
Bispecific T Cell Engagers for the Treatment of Multiple Myeloma: Achievements and Challenges

 , , , , , ,
, , , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
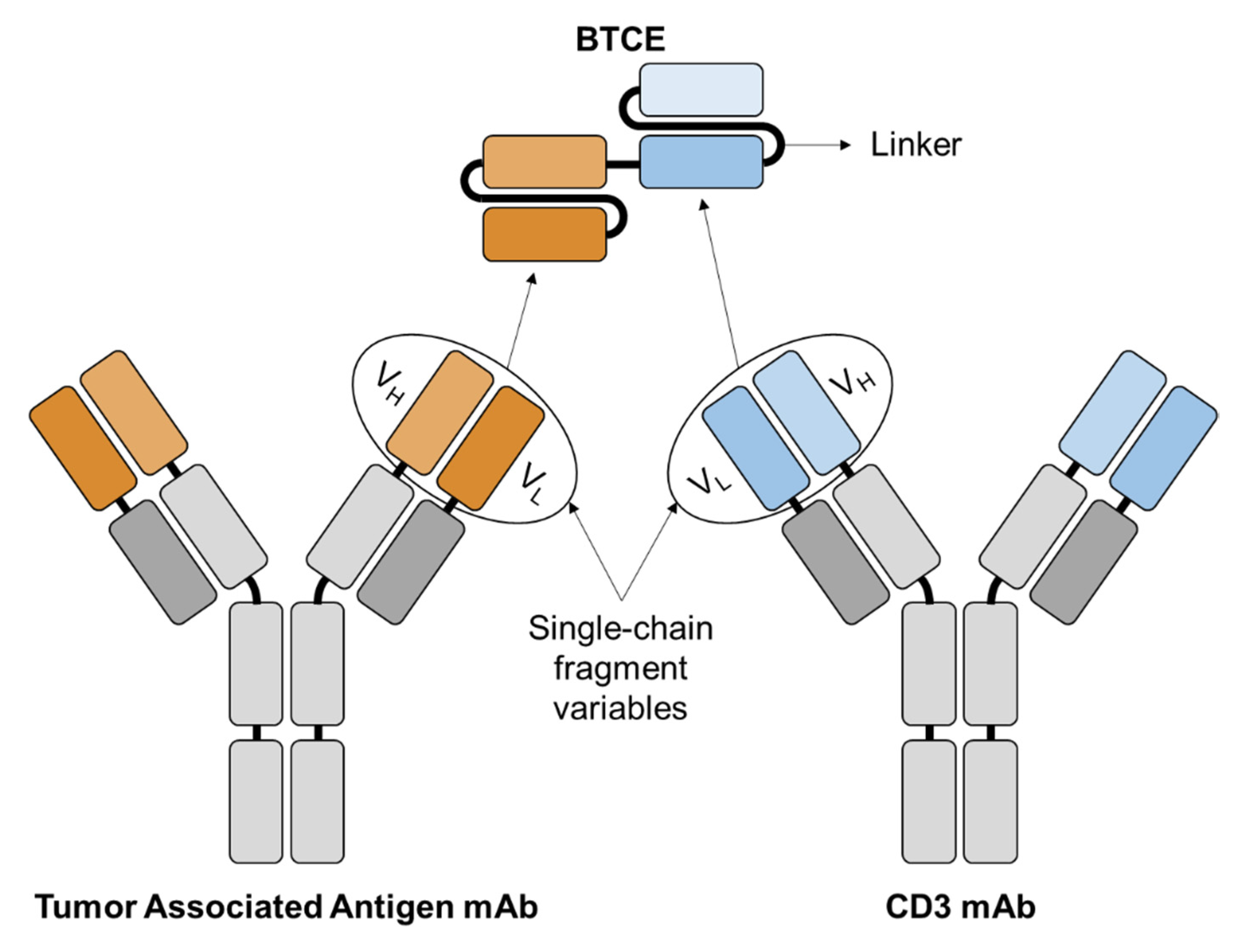
2. Bispecific T Cell Engagers
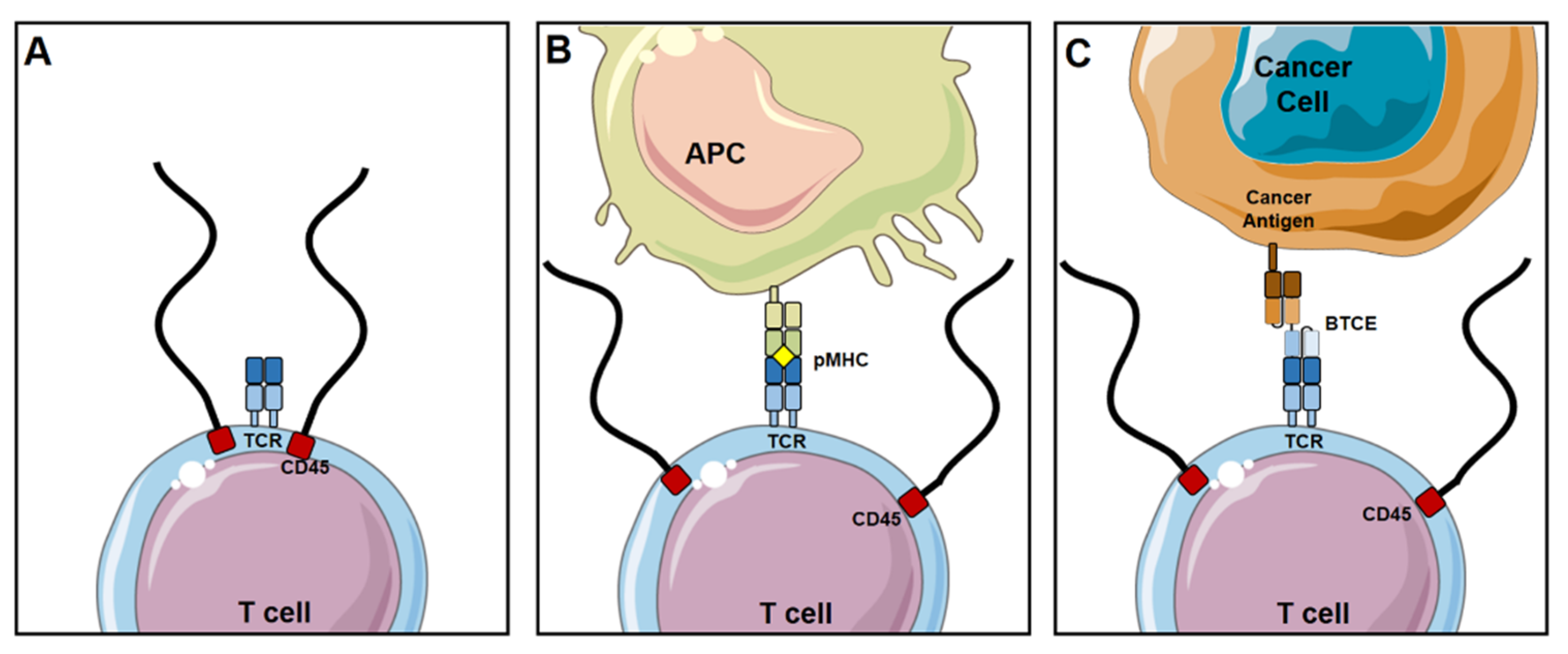
2.1. Mechanism of Action
2.2. Advantages
2.2.1. High Potency and Efficacy
2.2.2. Safety
2.2.3. Availability Off-the-Shelf
2.2.4. Lower Cost
2.3. Challenges
2.3.1. Poor Pharmacokinetic Profile
2.3.2. Laborious and Cumbersome to Produce
2.3.3. Inability to Target Multiple Antigens
3. BTCEs for the Treatment of MM
3.1. BCMA
3.2. CD38
3.3. FcRH5
3.4. CD19
3.5. CD138
3.6. Novel TCE Strategies
3.7. Toxicities
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Laubach, J.; Richardson, P.; Anderson, K. Multiple myeloma. Annu. Rev. Med. 2011, 62, 249–264. [Google Scholar] [CrossRef] [Green Version]
- Richardson, P.G.; San Miguel, J.F.; Moreau, P.; Hajek, R.; Dimopoulos, M.A.; Laubach, J.P.; Palumbo, A.; Luptakova, K.; Romanus, D.; Skacel, T.; et al. Interpreting clinical trial data in multiple myeloma: Translating findings to the real-world setting. Blood Cancer J. 2018, 8, 109. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Minnie, S.A.; Hill, G.R. Immunotherapy of multiple myeloma. J. Clin. Investig. 2020, 130, 1565–1575. [Google Scholar] [CrossRef]
- Sadelain, M.; Rivière, I.; Riddell, S. Therapeutic T cell engineering. Nature 2017, 545, 423–431. [Google Scholar] [CrossRef] [Green Version]
- Rosenblatt, J.; Avigan, D. Targeting the PD-1/PD-L1 axis in multiple myeloma: A dream or a reality? Blood 2017, 129, 275–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkilineni, L.; Kochenderfer, J.N. Chimeric antigen receptor T-cell therapies for multiple myeloma. Blood 2017, 130, 2594–2602. [Google Scholar] [CrossRef] [Green Version]
- BiTE Therapy Active in Multiple Myeloma. Cancer Discov. 2019, 9, 157–158. [CrossRef] [PubMed] [Green Version]
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T-cell engagers for cancer immunotherapy. Immunol. Cell Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedykh, S.E.; Prinz, V.V.; Buneva, V.N.; Nevinsky, G.A. Bispecific antibodies: Design, therapy, perspectives. Drug Des. Dev. Ther. 2018, 12, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Chen, Y.; Park, J.; Liu, X.; Hu, Y.; Wang, T.; McFarland, K.; Betenbaugh, M.J. Design and Production of Bispecific Antibodies. Antibodies 2019, 8, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inbar, D.; Hochman, J.; Givol, D. Localization of antibody-combining sites within the variable portions of heavy and light chains. Proc. Natl. Acad. Sci. USA 1972, 69, 2659–2662. [Google Scholar] [CrossRef] [Green Version]
- Baeuerle, P.A.; Reinhardt, C.; Kufer, P. BiTE: A new class of antibodies that recruit T-cells. Drugs Future 2008, 33, 1–11. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Kufer, P.; Bargou, R. BiTE: Teaching antibodies to engage T-cells for cancer therapy. Curr. Opin. Mol. Ther. 2009, 11, 22–30. [Google Scholar]
- Baeuerle, P.A.; Reinhardt, C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 2009, 69, 4941–4944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreier, T.; Lorenczewski, G.; Brandl, C.; Hoffmann, P.; Syring, U.; Hanakam, F.; Kufer, P.; Riethmuller, G.; Bargou, R.; Baeuerle, P.A. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int. J. Cancer 2002, 100, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.J.; van der Merwe, P.A. The kinetic-segregation model: TCR triggering and beyond. Nat. Immunol. 2006, 7, 803–809. [Google Scholar] [CrossRef]
- Mustelin, T.; Coggeshall, K.M.; Altman, A. Rapid activation of the T-cell tyrosine protein kinase pp56lck by the CD45 phosphotyrosine phosphatase. Proc. Natl. Acad. Sci. USA 1989, 86, 6302–6306. [Google Scholar] [CrossRef] [Green Version]
- Trowbridge, I.S.; Thomas, M.L. CD45: An emerging role as a protein tyrosine phosphatase required for lymphocyte activation and development. Annu. Rev. Immunol. 1994, 12, 85–116. [Google Scholar] [CrossRef]
- Cordoba, S.P.; Choudhuri, K.; Zhang, H.; Bridge, M.; Basat, A.B.; Dustin, M.L.; van der Merwe, P.A. The large ectodomains of CD45 and CD148 regulate their segregation from and inhibition of ligated T-cell receptor. Blood 2013, 121, 4295–4302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, M.M.; Boniface, J.J.; Reich, Z.; Lyons, D.; Hampl, J.; Arden, B.; Chien, Y. Ligand recognition by alpha beta T cell receptors. Annu. Rev. Immunol. 1998, 16, 523–544. [Google Scholar] [CrossRef] [PubMed]
- Offner, S.; Hofmeister, R.; Romaniuk, A.; Kufer, P.; Baeuerle, P.A. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 2006, 43, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Burroughs, N.J.; Lazic, Z.; van der Merwe, P.A. Ligand detection and discrimination by spatial relocalization: A kinase-phosphatase segregation model of TCR activation. Biophys. J. 2006, 91, 1619–1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Stagg, N.J.; Johnston, J.; Harris, M.J.; Menzies, S.A.; DiCara, D.; Clark, V.; Hristopoulos, M.; Cook, R.; Slaga, D.; et al. Membrane-Proximal Epitope Facilitates Efficient T Cell Synapse Formation by Anti-FcRH5/CD3 and Is a Requirement for Myeloma Cell Killing. Cancer Cell 2017, 31, 383–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellerman, D. Bispecific T-cell engagers: Towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods 2019, 154, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Slaney, C.Y.; Wang, P.; Darcy, P.K.; Kershaw, M.H. CARs versus BiTEs: A Comparison between T Cell-Redirection Strategies for Cancer Treatment. Cancer Discov. 2018, 8, 924–934. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.D.; Raje, N.; Fowler, J.A.; Mezzi, K.; Scott, E.C.; Dhodapkar, M.V. How to Train Your T Cells: Overcoming Immune Dysfunction in Multiple Myeloma. Clin. Cancer Res. 2020, 26, 1541–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuraszeck, T.; Kasichayanula, S.; Benjamin, J.E. Translation and Clinical Development of Bispecific T-cell Engaging Antibodies for Cancer Treatment. Clin. Pharmacol. Ther. 2017, 101, 634–645. [Google Scholar] [CrossRef] [Green Version]
- Hipp, S.; Tai, Y.T.; Blanset, D.; Deegen, P.; Wahl, J.; Thomas, O.; Rattel, B.; Adam, P.J.; Anderson, K.C.; Friedrich, M. A novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo. Leukemia 2017, 31, 1743–1751. [Google Scholar] [CrossRef]
- Stieglmaier, J.; Benjamin, J.; Nagorsen, D. Utilizing the BiTE (bispecific T-cell engager) platform for immunotherapy of cancer. Expert Opin. Biol. Ther. 2015, 15, 1093–1099. [Google Scholar] [CrossRef]
- Trinklein, N.D.; Pham, D.; Schellenberger, U.; Buelow, B.; Boudreau, A.; Choudhry, P.; Clarke, S.C.; Dang, K.; Harris, K.E.; Iyer, S.; et al. Efficient tumor killing and minimal cytokine release with novel T-cell agonist bispecific antibodies. MAbs 2019, 11, 639–652. [Google Scholar] [CrossRef]
- Zhukovsky, E.A.; Morse, R.J.; Maus, M.V. Bispecific antibodies and CARs: Generalized immunotherapeutics harnessing T cell redirection. Curr. Opin. Immunol. 2016, 40, 24–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajkumar, S.V. Value and Cost of Myeloma Therapy. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 662–666. [Google Scholar] [CrossRef]
- Anderson, S. Blenrep® for Relapsed or Refractory Multiple Myeloma. In Cleveland Clinic Clinical Rx Forum; Cleveland Clinic Department of Pharmacy: Lyndhurst, OH, USA, 2021; Volume 9. [Google Scholar]
- Strohl, W.R.; Naso, M. Bispecific T-Cell Redirection versus Chimeric Antigen Receptor (CAR)-T Cells as Approaches to Kill Cancer Cells. Antibodies 2019, 8, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhallak, K.; Sun, J.; Muz, B.; Azab, A.K. Biomaterials for cancer immunotherapy. In Biomaterials for Cancer Therapeutics; Woodhead Publishing, Elsevier: Cambridge, UK, 2020; pp. 499–526. [Google Scholar] [CrossRef]
- Lobo, E.D.; Hansen, R.J.; Balthasar, J.P. Antibody pharmacokinetics and pharmacodynamics. J. Pharm. Sci. 2004, 93, 2645–2668. [Google Scholar] [CrossRef]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef]
- Zhu, M.; Wu, B.; Brandl, C.; Johnson, J.; Wolf, A.; Chow, A.; Doshi, S. Blinatumomab, a Bispecific T-cell Engager (BiTE®) for CD-19 Targeted Cancer Immunotherapy: Clinical Pharmacology and Its Implications. Clin. Pharmacokinet. 2016, 55, 1271–1288. [Google Scholar] [CrossRef]
- Wesche, H.; Aaron, W.; Austin, R.J.; Baeuerle, P.A.; Jones, A.; Lemon, B.; Sexton, K.; Yu, T. Abstract 3814: TriTACs Are Novel T Cell-Engaging Therapeutic Proteins Optimized for the Treatment of Solid Tumors and for Long Serum Half-Life. In Proceedings of the American Association for Cancer Research Annual Meeting 2018, Chicago, IL, USA, 14–18 April 2018; AACR: Philadelphia, PA, USA, 2018. [Google Scholar]
- Leenaars, M.; Hendriksen, C.F. Critical steps in the production of polyclonal and monoclonal antibodies: Evaluation and recommendations. Ilar J. 2005, 46, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Vazquez-Lombardi, R.; Nevoltris, D.; Luthra, A.; Schofield, P.; Zimmermann, C.; Christ, D. Transient expression of human antibodies in mammalian cells. Nat. Protoc. 2018, 13, 99–117. [Google Scholar] [CrossRef] [PubMed]
- Kuan, C.T.; Reist, C.J.; Foulon, C.F.; Lorimer, I.A.; Archer, G.; Pegram, C.N.; Pastan, I.; Zalutsky, M.R.; Bigner, D.D. 125I-labeled anti-epidermal growth factor receptor-vIII single-chain Fv exhibits specific and high-level targeting of glioma xenografts. Clin. Cancer Res. 1999, 5, 1539–1549. [Google Scholar]
- Clackson, T.; Hoogenboom, H.R.; Griffiths, A.D.; Winter, G. Making antibody fragments using phage display libraries. Nature 1991, 352, 624–628. [Google Scholar] [CrossRef]
- Mack, M.; Gruber, R.; Schmidt, S.; Riethmüller, G.; Kufer, P. Biologic properties of a bispecific single-chain antibody directed against 17-1A (EpCAM) and CD3: Tumor cell-dependent T cell stimulation and cytotoxic activity. J. Immunol. 1997, 158, 3965–3970. [Google Scholar]
- Choi, B.D.; Kuan, C.T.; Cai, M.; Archer, G.E.; Mitchell, D.A.; Gedeon, P.C.; Sanchez-Perez, L.; Pastan, I.; Bigner, D.D.; Sampson, J.H. Systemic administration of a bispecific antibody targeting EGFRvIII successfully treats intracerebral glioma. Proc. Natl. Acad. Sci. USA 2013, 110, 270–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foltz, S.M.; Gao, Q.; Yoon, C.J.; Sun, H.; Yao, L.; Li, Y.; Jayasinghe, R.G.; Cao, S.; King, J.; Kohnen, D.R.; et al. Evolution and structure of clinically relevant gene fusions in multiple myeloma. Nat. Commun. 2020, 11, 2666. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.D.; Garfall, A.L.; Stadtmauer, E.A.; Melenhorst, J.J.; Lacey, S.F.; Lancaster, E.; Vogl, D.T.; Weiss, B.M.; Dengel, K.; Nelson, A.; et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J. Clin. Investig. 2019, 129, 2210–2221. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; Ledbetter, J.A.; Linsley, P.S.; Thompson, C.B. Role of the CD28 receptor in T-cell activation. Immunol. Today 1990, 11, 211–216. [Google Scholar] [CrossRef] [Green Version]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Seung, E.; Xu, L.; Rao, E.; Lord, D.M.; Wei, R.R.; Cortez-Retamozo, V.; Ospina, B.; Posternak, V.; Ulinski, G. Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nat. Cancer 2020, 1, 86–98. [Google Scholar] [CrossRef]
- Maura, F.; Bolli, N.; Angelopoulos, N.; Dawson, K.J.; Leongamornlert, D.; Martincorena, I.; Mitchell, T.J.; Fullam, A.; Gonzalez, S.; Szalat, R.; et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat. Commun. 2019, 10, 3835. [Google Scholar] [CrossRef] [Green Version]
- Muz, B.; de la Puente, P.; Azab, F.; Luderer, M.J.; King, J.; Vij, R.; Azab, A.K. A CD138-independent strategy to detect minimal residual disease and circulating tumour cells in multiple myeloma. Br. J. Haematol. 2016, 173, 70–81. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.; Asmann, Y.; Cattaneo, L.; Braggio, E.; Keats, J.; Auclair, D.; Lonial, S.; Russell, S.J.; Stewart, A.K. High somatic mutation and neoantigen burden are correlated with decreased progression-free survival in multiple myeloma. Blood Cancer J. 2017, 7, e612. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.F.; Anderson, K.C.; Tai, Y.T. Targeting B Cell Maturation Antigen (BCMA) in Multiple Myeloma: Potential Uses of BCMA-Based Immunotherapy. Front. Immunol. 2018, 9, 1821. [Google Scholar] [CrossRef]
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. BCMA is essential for the survival of long-lived bone marrow plasma cells. J. Exp. Med. 2004, 199, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.-T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.; Gliddon, L.; Fieles, W.; et al. Novel anti–B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef] [PubMed]
- Seckinger, A.; Delgado, J.A.; Moser, S.; Moreno, L.; Neuber, B.; Grab, A.; Lipp, S.; Merino, J.; Prosper, F.; Emde, M.; et al. Target Expression, Generation, Preclinical Activity, and Pharmacokinetics of the BCMA-T Cell Bispecific Antibody EM801 for Multiple Myeloma Treatment. Cancer Cell 2017, 31, 396–410. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.; Chari, A.; Scott, E.; Mezzi, K.; Usmani, S.Z. B-cell maturation antigen (BCMA) in multiple myeloma: Rationale for targeting and current therapeutic approaches. Leukemia 2020, 34, 985–1005. [Google Scholar] [CrossRef]
- Ghermezi, M.; Li, M.; Vardanyan, S.; Harutyunyan, N.M.; Gottlieb, J.; Berenson, A.; Spektor, T.M.; Andreu-Vieyra, C.; Petraki, S.; Sanchez, E.; et al. Serum B-cell maturation antigen: A novel biomarker to predict outcomes for multiple myeloma patients. Haematologica 2017, 102, 785–795. [Google Scholar] [CrossRef] [Green Version]
- Schuh, E.; Musumeci, A.; Thaler, F.S.; Laurent, S.; Ellwart, J.W.; Hohlfeld, R.; Krug, A.; Meinl, E. Human Plasmacytoid Dendritic Cells Display and Shed B Cell Maturation Antigen upon TLR Engagement. J. Immunol. 2017, 198, 3081–3088. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Hou, J. Immunotherapeutic strategies targeting B cell maturation antigen in multiple myeloma. Mil. Med. Res. 2021, 8, 9. [Google Scholar] [CrossRef]
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Krönke, J.; Facon, T.; Salnikov, A.V.; Lesley, R.; et al. Anti-B-Cell Maturation Antigen BiTE Molecule AMG 420 Induces Responses in Multiple Myeloma. J. Clin. Oncol. 2020, 38, 775–783. [Google Scholar] [CrossRef]
- Goldstein, R.L.; Goyos, A.; Li, C.M.; Deegen, P.; Bogner, P.; Sternjak, A.; Thomas, O.; Klinger, M.; Wahl, J.; Friedrich, M.; et al. AMG 701 induces cytotoxicity of multiple myeloma cells and depletes plasma cells in cynomolgus monkeys. Blood Adv. 2020, 4, 4180–4194. [Google Scholar] [CrossRef]
- Cooper, D.; Madduri, D.; Lentzsch, S.; Jagannath, S.; Li, J.; Boyapati, A.; Adriaens, L.; Chokshi, D.; Zhu, M.; Lowy, I. Safety and Preliminary Clinical Activity of REGN5458, an Anti-Bcma x Anti-CD3 Bispecific Antibody. In Patients with Relapsed/Refractory Multiple Myeloma; American Society of Hematology: Washington, DC, USA, 2019. [Google Scholar]
- Law, C.-L.; Aaron, W.; Austin, R.; Barath, M.; Callihan, E.; Evans, T.; Gamez Guerrero, M.; Hemmati, G.; Jones, A.; Kwant, K. Preclinical and nonclinical characterization of HPN217: A Tri-Specific T Cell Activating Construct (TriTAC) targeting B cell maturation antigen (BCMA) for the treatment of multiple myeloma. Blood 2018, 132, 3225. [Google Scholar] [CrossRef]
- Costa, F.; Dalla Palma, B.; Giuliani, N. CD38 Expression by Myeloma Cells and Its Role in the Context of Bone Marrow Microenvironment: Modulation by Therapeutic Agents. Cells 2019, 8, 1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chillemi, A.; Zaccarello, G.; Quarona, V.; Lazzaretti, M.; Martella, E.; Giuliani, N.; Ferracini, R.; Pistoia, V.; Horenstein, A.L.; Malavasi, F. CD38 and bone marrow microenvironment. Front. Biosci. 2014, 19, 152–162. [Google Scholar] [CrossRef]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef] [Green Version]
- Lonial, S.; Weiss, B.M.; Usmani, S.Z.; Singhal, S.; Chari, A.; Bahlis, N.J.; Belch, A.; Krishnan, A.; Vescio, R.A.; Mateos, M.V.; et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): An open-label, randomised, phase 2 trial. Lancet 2016, 387, 1551–1560. [Google Scholar] [CrossRef]
- Frerichs, K.A.; Nagy, N.A.; Lindenbergh, P.L.; Bosman, P.; Marin Soto, J.; Broekmans, M.; Groen, R.W.J.; Themeli, M.; Nieuwenhuis, L.; Stege, C.; et al. CD38-targeting antibodies in multiple myeloma: Mechanisms of action and clinical experience. Expert Rev. Clin. Immunol. 2018, 14, 197–206. [Google Scholar] [CrossRef]
- Zuch de Zafra, C.L.; Fajardo, F.; Zhong, W.; Bernett, M.J.; Muchhal, U.S.; Moore, G.L.; Stevens, J.; Case, R.; Pearson, J.T.; Liu, S.; et al. Targeting Multiple Myeloma with AMG 424, a Novel Anti-CD38/CD3 Bispecific T-cell-recruiting Antibody Optimized for Cytotoxicity and Cytokine Release. Clin. Cancer Res. 2019, 25, 3921–3933. [Google Scholar] [CrossRef] [Green Version]
- Richter, J.R.; Landgren, C.O.; Kauh, J.S.; Back, J.; Salhi, Y.; Reddy, V.; Bayever, E.; BERDEJA, J. Phase 1, multicenter, open-label study of single-agent bispecific antibody T-cell engager GBR 1342 in relapsed/refractory multiple myeloma. J. Clin. Oncol. 2018, 36, TPS3132. [Google Scholar] [CrossRef]
- Ollier, R.; Hou, S.; Lissilaa, R.; Skegro, D.; Back, J. CD3/CD38 T Cell Retargeting Hetero-Dimeric Immunoglobulins and Methods of Their Production. U.S. Patent Application 16/159,231, 12 October 2019. [Google Scholar]
- Elkins, K.; Zheng, B.; Go, M.; Slaga, D.; Du, C.; Scales, S.J.; Yu, S.F.; McBride, J.; de Tute, R.; Rawstron, A.; et al. FcRL5 as a target of antibody-drug conjugates for the treatment of multiple myeloma. Mol. Cancer Ther. 2012, 11, 2222–2232. [Google Scholar] [CrossRef] [Green Version]
- Polson, A.G.; Zheng, B.; Elkins, K.; Chang, W.; Du, C.; Dowd, P.; Yen, L.; Tan, C.; Hongo, J.A.; Koeppen, H.; et al. Expression pattern of the human FcRH/IRTA receptors in normal tissue and in B-chronic lymphocytic leukemia. Int. Immunol. 2006, 18, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- Ise, T.; Nagata, S.; Kreitman, R.J.; Wilson, W.H.; Wayne, A.S.; Stetler-Stevenson, M.; Bishop, M.R.; Scheinberg, D.A.; Rassenti, L.; Kipps, T.J.; et al. Elevation of soluble CD307 (IRTA2/FcRH5) protein in the blood and expression on malignant cells of patients with multiple myeloma, chronic lymphocytic leukemia, and mantle cell lymphoma. Leukemia 2007, 21, 169–174. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Smith, E.L.; Liu, C. Chimeric Antigen Receptors Targeting fc Receptor-Like 5 and Uses Thereof. U.S. Patent Application US15/614,108, 5 June 2017. [Google Scholar]
- Ovacik, A.M.; Li, J.; Lemper, M.; Danilenko, D.; Stagg, N.; Mathieu, M.; Ellerman, D.; Gupta, V.; Kalia, N.; Nguy, T.; et al. Single cell-produced and in vitro-assembled anti-FcRH5/CD3 T-cell dependent bispecific antibodies have similar in vitro and in vivo properties. MAbs 2019, 11, 422–433. [Google Scholar] [CrossRef] [Green Version]
- Depoil, D.; Fleire, S.; Treanor, B.L.; Weber, M.; Harwood, N.E.; Marchbank, K.L.; Tybulewicz, V.L.; Batista, F.D. CD19 is essential for B cell activation by promoting B cell receptor-antigen microcluster formation in response to membrane-bound ligand. Nat. Immunol. 2008, 9, 63–72. [Google Scholar] [CrossRef]
- Hajek, R.; Okubote, S.A.; Svachova, H. Myeloma stem cell concepts, heterogeneity and plasticity of multiple myeloma. Br. J. Haematol. 2013, 163, 551–564. [Google Scholar] [CrossRef]
- Tembhare, P.R.; Yuan, C.M.; Venzon, D.; Braylan, R.; Korde, N.; Manasanch, E.; Zuchlinsky, D.; Calvo, K.; Kurlander, R.; Bhutani, M.; et al. Flow cytometric differentiation of abnormal and normal plasma cells in the bone marrow in patients with multiple myeloma and its precursor diseases. Leuk. Res. 2014, 38, 371–376. [Google Scholar] [CrossRef] [Green Version]
- Boucher, K.; Parquet, N.; Widen, R.; Shain, K.; Baz, R.; Alsina, M.; Koomen, J.; Anasetti, C.; Dalton, W.; Perez, L.E. Stemness of B-cell progenitors in multiple myeloma bone marrow. Clin. Cancer Res. 2012, 18, 6155–6168. [Google Scholar] [CrossRef] [Green Version]
- Jen, E.Y.; Xu, Q.; Schetter, A.; Przepiorka, D.; Shen, Y.L.; Roscoe, D.; Sridhara, R.; Deisseroth, A.; Philip, R.; Farrell, A.T.; et al. FDA Approval: Blinatumomab for Patients with B-cell Precursor Acute Lymphoblastic Leukemia in Morphologic Remission with Minimal Residual Disease. Clin. Cancer Res. 2019, 25, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Dufner, V.; Sayehli, C.M.; Chatterjee, M.; Hummel, H.D.; Gelbrich, G.; Bargou, R.C.; Goebeler, M.E. Long-term outcome of patients with relapsed/refractory B-cell non-Hodgkin lymphoma treated with blinatumomab. Blood Adv. 2019, 3, 2491–2498. [Google Scholar] [CrossRef] [Green Version]
- Gharbaran, R. Advances in the molecular functions of syndecan-1 (SDC1/CD138) in the pathogenesis of malignancies. Crit. Rev. Oncol./Hematol. 2015, 94, 1–17. [Google Scholar] [CrossRef]
- Hayashida, K.; Bartlett, A.H.; Chen, Y.; Park, P.W. Molecular and cellular mechanisms of ectodomain shedding. Anat. Rec. 2010, 293, 925–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhmetzyanova, I.; McCarron, M.J.; Parekh, S.; Chesi, M.; Bergsagel, P.L.; Fooksman, D.R. Dynamic CD138 surface expression regulates switch between myeloma growth and dissemination. Leukemia 2020, 34, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Chen, D.; Zong, Y.; Ye, S.; Tang, J.; Meng, H.; An, G.; Zhang, X.; Yang, L. Immunotherapy based on bispecific T-cell engager with hIgG1 Fc sequence as a new therapeutic strategy in multiple myeloma. Cancer Sci. 2015, 106, 512–521. [Google Scholar] [CrossRef]
- Alhallak, K.; Sun, J.; Wasden, K.; Guenthner, N.; O’Neal, J.; Muz, B.; King, J.; Kohnen, D.; Vij, R.; Achilefu, S.; et al. Nanoparticle T-cell engagers as a modular platform for cancer immunotherapy. Leukemia 2021. [Google Scholar] [CrossRef]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef]
- Yan, Z.; Cao, J.; Cheng, H.; Qiao, J.; Zhang, H.; Wang, Y.; Shi, M.; Lan, J.; Fei, X.; Jin, L.; et al. A combination of humanised anti-CD19 and anti-BCMA CAR T cells in patients with relapsed or refractory multiple myeloma: A single-arm, phase 2 trial. Lancet. Haematol. 2019, 6, e521–e529. [Google Scholar] [CrossRef]
- Xu, J.; Chen, L.J.; Yang, S.S.; Sun, Y.; Wu, W.; Liu, Y.F.; Xu, J.; Zhuang, Y.; Zhang, W.; Weng, X.Q.; et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proc. Natl. Acad. Sci. USA 2019, 116, 9543–9551. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.A.; Shi, V.; Maric, I.; Wang, M.; Stroncek, D.F.; Rose, J.J.; Brudno, J.N.; Stetler-Stevenson, M.; Feldman, S.A.; Hansen, B.G.; et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016, 128, 1688–1700. [Google Scholar] [CrossRef]
- Zhao, W.H.; Liu, J.; Wang, B.Y.; Chen, Y.X.; Cao, X.M.; Yang, Y.; Zhang, Y.L.; Wang, F.X.; Zhang, P.Y.; Lei, B.; et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J. Hematol. Oncol. 2018, 11, 141. [Google Scholar] [CrossRef]
- Ding, L.; Hu, Y.; Huang, H. Novel progresses of chimeric antigen receptor (CAR) T cell therapy in multiple myeloma. Stem Cell Investig. 2021, 8, 1. [Google Scholar] [CrossRef]
- Bensinger, W.I.; Hege, K.; Burgess, M.R.; Kelly, L.M.; Boss, I.; Gaudy, A.; Lardelli, P.; Sarmiento, R.; Li, S.; San-Miguel, J.; et al. First Clinical Study of the B-Cell Maturation Antigen (BCMA) 2+1 T Cell Engager (TCE) CC-93269 in Patients (Pts) with Relapsed/Refractory Multiple Myeloma (RRMM): Interim Results of a Phase 1 Multicenter Trial. Blood 2019, 134, 143. [Google Scholar] [CrossRef]
- Raje, N.S.; Jakubowiak, A.; Gasparetto, C.; Cornell, R.F.; Krupka, H.I.; Navarro, D.; Forgie, A.J.; Udata, C.; Basu, C.; Chou, J.; et al. Safety, Clinical Activity, Pharmacokinetics, and Pharmacodynamics from a Phase I Study of PF-06863135, a B-Cell Maturation Antigen (BCMA)-CD3 Bispecific Antibody, in Patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2019, 134, 1869. [Google Scholar] [CrossRef]
- Healthcare, G. Trials of Pfizer’s Promising Bispecific Antibody Paused Due to Side Effect Concerns. Available online: https://www.clinicaltrialsarena.com/comment/pfizer-bispecific-antibody-multiple-myeloma (accessed on 25 May 2021).
- Wudhikarn, K.; Palomba, M.L.; Pennisi, M.; Garcia-Recio, M.; Flynn, J.R.; Devlin, S.M.; Afuye, A.; Silverberg, M.L.; Maloy, M.A.; Shah, G.L.; et al. Infection during the first year in patients treated with CD19 CAR T cells for diffuse large B cell lymphoma. Blood Cancer J. 2020, 10, 79. [Google Scholar] [CrossRef]
- Martinelli, G.; Boissel, N.; Chevallier, P.; Ottmann, O.; Gökbuget, N.; Topp, M.S.; Fielding, A.K.; Rambaldi, A.; Ritchie, E.K.; Papayannidis, C.; et al. Complete Hematologic and Molecular Response in Adult Patients With Relapsed/Refractory Philadelphia Chromosome–Positive B-Precursor Acute Lymphoblastic Leukemia Following Treatment With Blinatumomab: Results From a Phase II, Single-Arm, Multicenter Study. J. Clin. Oncol. 2017, 35, 1795–1802. [Google Scholar] [CrossRef] [PubMed]
- Lum, L.G.; Thakur, A.; Elhakiem, A.; Alameer, L.; Dinning, E.; Huang, M. Anti-CS1 x Anti-CD3 Bispecific Antibody (BiAb)-Armed Anti-CD3 Activated T Cells (CS1-BATs) Kill CS1(+) Myeloma Cells and Release Type-1 Cytokines. Front. Oncol. 2020, 10, 544. [Google Scholar] [CrossRef] [PubMed]
- Horn, L.A.; Ciavattone, N.G.; Atkinson, R.; Woldergerima, N.; Wolf, J.; Clements, V.K.; Sinha, P.; Poudel, M.; Ostrand-Rosenberg, S. CD3xPDL1 bi-specific T cell engager (BiTE) simultaneously activates T cells and NKT cells, kills PDL1(+) tumor cells, and extends the survival of tumor-bearing humanized mice. Oncotarget 2017, 8, 57964–57980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lameris, R.; Shahine, A.; Pellicci, D.G.; Uldrich, A.P.; Gras, S.; Le Nours, J.; Groen, R.W.J.; Vree, J.; Reddiex, S.J.J.; Quiñones-Parra, S.M.; et al. A single-domain bispecific antibody targeting CD1d and the NKT T-cell receptor induces a potent antitumor response. Nat. Cancer 2020, 1, 1054–1065. [Google Scholar] [CrossRef]
- Reusch, U.; Duell, J.; Ellwanger, K.; Herbrecht, C.; Knackmuss, S.H.; Fucek, I.; Eser, M.; McAleese, F.; Molkenthin, V.; Gall, F.L.; et al. A tetravalent bispecific TandAb (CD19/CD3), AFM11, efficiently recruits T cells for the potent lysis of CD19(+) tumor cells. MAbs 2015, 7, 584–604. [Google Scholar] [CrossRef] [Green Version]
- Chichili, G.R.; Huang, L.; Li, H.; Burke, S.; He, L.; Tang, Q.; Jin, L.; Gorlatov, S.; Ciccarone, V.; Chen, F.; et al. A CD3xCD123 bispecific DART for redirecting host T cells to myelogenous leukemia: Preclinical activity and safety in nonhuman primates. Sci. Transl. Med. 2015, 7, 289ra82. [Google Scholar] [CrossRef]
- Zhong, X.; D’Antona, A.M. Recent Advances in the Molecular Design and Applications of Multispecific Biotherapeutics. Antibodies 2021, 10, 13. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| BiTE Name | BiTE Target | Phase | Status | Clinical Trial Number | Completion Date |
|---|---|---|---|---|---|
| Blinatumomab | CD19 | I | Terminated | NCT03173430 | 2019 |
| AMG 424 | CD38 | I/II | Recruiting | NCT03445663 | 2022 |
| GBR 1342 | CD38 | I/II | Recruiting | NCT03309111 | 2021 |
| BFCR4350A | FcRH5 | I | Recruiting | NCT03275103 | 2022 |
| AMG 420 | BCMA | I | Completed/ Active | NCT02514239/ NCT03836053 | 2020/ 2025 |
| AMG 701 | BCMA | I/II | Recruiting | NCT03287908 | 2025 |
| CC-93269 | BCMA | I | Recruiting | NCT03486067 | 2026 |
| Elranatamab (PF-06863135) | BCMA | I/II | Recruiting/ Active | NCT03269136/ NCT04649359 | 2023 |
| REGN5458 | BCMA | I/II | Recruiting | NCT03761108 | 2022 |
| TNB-383B | BCMA | I | Recruiting | NCT03933735 | 2021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhallak, K.; Sun, J.; Jeske, A.; Park, C.; Yavner, J.; Bash, H.; Lubben, B.; Adebayo, O.; Khaskiah, A.; Azab, A.K. Bispecific T Cell Engagers for the Treatment of Multiple Myeloma: Achievements and Challenges. Cancers 2021, 13, 2853. https://doi.org/10.3390/cancers13122853
Alhallak K, Sun J, Jeske A, Park C, Yavner J, Bash H, Lubben B, Adebayo O, Khaskiah A, Azab AK. Bispecific T Cell Engagers for the Treatment of Multiple Myeloma: Achievements and Challenges. Cancers. 2021; 13(12):2853. https://doi.org/10.3390/cancers13122853
Chicago/Turabian StyleAlhallak, Kinan, Jennifer Sun, Amanda Jeske, Chaelee Park, Jessica Yavner, Hannah Bash, Berit Lubben, Ola Adebayo, Ayah Khaskiah, and Abdel Kareem Azab. 2021. "Bispecific T Cell Engagers for the Treatment of Multiple Myeloma: Achievements and Challenges" Cancers 13, no. 12: 2853. https://doi.org/10.3390/cancers13122853
APA StyleAlhallak, K., Sun, J., Jeske, A., Park, C., Yavner, J., Bash, H., Lubben, B., Adebayo, O., Khaskiah, A., & Azab, A. K. (2021). Bispecific T Cell Engagers for the Treatment of Multiple Myeloma: Achievements and Challenges. Cancers, 13(12), 2853. https://doi.org/10.3390/cancers13122853