancer: EORTC study 16023. Cancer Chemother. Pharmacol. 2013, 71, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Morgillo, F.; Lee, H.Y. Lonafarnib in cancer therapy. Expert Opin. Investig. Drugs 2006, 15, 709–719. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, C.; Feldman, M.J.; Wang, H.; Pang, Y.; Maggio, D.M.; Zhu, D.; Nesvick, C.L.; Dmitriev, P.; Bullova, P.; et al. Vorinostat suppresses hypoxia signaling by modulating nuclear translocation of hypoxia inducible factor 1 alpha. Oncotarget 2017, 8, 56110–56125. [Google Scholar] [CrossRef] [Green Version]
- Hutt, D.M.; Roth, D.M.; Vignaud, H.; Cullin, C.; Bouchecareilh, M. The histone deacetylase inhibitor, Vorinostat, represses hypoxia inducible factor 1 alpha expression through translational inhibition. PLoS ONE 2014, 9, e106224. [Google Scholar] [CrossRef] [Green Version]
- Richon, V.M.; Garcia-Vargas, J.; Hardwick, J.S. Development of vorinostat: Current applications and future perspectives for cancer therapy. Cancer Lett. 2009, 280, 201–210. [Google Scholar] [CrossRef]
- Mie Lee, Y.; Kim, S.H.; Kim, H.S.; Jin Son, M.; Nakajima, H.; Jeong Kwon, H.; Kim, K.W. Inhibition of hypoxia-induced angiogenesis by FK228, a specific histone deacetylase inhibitor, via suppression of HIF-1alpha activity. Biochem. Biophys. Res. Commun. 2003, 300, 241–246. [Google Scholar] [CrossRef]
- Schoepflin, Z.R.; Shapiro, I.M.; Risbud, M.V. Class I and IIa HDACs Mediate HIF-1alpha Stability Through PHD2-Dependent Mechanism, While HDAC6, a Class IIb Member, Promotes HIF-1alpha Transcriptional Activity in Nucleus Pulposus Cells of the Intervertebral Disc. J. Bone Miner. Res. 2016, 31, 1287–1299. [Google Scholar] [CrossRef] [Green Version]
- Kang, F.W.; Que, L.; Wu, M.; Wang, Z.L.; Sun, J. Effects of trichostatin A on HIF-1alpha and VEGF expression in human tongue squamous cell carcinoma cells in vitro. Oncol. Rep. 2012, 28, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Kwon, H.J.; Lee, Y.M.; Baek, J.H.; Jang, J.E.; Lee, S.W.; Moon, E.J.; Kim, H.S.; Lee, S.K.; Chung, H.Y.; et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat. Med. 2001, 7, 437–443. [Google Scholar] [CrossRef]
- Lee, K.; Zhang, H.; Qian, D.Z.; Rey, S.; Liu, J.O.; Semenza, G.L. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc. Natl. Acad. Sci. USA 2009, 106, 17910–17915. [Google Scholar] [CrossRef] [Green Version]
- Zeng, M.; Shen, J.; Liu, Y.; Lu, L.Y.; Ding, K.; Fortmann, S.D.; Khan, M.; Wang, J.; Hackett, S.F.; Semenza, G.L.; et al. The HIF-1 antagonist acriflavine: Visualization in retina and suppression of ocular neovascularization. J. Mol. Med. 2017, 95, 417–429. [Google Scholar] [CrossRef] [Green Version]
- Nehme, R.; Hallal, R.; El Dor, M.; Kobeissy, F.; Gouilleux, F.; Mazurier, F.; Zibara, K. Repurposing of Acriflavine to target Chronic Myeloid Leukemia treatment. Curr. Med. Chem. 2020, 28, 2218–2233. [Google Scholar] [CrossRef]
- Kong, D.; Park, E.J.; Stephen, A.G.; Calvani, M.; Cardellina, J.H.; Monks, A.; Fisher, R.J.; Shoemaker, R.H.; Melillo, G. Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Res. 2005, 65, 9047–9055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, A.Y.; Kim, K.; Boucher, H.; Bonomi, P.; Stewart, J.A.; Karp, D.D.; Blum, R.H. A randomized phase II trial of echinomycin, trimetrexate, and cisplatin plus etoposide in patients with metastatic nonsmall cell lung carcinoma: An Eastern Cooperative Oncology Group Study (E1587). Cancer 1998, 82, 292–300. [Google Scholar] [CrossRef]
- Gradishar, W.J.; Vogelzang, N.J.; Kilton, L.J.; Leibach, S.J.; Rademaker, A.W.; French, S.; Benson, A.B., 3rd. A phase II clinical trial of echinomycin in metastatic soft tissue sarcoma. An Illinois Cancer Center Study. Investig. New Drugs 1995, 13, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Koike, K.; Nagano, M.; Ebihara, M.; Hirayama, T.; Tsuji, M.; Suga, H.; Nagasawa, H. Design, Synthesis, and Conformation-Activity Study of Unnatural Bridged Bicyclic Depsipeptides as Highly Potent Hypoxia Inducible Factor-1 Inhibitors and Antitumor Agents. J. Med. Chem. 2020, 63, 4022–4046. [Google Scholar] [CrossRef]
- Bailey, C.M.; Liu, Y.; Peng, G.; Zhang, H.; He, M.; Sun, D.; Zheng, P.; Liu, Y.; Wang, Y. Liposomal formulation of HIF-1alpha inhibitor echinomycin eliminates established metastases of triple-negative breast cancer. Nanomedicine 2020, 29, 102278. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Yamaguchi, J.; Shoji, K.; Nangaku, M. Anthracycline inhibits recruitment of hypoxia-inducible transcription factors and suppresses tumor cell migration and cardiac angiogenic response in the host. J. Biol. Chem. 2012, 287, 34866–34882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, T.W.; Akinaga, S.; Murakata, T.; Agatsuma, T.; Sugimoto, S.; Nakano, H.; Lee, Y.S.; Simen, B.B.; Argon, Y.; Felts, S.; et al. Interaction of radicicol with members of the heat shock protein 90 family of molecular chaperones. Mol. Endocrinol. 1999, 13, 1435–1448. [Google Scholar] [CrossRef]
- Hur, E.; Kim, H.H.; Choi, S.M.; Kim, J.H.; Yim, S.; Kwon, H.J.; Choi, Y.; Kim, D.K.; Lee, M.O.; Park, H. Reduction of hypoxia-induced transcription through the repression of hypoxia-inducible factor-1alpha/aryl hydrocarbon receptor nuclear translocator DNA binding by the 90-kDa heat-shock protein inhibitor radicicol. Mol. Pharmacol. 2002, 62, 975–982. [Google Scholar] [CrossRef] [Green Version]
- Befani, C.D.; Vlachostergios, P.J.; Hatzidaki, E.; Patrikidou, A.; Bonanou, S.; Simos, G.; Papandreou, C.N.; Liakos, P. Bortezomib represses HIF-1alpha protein expression and nuclear accumulation by inhibiting both PI3K/Akt/TOR and MAPK pathways in prostate cancer cells. J. Mol. Med. 2012, 90, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Chun, Y.S.; Lee, D.S.; Huang, L.E.; Park, J.W. Bortezomib inhibits tumor adaptation to hypoxia by stimulating the FIH-mediated repression of hypoxia-inducible factor-1. Blood 2008, 111, 3131–3136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falchook, G.S.; Wheler, J.J.; Naing, A.; Jackson, E.F.; Janku, F.; Hong, D.; Ng, C.S.; Tannir, N.M.; Lawhorn, K.N.; Huang, M.; et al. Targeting hypoxia-inducible factor-1alpha (HIF-1alpha) in combination with antiangiogenic therapy: A phase I trial of bortezomib plus bevacizumab. Oncotarget 2014, 5, 10280–10292. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, H.; Khayat, D.; Giaccone, G.; Facon, T. Proteasome inhibition and its clinical prospects in the treatment of hematologic and solid malignancies. Cancer 2005, 104, 1794–1807. [Google Scholar] [CrossRef] [Green Version]
- Birle, D.C.; Hedley, D.W. Suppression of the hypoxia-inducible factor-1 response in cervical carcinoma xenografts by proteasome inhibitors. Cancer Res. 2007, 67, 1735–1743. [Google Scholar] [CrossRef] [Green Version]
- Koh, M.Y.; Spivak-Kroizman, T.; Venturini, S.; Welsh, S.; Williams, R.R.; Kirkpatrick, D.L.; Powis, G. Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1alpha. Mol. Cancer Ther. 2008, 7, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Welsh, S.; Williams, R.; Kirkpatrick, L.; Paine-Murrieta, G.; Powis, G. Antitumor activity and pharmacodynamic properties of PX-478, an inhibitor of hypoxia-inducible factor-1alpha. Mol. Cancer Ther. 2004, 3, 233–244. [Google Scholar]
- Lou, J.J.; Chua, Y.L.; Chew, E.H.; Gao, J.; Bushell, M.; Hagen, T. Inhibition of hypoxia-inducible factor-1alpha (HIF-1alpha) protein synthesis by DNA damage inducing agents. PLoS ONE 2010, 5, e10522. [Google Scholar] [CrossRef]
- Rapisarda, A.; Uranchimeg, B.; Sordet, O.; Pommier, Y.; Shoemaker, R.H.; Melillo, G. Topoisomerase I-mediated inhibition of hypoxia-inducible factor 1: Mechanism and therapeutic implications. Cancer Res. 2004, 64, 1475–1482. [Google Scholar] [CrossRef] [Green Version]
- Sapra, P.; Kraft, P.; Pastorino, F.; Ribatti, D.; Dumble, M.; Mehlig, M.; Wang, M.; Ponzoni, M.; Greenberger, L.M.; Horak, I.D. Potent and sustained inhibition of HIF-1alpha and downstream genes by a polyethyleneglycol-SN38 conjugate, EZN-2208, results in anti-angiogenic effects. Angiogenesis 2011, 14, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Rapisarda, A.; Hollingshead, M.; Uranchimeg, B.; Bonomi, C.A.; Borgel, S.D.; Carter, J.P.; Gehrs, B.; Raffeld, M.; Kinders, R.J.; Parchment, R.; et al. Increased antitumor activity of bevacizumab in combination with hypoxia inducible factor-1 inhibition. Mol. Cancer Ther. 2009, 8, 1867–1877. [Google Scholar] [CrossRef] [Green Version]
- Puppo, M.; Battaglia, F.; Ottaviano, C.; Delfino, S.; Ribatti, D.; Varesio, L.; Bosco, M.C. Topotecan inhibits vascular endothelial growth factor production and angiogenic activity induced by hypoxia in human neuroblastoma by targeting hypoxia-inducible factor-1alpha and -2alpha. Mol. Cancer Ther. 2008, 7, 1974–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, E.; Birrer, M.J.; Eliasof, S.; Garmey, E.G.; Lazarus, D.; Lee, C.R.; Man, S.; Matulonis, U.A.; Peters, C.G.; Xu, P.; et al. Translational impact of nanoparticle-drug conjugate CRLX101 with or without bevacizumab in advanced ovarian cancer. Clin. Cancer Res. 2015, 21, 808–818. [Google Scholar] [CrossRef] [Green Version]
- Pham, E.; Yin, M.; Peters, C.G.; Lee, C.R.; Brown, D.; Xu, P.; Man, S.; Jayaraman, L.; Rohde, E.; Chow, A.; et al. Preclinical Efficacy of Bevacizumab with CRLX101, an Investigational Nanoparticle-Drug Conjugate, in Treatment of Metastatic Triple-Negative Breast Cancer. Cancer Res. 2016, 76, 4493–4503. [Google Scholar] [CrossRef] [Green Version]
- Keefe, S.M.; Hoffman-Censits, J.; Cohen, R.B.; Mamtani, R.; Heitjan, D.; Eliasof, S.; Nixon, A.; Turnbull, B.; Garmey, E.G.; Gunnarsson, O.; et al. Efficacy of the nanoparticle-drug conjugate CRLX101 in combination with bevacizumab in metastatic renal cell carcinoma: Results of an investigator-initiated phase I-IIa clinical trial. Ann. Oncol. 2016, 27, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Voss, M.H.; Hussain, A.; Vogelzang, N.; Lee, J.L.; Keam, B.; Rha, S.Y.; Vaishampayan, U.; Harris, W.B.; Richey, S.; Randall, J.M.; et al. A randomized phase II trial of CRLX101 in combination with bevacizumab versus standard of care in patients with advanced renal cell carcinoma. Ann. Oncol. 2017, 28, 2754–2760. [Google Scholar] [CrossRef] [PubMed]
- Chuan, L.; Huang, X.; Fan, C.; Wen, S.; Yang, X.; Wang, J.; Ren, J.; Ru, J.; Ding, L. Metformin ameliorates brain damage caused by cardiopulmonary resuscitation via targeting endoplasmic reticulum stress-related proteins GRP78 and XBP1. Eur. J. Pharmacol. 2021, 891, 173716. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, J.; Yi, G.; Deng, M.; Liu, H.; Liang, M.; Shi, B.; Fu, X.; Chen, Y.; Chen, L.; et al. Metformin suppresses hypoxia-induced stabilization of HIF-1alpha through reprogramming of oxygen metabolism in hepatocellular carcinoma. Oncotarget 2016, 7, 873–884. [Google Scholar] [CrossRef] [Green Version]
- Kitson, S.J.; Maskell, Z.; Sivalingam, V.N.; Allen, J.L.; Ali, S.; Burns, S.; Gilmour, K.; Latheef, R.; Slade, R.J.; Pemberton, P.W.; et al. PRE-surgical Metformin In Uterine Malignancy (PREMIUM): A Multi-Center, Randomized Double-Blind, Placebo-Controlled Phase III Trial. Clin. Cancer Res. 2019, 25, 2424–2432. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Buart, S.; Van Pelt, J.; Richon, C.; Hasmim, M.; Leleu, N.; Suchorska, W.M.; Jalil, A.; Lecluse, Y.; El Hage, F.; et al. The cooperative induction of hypoxia-inducible factor-1 alpha and STAT3 during hypoxia induced an impairment of tumor susceptibility to CTL-mediated cell lysis. J. Immunol. 2009, 182, 3510–3521. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, J.; Huang, H. Recent agents targeting HIF-1alpha for cancer therapy. J. Cell Biochem. 2013, 114, 498–509. [Google Scholar] [CrossRef]
- Zeidner, J.F.; Foster, M.C.; Blackford, A.L.; Litzow, M.R.; Morris, L.E.; Strickland, S.A.; Lancet, J.E.; Bose, P.; Levy, M.Y.; Tibes, R.; et al. Final results of a randomized multicenter phase II study of alvocidib, cytarabine, and mitoxantrone versus cytarabine and daunorubicin (7 + 3) in newly diagnosed high-risk acute myeloid leukemia (AML). Leuk. Res. 2018, 72, 92–95. [Google Scholar] [CrossRef]
- Samec, M.; Liskova, A.; Koklesova, L.; Mersakova, S.; Strnadel, J.; Kajo, K.; Pec, M.; Zhai, K.; Smejkal, K.; Mirzaei, S.; et al. Flavonoids Targeting HIF-1: Implications on Cancer Metabolism. Cancers 2021, 13, 130. [Google Scholar] [CrossRef]
- Chan, W.K.; Yao, G.; Gu, Y.Z.; Bradfield, C.A. Cross-talk between the aryl hydrocarbon receptor and hypoxia inducible factor signaling pathways. Demonstration of competition and compensation. J. Biol. Chem. 1999, 274, 12115–12123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, M.; Blankenship, A.L.; Giesy, J.P. Interactions between aryl hydrocarbon receptor (AhR) and hypoxia signaling pathways. Environ. Toxicol. Pharmacol. 2001, 10, 17–27. [Google Scholar] [CrossRef]
- Wigerup, C.; Pahlman, S.; Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Contratto, M.; Shanbhogue, K.P.; Manji, G.A.; O’Neil, B.H.; Noonan, A.; Tudor, R.; Lee, R. Evaluation of a locked nucleic acid form of antisense oligo targeting HIF-1alpha in advanced hepatocellular carcinoma. World J. Clin. Oncol. 2019, 10, 149–160. [Google Scholar] [CrossRef]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar]
- Faubert, B.; Vincent, E.E.; Griss, T.; Samborska, B.; Izreig, S.; Svensson, R.U.; Mamer, O.A.; Avizonis, D.; Shackelford, D.B.; Shaw, R.J.; et al. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1alpha. Proc. Natl. Acad. Sci. USA 2014, 111, 2554–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef] [Green Version]
- Pore, N.; Jiang, Z.; Gupta, A.; Cerniglia, G.; Kao, G.D.; Maity, A. EGFR tyrosine kinase inhibitors decrease VEGF expression by both hypoxia-inducible factor (HIF)-1-independent and HIF-1-dependent mechanisms. Cancer Res. 2006, 66, 3197–3204. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.H.; Jiang, G.; Zheng, J.Z.; Lu, Z.; Hunter, T.; Vogt, P.K. Phosphatidylinositol 3-kinase signaling controls levels of hypoxia-inducible factor 1. Cell Growth Differ. 2001, 12, 363–369. [Google Scholar] [PubMed]
- Janku, F. Phosphoinositide 3-kinase (PI3K) pathway inhibitors in solid tumors: From laboratory to patients. Cancer Treat. Rev. 2017, 59, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Fallone, F.; Britton, S.; Nieto, L.; Salles, B.; Muller, C. ATR controls cellular adaptation to hypoxia through positive regulation of hypoxia-inducible factor 1 (HIF-1) expression. Oncogene 2013, 32, 4387–4396. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, A.; Hall, S.; Curtin, N.; Drew, Y. Targeting ATR as Cancer Therapy: A new era for synthetic lethality and synergistic combinations? Pharmacol. Ther. 2020, 207, 107450. [Google Scholar] [CrossRef]
- Sundar, R.; Brown, J.; Ingles Russo, A.; Yap, T.A. Targeting ATR in cancer medicine. Curr. Probl. Cancer 2017, 41, 302–315. [Google Scholar] [CrossRef]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, B.D.; Snyder, S.H. H2S: A Novel Gasotransmitter that Signals by Sulfhydration. Trends Biochem. Sci. 2015, 40, 687–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neckers, L.; Ivy, S.P. Heat shock protein 90. Curr. Opin. Oncol. 2003, 15, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef]
- Isaacs, J.S.; Jung, Y.J.; Mimnaugh, E.G.; Martinez, A.; Cuttitta, F.; Neckers, L.M. Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1 alpha-degradative pathway. J. Biol. Chem. 2002, 277, 29936–29944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.Y.; Oh, S.H.; Morgillo, F.; Myers, J.N.; Kim, E.; Hong, W.K.; Lee, H.Y. Hypoxia-inducible factor 1alpha and antiangiogenic activity of farnesyltransferase inhibitor SCH66336 in human aerodigestive tract cancer. J. Natl. Cancer Inst. 2005, 97, 1272–1286. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Jeong, J.W.; Park, J.A.; Lee, J.W.; Seo, J.H.; Jung, B.K.; Bae, M.K.; Kim, K.W. Regulation of the HIF-1alpha stability by histone deacetylases. Oncol. Rep. 2007, 17, 647–651. [Google Scholar]
- Huang, X.; Liu, Y.; Wang, Y.; Bailey, C.; Zheng, P.; Liu, Y. Dual Targeting Oncoproteins MYC and HIF1alpha Regresses Tumor Growth of Lung Cancer and Lymphoma. Cancers 2021, 13, 694. [Google Scholar] [CrossRef] [PubMed]
- Sritharan, S.; Sivalingam, N. A comprehensive review on time-tested anticancer drug doxorubicin. Life Sci. 2021, 278, 119527. [Google Scholar] [CrossRef]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [Google Scholar] [CrossRef]
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000, 14, 34–44. [Google Scholar]
- Yeo, E.J. Hypoxia and aging. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef]
- Harada, H. Hypoxia-inducible factor 1-mediated characteristic features of cancer cells for tumor radioresistance. J. Radiat. Res. 2016, 57, i99–i105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, H.; Inoue, M.; Itasaka, S.; Hirota, K.; Morinibu, A.; Shinomiya, K.; Zeng, L.; Ou, G.; Zhu, Y.; Yoshimura, M.; et al. Cancer cells that survive radiation therapy acquire HIF-1 activity and translocate towards tumour blood vessels. Nat. Commun. 2012, 3, 783. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| HIF-1 Inhibitors | Target Molecules | Tumors/Cell Lines |
|---|---|---|
| Transcriptional initiation | ||
| Flavopiridol | blocking phosphorylation of RNA polymerase II’s COOH-terminal domain | acute myeloid leukemia, advanced renal cell carcinoma |
| Aminoflavone | AhR | breast cancer cells (MCF7), human renal cell lines |
| mRNA stabilization | ||
| EZN-2968 | HIF-1α mRNA | prostate (15PC3, PC3, and DU145), glioblastoma (U373) cells, duodenal neuroendocrine tumor |
| Translational initiation | ||
| Rapamycin | mTOR | prostate cancer cells (PC-3) |
| Cetuximab | EGFR | HNSCC cells (FaDu, HN5, UMSCC1, and OSC19) |
| Buparlisib | PI3K | breast cancer |
| KC7F2 | mTOR, S6K, 4EBP1 | U251MG, MCF7, PC3, LNZ308, LN229 cells |
| 2-MEs | tubulin | MCF7, MDA-MB-231, PC-3, ovarian cancer, multiple myeloma |
| Digoxin | Na+/K+ ATPase | Hep3B, 293, 293T, PC3, P493 |
| AZD6738, VX-970 | ATR | esophageal cancer cells (OE21, FLO-1) (VX-970) |
| Protein stabilization | ||
| Melatonin, NB-5-MT | GSK-3β, activating PHDs | colon cancer cells (HCT116), prostate cancer cells (DU145, PC-3, LNCaP), breast cancer cells (MCF-7, MDA-MB-231), renal cell carcinoma (RenCa) |
| LW6 | inducing pVHL expression | HCT116 |
| GYY4137 | slow-releasing H2S donor | HeLa, HCT-116, Hep G2, HL-60, MCF-7, MV4-11, U2OS |
| Ganetespib | ATP-binding site of HSP90 | NSCLC (NCI-H1975, HCC827) cells, colorectal cancer (HCT116, HT29), non-small cell lung cancer |
| Apigenin | CK2 activity | multiple myeloma cells, pancreatic cancer cells (S2-013, CD18) |
| Lonafarnib | interaction between HSP90 and HIF-1α | non-small cell lung carcinoma, breast cancer |
| Vorinostat | increasing acetylation of HSP90 | Hep3B, HepG2, U2OS, MG63, U87 MG, HuH7 |
| Romidepsin (FK228) | HDAC | Lewis lung carcinoma (LLC), HT1080 |
| Trichostatin | HDAC | tongue squamous cell carcinoma SCC-6, BAECs, 786-0 (VHL-negative), HEK 293 cells, Lewis lung carcinoma cells, HepG2 |
| Heterodimerization | ||
| Acriflavine | PAS-B domain of HIF-1α | HEK293, HeLa, Hep3B-c1, PC-3, P493 |
| DNA binding | ||
| Echinomycin | binding to HRE sequences | U251, MCF-7, non-small cell lung carcinoma, soft tissue sarcoma |
| Anthracycline | HIF-HRE binding | HK-2. HeLa, A498, KMRC-3 VHL-defective renal cell carcinoma (RCC) cells, Caki-1 VHL-competent renal cell carcinoma cells |
| Radicicol | HIF-DNA binding, HSP90 | SKBR3, Hep 3B |
| Transactivation | ||
| Bortezomib | PI3K/Akt/mTOR, MAPK, p300 recruitment | prostate cancer cells, Hep3B, HEK293, ARH77, U299 cells, RCC, breast cancer, rectal carcinoma, nasopharyngeal, neuroendocrine carcinoma, prostate cancer |
| Other steps | ||
| PX-478 | inhibiting HIF-1α deubiquitination, reducing HIF-1α mRNA and HIF-1α translation | PC-3, MCF-7, HT-29, Panc-1, BxPC-3 cells, RCC4, RCC4/VHL, HCT116 |
| Camptothecin analogues (TPT, EZN-2208) | topoisomerase-1 | HEK293, U251, leukemia cell lines, neuroblastoma cells (GI-LI-N, LAN-5) derived from metastatic |
| CRLX101 | topoisomerase-1 | ovarian cancer, breast cancer, renal cell carcinoma |
| Metformin | Mitochondria complex I, XBP1 | HCT116, HCC, NSCLC, etc. |
| HIF-1 Inhibitors | Animal Models | Clinical Trials | References |
|---|---|---|---|
| Transcriptional initiation | |||
| Flavopiridol | No animal model looking at effect on hypoxia/HIF-1 | Not in clinical use | [35,36,37,38,39] |
| Aminoflavone (AF) | MCF-7 xenografts | Phase 1 (Completed) | [40,41,42,43] |
| mRNA stabilization | |||
| EZN-2968 | DU145 xenograft | Phase 1 (Completed) | [44,45] |
| Translational initiation | |||
| Rapamycin | LKB1 ± mice (Peutz-Jeghers syndrome model), ACHN xenograft, SN12C xenograft | FDA-approved for treatment for renal cell carcinoma (RCC) | [46,47,48] |
| Cetuximab | Tgfb1/Pten deletion 2cKO mice, CAL27 xenograft | FDA-approved for head and neck squamous cell carcinoma (HNSCC), colorectal cancer (CRC) | [49,50] |
| Buparlisib | No animal model looking at effect on hypoxia/HIF-1 | Phase 3 (Completed, Terminated) | [51] |
| KC7F2 | No animal model looking at effect on hypoxia/HIF-1 | Not in clinical use | [52] |
| 2-MEs | LLC xenograft/MDA-MB-231 xenograft | Phase 2 (Completed) | [53,54,55,56,57] |
| Digoxin | P493 xenograft/PC3 xenograft | Phase 2 (Completed, Terminated) | [58,59] |
| AZD6738, VX-970 | OE21 xenograft (VX-970) | AZD6738: Phase 2 (In progress) | [60] |
| VX-970: Phase 2 (In progress) | |||
| Protein stabilization | |||
| Melatonin, NB-5-MT | Tumor xenograft models with breast, kidney, prostate, and colon cancer cells in mouse and zebrafish | Not in clinical use | [61,62,63,64,65,66] |
| LW6 | HCT116 xenograft | Not in clinical use | [67] |
| GYY4137 | No animal model looking at effect on hypoxia/HIF-1 | Not in clinical use | [68,69] |
| Ganetespib | Orthotopic implantation of MDA-MB-231, MDA-MB-435 | Phase 2 (Completed) Phase 3 (Terminated) | [70,71,72,73] |
| Apigenin | PC-3 and OVCAR3 Matrigel plug assay | Phase 2 (Terminated) | [74,75,76,77,78] |
| Lonafarnib | Orthotopic implantation of UMSCC38 | Phase 2 (Completed) Phase 3 (Terminated) | [79,80,81,82,83] |
| Vorinostat | Hep3B xenograft | FDA-approved for cutaneous T-cell lymphoma Phase 2 with results for other cancer types | [84,85,86] |
| Romidepsin (FK228) | Orthotopic implantation of LLC | FDA-approved for cutaneous T-cell lymphoma Phase 2 with results for other cancer types | [87] |
| Trichostatin | No animal model looking at effect on hypoxia/HIF-1 | Phase 1 (In progress) | [88,89,90] |
| Heterodimerization | |||
| Acriflavine | PC-3 xenografts | Not in clinical use | [91,92,93] |
| DNA binding | |||
| Echinomycin | MDA-MB-231 and SUM-159 xenografts | Phase 2 (Completed) | [94,95,96,97,98] |
| Anthracycline | HeLa xenografts | Phase 2 (Completed) | [99] |
| Radicicol | No animal model looking at effect on hypoxia/HIF-1 | Not in clinical use | [100,101] |
| Transactivation | |||
| Bortezomib | SiHa xenografts | Phase 1 (Completed) | [102,103,104,105,106] |
| Other steps | |||
| PX-478 | HT-29 xenografts | Phase 1 (Completed) | [107,108] |
| Camptothecin analogues (TPT, EZN-2208) | U251 glioma xenografts | TPT: Phase 1 (Completed) | [109,110,111,112,113] |
| EZN-2208 combination with other drugs: Phase 1 (Completed) | |||
| CRLX101 | orthotopic primary triple-negative breast cancer xenografts | Phase 2 (Completed) | [114,115,116,117] |
| Metformin | Various types of tumors, e.g., CRC and HCC | Phase 3 (Completed) | [118,119,120,121] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shirai, Y.; Chow, C.C.T.; Kambe, G.; Suwa, T.; Kobayashi, M.; Takahashi, I.; Harada, H.; Nam, J.-M. An Overview of the Recent Development of Anticancer Agents Targeting the HIF-1 Transcription Factor. Cancers 2021, 13, 2813. https://doi.org/10.3390/cancers13112813
Shirai Y, Chow CCT, Kambe G, Suwa T, Kobayashi M, Takahashi I, Harada H, Nam J-M. An Overview of the Recent Development of Anticancer Agents Targeting the HIF-1 Transcription Factor. Cancers. 2021; 13(11):2813. https://doi.org/10.3390/cancers13112813
Chicago/Turabian StyleShirai, Yukari, Christalle C. T. Chow, Gouki Kambe, Tatsuya Suwa, Minoru Kobayashi, Itsuki Takahashi, Hiroshi Harada, and Jin-Min Nam. 2021. "An Overview of the Recent Development of Anticancer Agents Targeting the HIF-1 Transcription Factor" Cancers 13, no. 11: 2813. https://doi.org/10.3390/cancers13112813
APA StyleShirai, Y., Chow, C. C. T., Kambe, G., Suwa, T., Kobayashi, M., Takahashi, I., Harada, H., & Nam, J. -M. (2021). An Overview of the Recent Development of Anticancer Agents Targeting the HIF-1 Transcription Factor. Cancers, 13(11), 2813. https://doi.org/10.3390/cancers13112813