
ctDNA to Guide Adjuvant Therapy in Localized Colorectal Cancer (CRC)


Abstract
:Simple Summary
Abstract
1. Introduction
Current Perspectives of Liquid Biopsy in Colorectal Cancer (CRC)
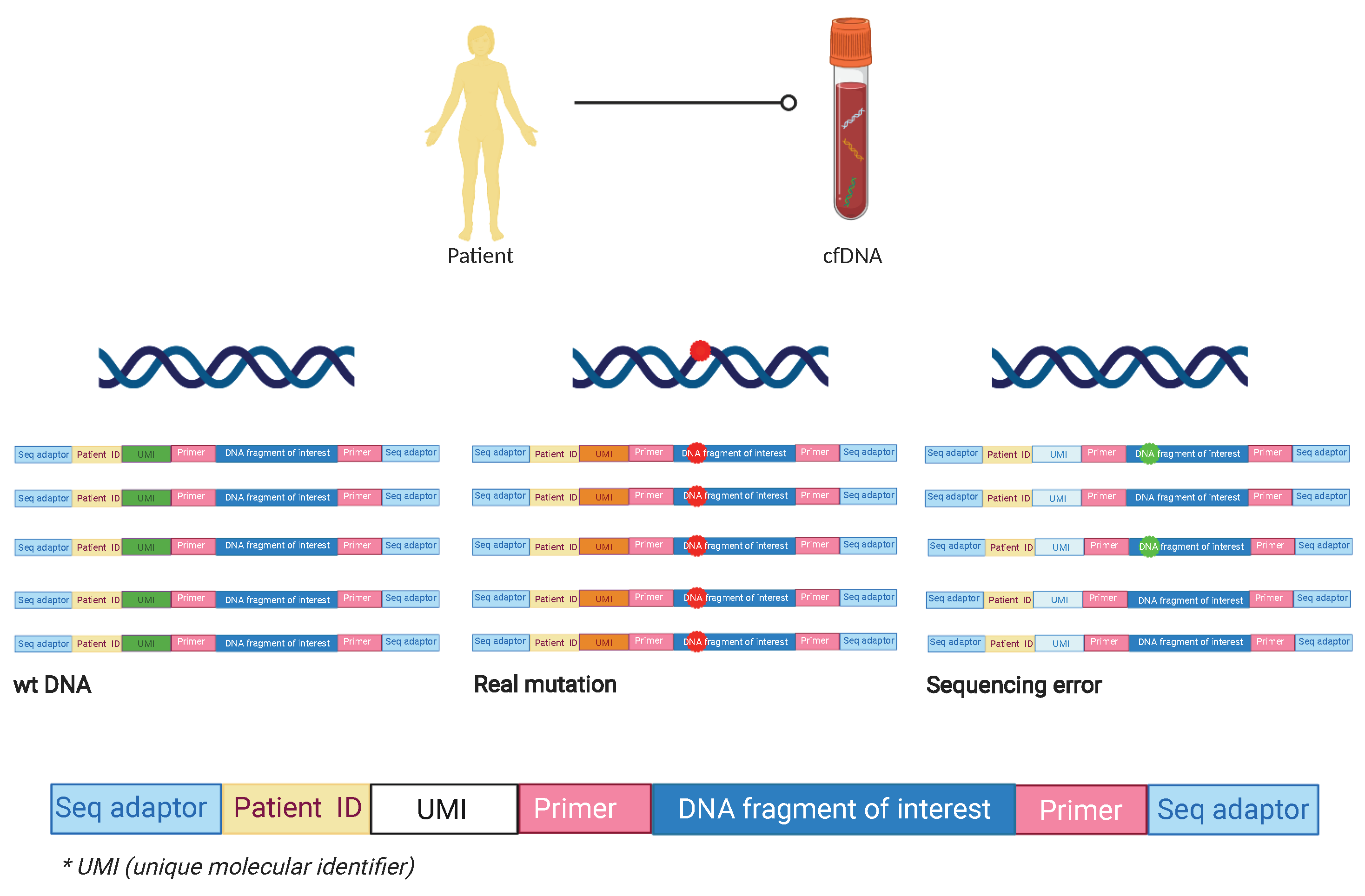
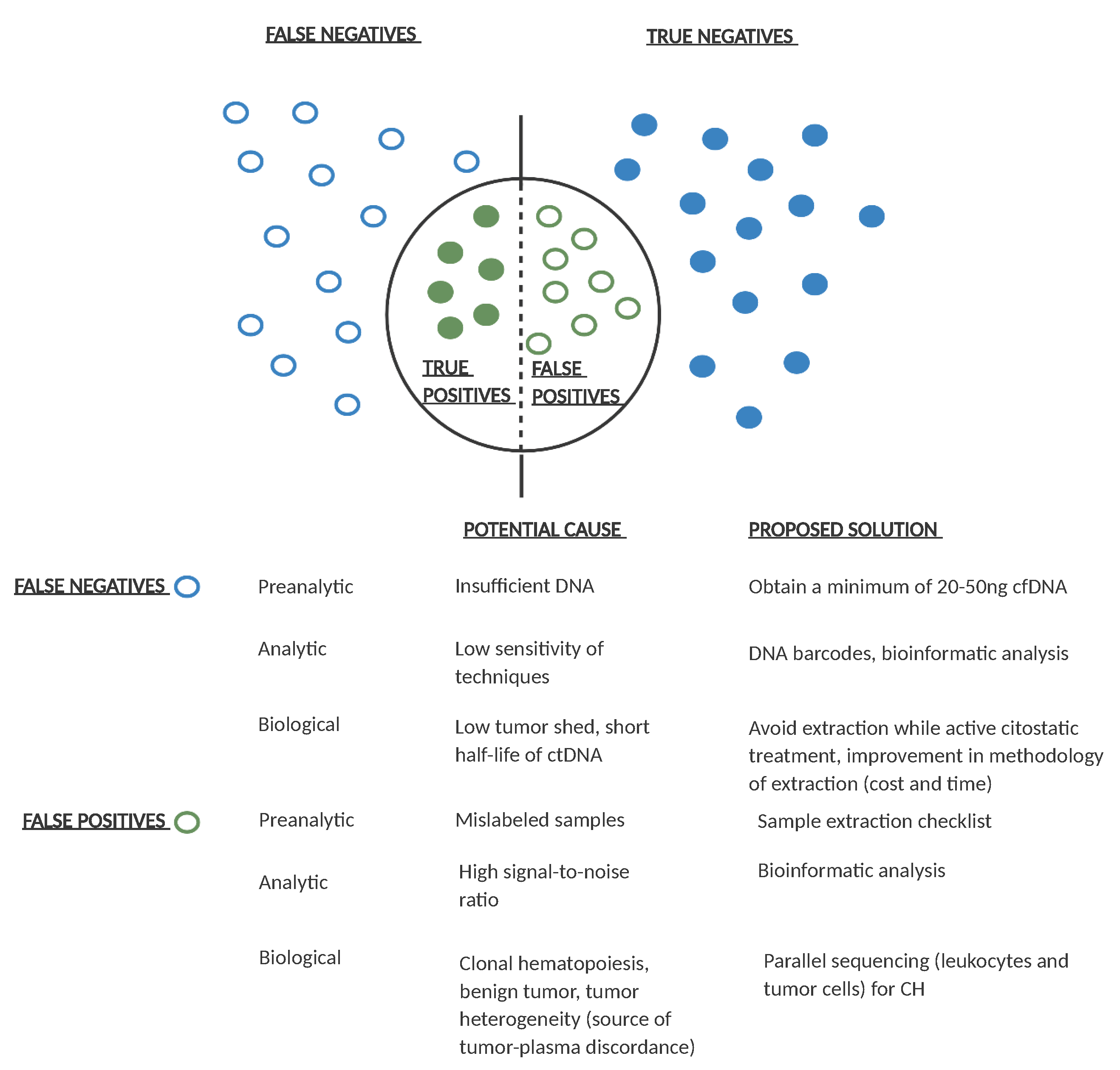
2. Technical Approaches in Detecting MRD
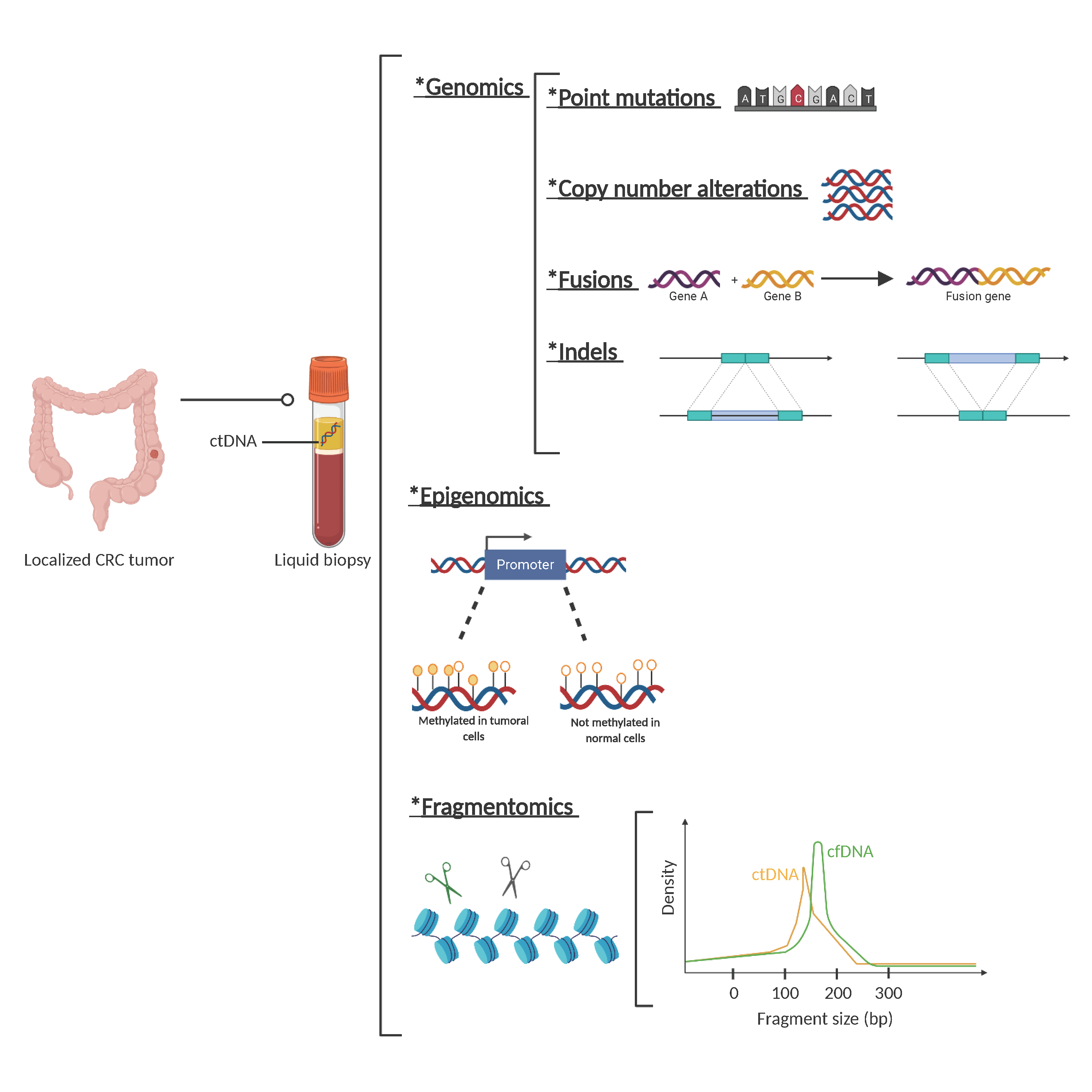
3. ctDNA Detection in Localized CRC
3.1. Stage II Colon Cancer
3.2. Stage III Colon Cancer
3.3. Locally Advanced Rectal Cancer (LARC)
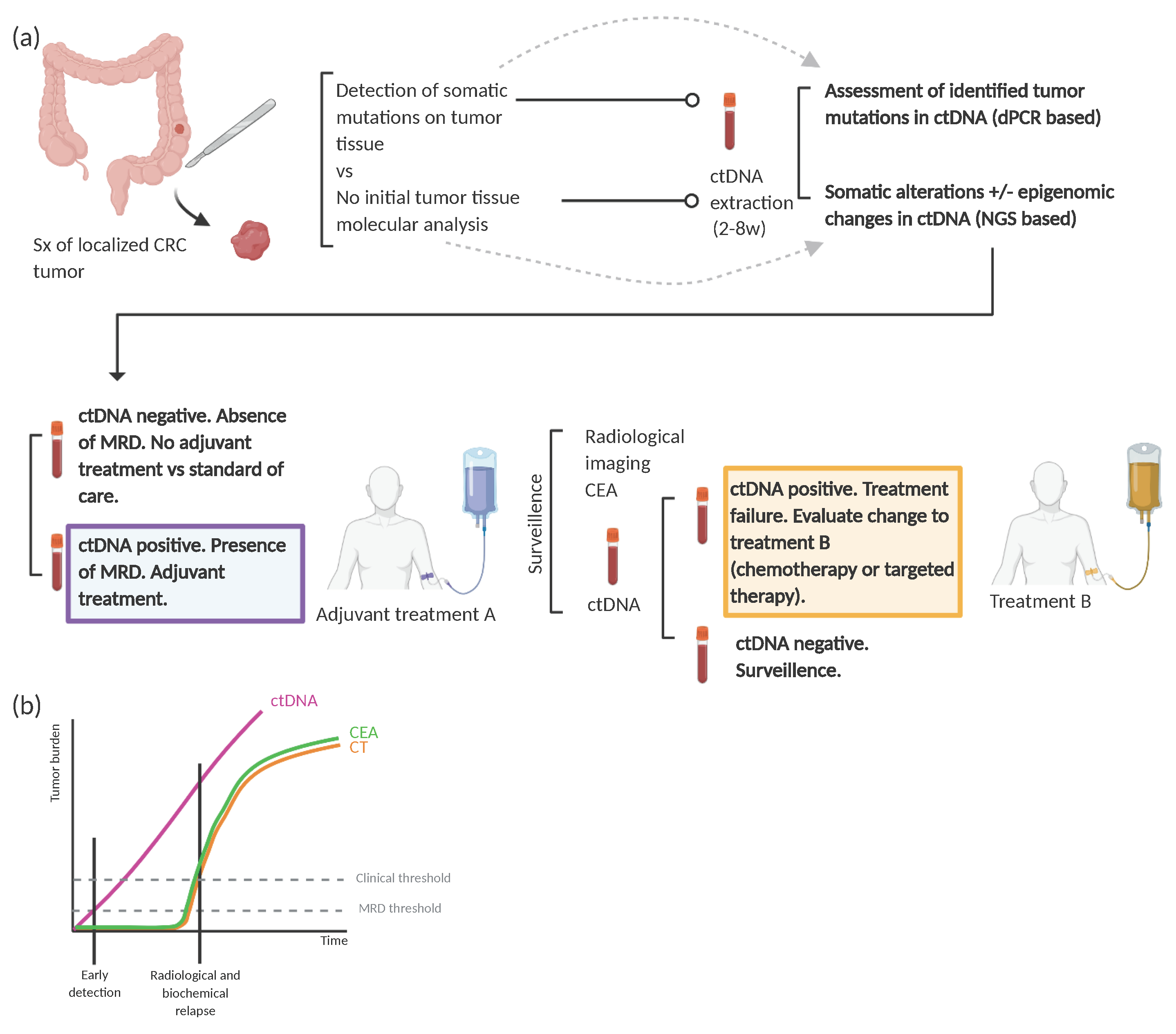
4. ctDNA as a Post-Treatment Surveillance Strategy
5. ctDNA Based Clinical Trials
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argilés, G.; Tabernero, J.; Labianca, R.; Hochhauser, D.; Salazar, R.; Iveson, T.; Laurent-Puig, P.; Quirke, P.; Yoshino, T.; Taieb, J.; et al. Localised Colon Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2020, S0923753420399324. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; Atkin, W.; Lenz, H.-J.; Lynch, H.T.; Minsky, B.; Nordlinger, B.; Starling, N. Colorectal Cancer. Lancet 2010, 375, 18. [Google Scholar] [CrossRef]
- Schmoll, H.-J.; Twelves, C.; Sun, W.; O’Connell, M.J.; Cartwright, T.; McKenna, E.; Saif, M.; Lee, S.; Yothers, G.; Haller, D. Effect of Adjuvant Capecitabine or Fluorouracil, with or without Oxaliplatin, on Survival Outcomes in Stage III Colon Cancer and the Effect of Oxaliplatin on Post-Relapse Survival: A Pooled Analysis of Individual Patient Data from Four Randomised Controlled Trials. Lancet Oncol. 2014, 15, 1481–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaniboni, A.; Labianca, R.; Marsoni, S.; Torri, V.; Mosconi, P.; Grilli, R.; Apolone, G.; Cifani, S.; Tinazzi, A. A Randomized Trial of Adjuvant 5-Fluorouracil and Folinic Acid Administered to Patients with Colon Carcinoma—Long Term Results and Evaluation of the Indicators of Health-Related Quality of Life. Cancer 1998, 82, 2135–2144. [Google Scholar] [CrossRef]
- International Multicentre Pooled Analysis of Colon Cancer Trials (IMPACT) Investigators. Efficacy of Adjuvant Fluorouracil and Folinic Acid in Colon Cancer. Lancet 1995, 345, 939–944. [Google Scholar] [CrossRef]
- Bahadoer, R.R.; Dijkstra, E.A.; van Etten, B.; Marijnen, C.A.M.; Putter, H.; Kranenbarg, E.M.-K.; Roodvoets, A.G.H.; Nagtegaal, I.D.; Beets-Tan, R.G.H.; Blomqvist, L.K.; et al. Short-Course Radiotherapy Followed by Chemotherapy before Total Mesorectal Excision (TME) versus Preoperative Chemoradiotherapy, TME, and Optional Adjuvant Chemotherapy in Locally Advanced Rectal Cancer (RAPIDO): A Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2021, 22, 29–42. [Google Scholar] [CrossRef]
- Conroy, T.; Lamfichekh, N.; Etienne, P.-L.; Rio, E.; Francois, E.; Mesgouez-Nebout, N.; Vendrely, V.; Artignan, X.; Bouché, O.; Gargot, D.; et al. Total Neoadjuvant Therapy with MFOLFIRINOX versus Preoperative Chemoradiation in Patients with Locally Advanced Rectal Cancer: Final Results of PRODIGE 23 Phase III Trial, a UNICANCER GI Trial. J. Clin. Oncol. 2020, 38, 4007. [Google Scholar] [CrossRef]
- Hong, Y.S.; Kim, S.Y.; Lee, J.S.; Nam, B.-H.; Kim, K.; Kim, J.E.; Park, Y.S.; Park, J.O.; Baek, J.Y.; Kim, T.-Y.; et al. Oxaliplatin-Based Adjuvant Chemotherapy for Rectal Cancer After Preoperative Chemoradiotherapy (ADORE): Long-Term Results of a Randomized Controlled Trial. J. Clin. Oncol. 2019, 37, 3111–3123. [Google Scholar] [CrossRef] [PubMed]
- Breugom, A.J.; Swets, M.; Bosset, J.-F.; Collette, L.; Sainato, A.; Cionini, L.; Glynne-Jones, R.; Counsell, N.; Bastiaannet, E.; van den Broek, C.B.M.; et al. Adjuvant Chemotherapy after Preoperative (Chemo)Radiotherapy and Surgery for Patients with Rectal Cancer: A Systematic Review and Meta-Analysis of Individual Patient Data. Lancet Oncol. 2015, 16, 200–207. [Google Scholar] [CrossRef]
- Henricks, L.M.; Lunenburg, C.A.T.C.; de Man, F.M.; Meulendijks, D.; Frederix, G.W.J.; Kienhuis, E.; Creemers, G.-J.; Baars, A.; Dezentjé, V.O.; Imholz, A.L.T.; et al. DPYD Genotype-Guided Dose Individualisation of Fluoropyrimidine Therapy in Patients with Cancer: A Prospective Safety Analysis. Lancet Oncol. 2018, 19, 1459–1467. [Google Scholar] [CrossRef]
- Argyriou, A.A.; Polychronopoulos, P.; Iconomou, G.; Chroni, E.; Kalofonos, H.P. A Review on Oxaliplatin-Induced Peripheral Nerve Damage. Cancer Treat. Rev. 2008, 34, 368–377. [Google Scholar] [CrossRef]
- Grothey, A. Oxaliplatin-Safety Profile: Neurotoxicity. Semin. Oncol. 2003, 30, 5–13. [Google Scholar] [CrossRef]
- Mouliere, F.; Robert, B.; Arnau Peyrotte, E.; Del Rio, M.; Ychou, M.; Molina, F.; Gongora, C.; Thierry, A.R. High Fragmentation Characterizes Tumour-Derived Circulating DNA. PLoS ONE 2011, 6, e23418. [Google Scholar] [CrossRef]
- Mouliere, F.; El Messaoudi, S.; Pang, D.; Dritschilo, A.; Thierry, A.R. Multi-Marker Analysis of Circulating Cell-Free DNA toward Personalized Medicine for Colorectal Cancer. Mol. Oncol. 2014, 8, 927–941. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Durkee, K.H.; Moore, K.J.; Goodman, S.N.; Shuber, A.P.; Kinzler, K.W.; Vogelstein, B. Analysis of Mutations in DNA Isolated from Plasma and Stool of Colorectal Cancer Patients. Gastroenterology 2008, 135, 489–498.e7. [Google Scholar] [CrossRef] [Green Version]
- De Mattos-Arruda, L.; Mayor, R.; Ng, C.K.Y.; Weigelt, B.; Martínez-Ricarte, F.; Torrejon, D.; Oliveira, M.; Arias, A.; Raventos, C.; Tang, J.; et al. Cerebrospinal Fluid-Derived Circulating Tumour DNA Better Represents the Genomic Alterations of Brain Tumours than Plasma. Nat. Commun. 2015, 6, 8839. [Google Scholar] [CrossRef] [Green Version]
- Kimura, H.; Fujiwara, Y.; Sone, T.; Kunitoh, H.; Tamura, T.; Kasahara, K.; Nishio, K. EGFR Mutation Status in Tumour-Derived DNA from Pleural Effusion Fluid Is a Practical Basis for Predicting the Response to Gefitinib. Br. J. Cancer 2006, 95, 1390–1395. [Google Scholar] [CrossRef]
- Wang, Y.; Springer, S.; Mulvey, C.L.; Silliman, N.; Schaefer, J.; Sausen, M.; James, N.; Rettig, E.M.; Guo, T.; Pickering, C.R.; et al. Detection of Somatic Mutations and HPV in the Saliva and Plasma of Patients with Head and Neck Squamous Cell Carcinomas. Sci. Transl. Med. 2015, 7, 293ra104. [Google Scholar] [CrossRef] [Green Version]
- Reckamp, K.L.; Melnikova, V.O.; Karlovich, C.; Sequist, L.V.; Camidge, D.R.; Wakelee, H.; Perol, M.; Oxnard, G.R.; Kosco, K.; Croucher, P.; et al. A Highly Sensitive and Quantitative Test Platform for Detection of NSCLC EGFR Mutations in Urine and Plasma. J. Thorac. Oncol. 2016, 11, 1690–1700. [Google Scholar] [CrossRef] [Green Version]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [Green Version]
- Vidal, J.; Muinelo, L.; Dalmases, A.; Jones, F.; Edelstein, D.; Iglesias, M.; Orrillo, M.; Abalo, A.; Rodríguez, C.; Brozos, E.; et al. Plasma CtDNA RAS Mutation Analysis for the Diagnosis and Treatment Monitoring of Metastatic Colorectal Cancer Patients. Ann. Oncol. 2017, 28, 1325–1332. [Google Scholar] [CrossRef]
- Tie, J.; Kinde, I.; Wang, Y.; Wong, H.L.; Roebert, J.; Christie, M.; Tacey, M.; Wong, R.; Singh, M.; Karapetis, C.S.; et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann. Oncol. 2015, 26, 1715–1722. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating Mutant DNA to Assess Tumor Dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Avanzini, S. A Mathematical Model of CtDNA Shedding Predicts Tumor Detection Size_Science Advances 2020. Sci. Adv. 2020, 6, eabc4308. [Google Scholar] [CrossRef]
- Holdhoff, M.; Schmidt, K.; Donehower, R.; Diaz, L.A. Analysis of Circulating Tumor DNA to Confirm Somatic KRAS Mutations. JNCI J. Natl. Cancer Inst. 2009, 101, 1284–1285. [Google Scholar] [CrossRef]
- Diehl, F.; Li, M.; Dressman, D.; He, Y.; Shen, D.; Szabo, S.; Diaz, L.A.; Goodman, S.N.; David, K.A.; Juhl, H.; et al. Detection and Quantification of Mutations in the Plasma of Patients with Colorectal Tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 16368–16373. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Schwarzenbach, H.; Stoehlmacher, J.; Pantel, K.; Goekkurt, E. Detection and Monitoring of Cell-Free DNA in Blood of Patients with Colorectal Cancer. Ann. N. Y. Acad. Sci. 2008, 1137, 190–196. [Google Scholar] [CrossRef]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.Y.; Kaper, F.; Dawson, S.-J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive Identification and Monitoring of Cancer Mutations by Targeted Deep Sequencing of Plasma DNA. Sci. Transl. Med. 2012, 4, 136ra68. [Google Scholar] [CrossRef]
- Kennedy, S.R.; Schmitt, M.W.; Fox, E.J.; Kohrn, B.F.; Salk, J.J.; Ahn, E.H.; Prindle, M.J.; Kuong, K.J.; Shen, J.-C.; Risques, R.-A.; et al. Detecting Ultralow-Frequency Mutations by Duplex Sequencing. Nat. Protoc. 2014, 9, 2586–2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czeiger, D.; Shaked, G.; Eini, H.; Vered, I.; Belochitski, O.; Avriel, A.; Ariad, S.; Douvdevani, A. Measurement of Circulating Cell-Free DNA Levels by a New Simple Fluorescent Test in Patients with Primary Colorectal Cancer. Am. J. Clin. Pathol. 2011, 135, 264–270. [Google Scholar] [CrossRef]
- Cristofanilli, M. Circulating Tumor Cells Revisited. JAMA 2010, 303, 1092. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jiang, J.; Mostert, B.; Sieuwerts, A.; Martens, J.W.M.; Sleijfer, S.; Foekens, J.A.; Wang, Y. Allele-Specific, Non-Extendable Primer Blocker PCR (AS-NEPB-PCR) for DNA Mutation Detection in Cancer. J. Mol. Diagn. 2013, 15, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Freidin, M.B.; Freydina, D.V.; Leung, M.; Montero Fernandez, A.; Nicholson, A.G.; Lim, E. Circulating Tumor DNA Outperforms Circulating Tumor Cells for KRAS Mutation Detection in Thoracic Malignancies. Clin. Chem. 2015, 61, 1299–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glenn, T.C. Field Guide to Next-Generation DNA Sequencers: Field Guide to Next-Gen Sequencers. Mol. Ecol. Resour. 2011, 11, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Meddeb, R.; Pisareva, E.; Thierry, A.R. Guidelines for the Preanalytical Conditions for Analyzing Circulating Cell-Free DNA. Clin. Chem. 2019, 65, 623–633. [Google Scholar] [CrossRef]
- Van Dessel, L.F.; Beije, N.; Helmijr, J.C.A.; Vitale, S.R.; Kraan, J.; Look, M.P.; de Wit, R.; Sleijfer, S.; Jansen, M.P.H.M.; Martens, J.W.M.; et al. Application of Circulating Tumor DNA in Prospective Clinical Oncology Trials—Standardization of Preanalytical Conditions. Mol. Oncol. 2017, 11, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, R.B.; Chabner, B.A. Application of Cell-Free DNA Analysis to Cancer Treatment. N. Engl. J. Med. 2018, 379, 1754–1765. [Google Scholar] [CrossRef] [Green Version]
- Elazezy, M.; Joosse, S.A. Techniques of Using Circulating Tumor DNA as a Liquid Biopsy Component in Cancer Management. Comput. Struct. Biotechnol. J. 2018, 16, 370–378. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal Hematopoiesis of Indeterminate Potential and Its Distinction from Myelodysplastic Syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Razavi, P.; Li, B.T.; Brown, D.N.; Jung, B.; Hubbell, E.; Shen, R.; Abida, W.; Juluru, K.; De Bruijn, I.; Hou, C.; et al. High-Intensity Sequencing Reveals the Sources of Plasma Circulating Cell-Free DNA Variants. Nat. Med. 2019, 25, 1928–1937. [Google Scholar] [CrossRef]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-Free DNA Comprises an In Vivo Nucleosome Footprint That Informs Its Tissues-Of-Origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive Human Cell-Type Methylation Atlas Reveals Origins of Circulating Cell-Free DNA in Health and Disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef] [Green Version]
- Garrigou, S.; Perkins, G.; Garlan, F.; Normand, C.; Didelot, A.; Le Corre, D.; Peyvandi, S.; Mulot, C.; Niarra, R.; Aucouturier, P.; et al. A Study of Hypermethylated Circulating Tumor DNA as a Universal Colorectal Cancer Biomarker. Clin. Chem. 2016, 62, 1129–1139. [Google Scholar] [CrossRef] [Green Version]
- Taieb, J.; Taly, V.; Vernerey, D.; Bourreau, C.; Bennouna, J.; Faroux, R.; Desrame, J.; Bouche, O.; Borg, C.; Egreteau, J.; et al. Analysis of Circulating Tumour DNA (CtDNA) from Patients Enrolled in the IDEA-FRANCE Phase III Trial: Prognostic and Predictive Value for Adjuvant Treatment Duration. Ann. Oncol. 2019, 30, v867. [Google Scholar] [CrossRef]
- Artieri, C.; Axelrod, H.; Baca, A.; Burke, J.; Chudova, D.; Dahdouli, M.; Ghadiri, F.; Hartwig, A.; He, Y.; Hite, D.; et al. Analytical Validation of a Tissue Agnostic CtDNA MRD Assay Using Tumor Specific Methylation and Somatic Variant Profiles in Early-Stage CRC. J. Clin. Oncol. 2020, 38, e15549. [Google Scholar] [CrossRef]
- Keller, L.; Belloum, Y.; Wikman, H.; Pantel, K. Clinical Relevance of Blood-Based CtDNA Analysis: Mutation Detection and Beyond. Br. J. Cancer 2021, 124, 345–358. [Google Scholar] [CrossRef]
- Danyi, A.; Jager, M.; de Ridder, J. Cancer Type Classification in Liquid Biopsies Based on Sparse Mutational Profiles Enabled through Data Augmentation and Integration. Bioinformatics 2021, in press. [Google Scholar]
- Bachet, J.B.; Bouché, O.; Taieb, J.; Dubreuil, O.; Garcia, M.L.; Meurisse, A.; Normand, C.; Gornet, J.M.; Artru, P.; Louafi, S.; et al. RAS Mutation Analysis in Circulating Tumor DNA from Patients with Metastatic Colorectal Cancer: The AGEO RASANC Prospective Multicenter Study. Ann. Oncol. 2018, 29, 1211–1219. [Google Scholar] [CrossRef]
- García-Foncillas, J.; Tabernero, J.; Élez, E.; Aranda, E.; Benavides, M.; Camps, C.; Jantus-Lewintre, E.; López, R.; Muinelo-Romay, L.; Montagut, C.; et al. Prospective Multicenter Real-World RAS Mutation Comparison between OncoBEAM-Based Liquid Biopsy and Tissue Analysis in Metastatic Colorectal Cancer. Br. J. Cancer 2018, 119, 1464–1470. [Google Scholar] [CrossRef]
- Garcia-Murillas, I.; Schiavon, G.; Weigelt, B.; Ng, C.; Hrebien, S.; Cutts, R.J.; Cheang, M.; Osin, P.; Nerurkar, A.; Kozarewa, I.; et al. Mutation Tracking in Circulating Tumor DNA Predicts Relapse in Early Breast Cancer. Sci. Transl. Med. 2015, 7, 302ra133. [Google Scholar] [CrossRef]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An Ultrasensitive Method for Quantitating Circulating Tumor DNA with Broad Patient Coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Kruger, S.; Heinemann, V.; Ross, C.; Diehl, F.; Nagel, D.; Ormanns, S.; Liebmann, S.; Prinz-Bravin, I.; Westphalen, C.B.; Haas, M.; et al. Repeated MutKRAS CtDNA Measurements Represent a Novel and Promising Tool for Early Response Prediction and Therapy Monitoring in Advanced Pancreatic Cancer. Ann. Oncol. 2018, 29, 2348–2355. [Google Scholar] [CrossRef]
- Lee, R.J.; Gremel, G.; Marshall, A.; Myers, K.A.; Fisher, N.; Dunn, J.A.; Dhomen, N.; Corrie, P.G.; Middleton, M.R.; Lorigan, P.; et al. Circulating Tumor DNA Predicts Survival in Patients with Resected High-Risk Stage II/III Melanoma. Ann. Oncol. 2018, 29, 490–496. [Google Scholar] [CrossRef] [Green Version]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.-L.; Christie, M.; et al. Circulating Tumor DNA Analysis Detects Minimal Residual Disease and Predicts Recurrence in Patients with Stage II Colon Cancer. Sci. Transl. Med. 2016, 8, 346ra92. [Google Scholar] [CrossRef] [Green Version]
- The TRACERx Consortium; The PEACE Consortium; Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; et al. Phylogenetic CtDNA Analysis Depicts Early-Stage Lung Cancer Evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef]
- Dudley, J.C.; Schroers-Martin, J.; Lazzareschi, D.V.; Shi, W.Y.; Chen, S.B.; Esfahani, M.S.; Trivedi, D.; Chabon, J.J.; Chaudhuri, A.A.; Stehr, H.; et al. Detection and Surveillance of Bladder Cancer Using Urine Tumor DNA. Cancer Discov. 2019, 9, 500–509. [Google Scholar] [CrossRef]
- Wang, Y.; Li, L.; Cohen, J.D.; Kinde, I.; Ptak, J.; Popoli, M.; Schaefer, J.; Silliman, N.; Dobbyn, L.; Tie, J.; et al. Prognostic Potential of Circulating Tumor DNA Measurement in Postoperative Surveillance of Nonmetastatic Colorectal Cancer. JAMA Oncol. 2019, 5, 1118. [Google Scholar] [CrossRef]
- Edge, S.B.; Compton, C.C. The American Joint Committee on Cancer: The 7th Edition of the AJCC Cancer Staging Manual and the Future of TNM. Ann. Surg. Oncol. 2010, 17, 1471–1474. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.D.; Delorenzi, M.; Tejpar, S.; Yan, P.; Klingbiel, D.; Fiocca, R.; d’Ario, G.; Cisar, L.; Labianca, R.; Cunningham, D.; et al. Integrated Analysis of Molecular and Clinical Prognostic Factors in Stage II/III Colon Cancer. JNCI J. Natl. Cancer Inst. 2012, 104, 1635–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, K.O.; Hawkins, A.T.; Krishnamurthy, D.M.; Dharmarajan, S.; Glasgow, S.C.; Hunt, S.R.; Mutch, M.G.; Wise, P.; Silviera, M.L. Omission of Adjuvant Chemotherapy Is Associated with Increased Mortality in Patients With T3N0 Colon Cancer With Inadequate Lymph Node Harvest. Dis. Colon Rectum 2017, 60, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Benatti, P.; Gafà, R.; Barana, D.; Marino, M.; Scarselli, A.; Pedroni, M.; Maestri, I.; Guerzoni, L.; Roncucci, L.; Menigatti, M.; et al. Microsatellite Instability and Colorectal Cancer Prognosis. Clin. Cancer Res. 2005, 11, 8332–8340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jover, R.; Zapater, P.; Castells, A.; Llor, X.; Andreu, M.; Cubiella, J.; Balaguer, F.; Sempere, L.; Xicola, R.M.; Bujanda, L.; et al. The Efficacy of Adjuvant Chemotherapy with 5-Fluorouracil in Colorectal Cancer Depends on the Mismatch Repair Status. Eur. J. Cancer 2009, 45, 365–373. [Google Scholar] [CrossRef]
- Carethers, J.M.; Smith, E.J.; Behling, C.A.; Nguyen, L.; Tajima, A.; Doctolero, R.T.; Cabrera, B.L.; Goel, A.; Arnold, C.A.; Miyai, K.; et al. Use of 5-Fluorouracil and Survival in Patients with Microsatellite-Unstable Colorectal Cancer. Gastroenterology 2004, 126, 394–401. [Google Scholar] [CrossRef]
- Quah, H.-M.; Chou, J.F.; Gonen, M.; Shia, J.; Schrag, D.; Landmann, R.G.; Guillem, J.G.; Paty, P.B.; Temple, L.K.; Wong, W.D.; et al. Identification of Patients with High-Risk Stage II Colon Cancer for Adjuvant Therapy. Dis. Colon. Rectum 2008, 51, 503–507. [Google Scholar] [CrossRef]
- Niedzwiecki, D.; Bertagnolli, M.M.; Warren, R.S.; Compton, C.C.; Kemeny, N.E.; Benson, A.B.; Eckhardt, S.G.; Alberts, S.; Porjosh, G.N.; Kerr, D.J.; et al. Documenting the Natural History of Patients With Resected Stage II Adenocarcinoma of the Colon After Random Assignment to Adjuvant Treatment With Edrecolomab or Observation: Results From CALGB 9581. J. Clin. Oncol. 2011, 29, 3146–3152. [Google Scholar] [CrossRef] [Green Version]
- Bozic, I.; Reiter, J.G.; Allen, B.; Antal, T.; Chatterjee, K.; Shah, P.; Moon, Y.S.; Yaqubie, A.; Kelly, N.; Le, D.T.; et al. Evolutionary Dynamics of Cancer in Response to Targeted Combination Therapy. eLife 2013, 2, e00747. [Google Scholar] [CrossRef]
- Montagut, C.; Siravegna, G.; Bardelli, A. Liquid Biopsies to Evaluate Early Therapeutic Response in Colorectal Cancer. Ann. Oncol. 2015, 26, 1525–1527. [Google Scholar] [CrossRef]
- Thierry, A.; Corrado, B.; Lamia, M.-B.; Matilde, N.; Josep, T.; Tamas, H.; Clare, T.; Marta, Z.; Philip, C.; John, B.; et al. Oxaliplatin, Fluorouracil, and Leucovorin as Adjuvant Treatment for Colon Cancer. N. Engl. J. Med. 2004, 350, 2343–2351. [Google Scholar]
- Påhlman, L.A.; Hohenberger, W.M.; Matzel, K.; Sugihara, K.; Quirke, P.; Glimelius, B. Should the Benefit of Adjuvant Chemotherapy in Colon Cancer Be Re-Evaluated? J. Clin. Oncol. 2016, 34, 1297–1299. [Google Scholar] [CrossRef] [Green Version]
- Böckelman, C.; Engelmann, B.E.; Kaprio, T.; Hansen, T.F.; Glimelius, B. Risk of Recurrence in Patients with Colon Cancer Stage II and III: A Systematic Review and Meta-Analysis of Recent Literature. Acta Oncol. 2015, 54, 5–16. [Google Scholar] [CrossRef]
- Lash, T.L.; Riis, A.H.; Ostenfeld, E.B.; Erichsen, R.; Vyberg, M.; Ahern, T.P.; Thorlacius-Ussing, O. Associations of Statin Use With Colorectal Cancer Recurrence and Mortality in a Danish Cohort. Am. J. Epidemiol. 2017, 186, 679–687. [Google Scholar] [CrossRef]
- Osterman, E.; Glimelius, B. Recurrence Risk After Up-to-Date Colon Cancer Staging, Surgery, and Pathology: Analysis of the Entire Swedish Population. Dis. Colon Rectum 2018, 61, 1016–1025. [Google Scholar] [CrossRef]
- Grothey, A.; Sobrero, A.F.; Shields, A.F.; Yoshino, T.; Paul, J.; Taieb, J.; Souglakos, J.; Shi, Q.; Kerr, R.; Labianca, R.; et al. Duration of Adjuvant Chemotherapy for Stage III Colon Cancer. N. Engl. J. Med. 2018, 378, 1177–1188. [Google Scholar] [CrossRef]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.-T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients with Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124. [Google Scholar] [CrossRef] [Green Version]
- Tie, J.; Cohen, J.D.; Wang, Y.; Christie, M.; Simons, K.; Lee, M.; Wong, R.; Kosmider, S.; Ananda, S.; McKendrick, J.; et al. Circulating Tumor DNA Analyses as Markers of Recurrence Risk and Benefit of Adjuvant Therapy for Stage III Colon Cancer. JAMA Oncol. 2019, 5, 1710. [Google Scholar] [CrossRef]
- Tie, J. Prognostic Significance of Postsurgery CtDNA in Nonmetastatic CRC Individual Patient Pooled Analysis of Three Cohort Studies. Int. J. Cancer 2020, 148, 1014–1026. [Google Scholar] [CrossRef]
- Tarazona, N.; Gimeno-Valiente, F.; Gambardella, V.; Zuñiga, S.; Rentero-Garrido, P.; Huerta, M.; Roselló, S.; Martinez-Ciarpaglini, C.; Carbonell-Asins, J.A.; Carrasco, F.; et al. Targeted Next-Generation Sequencing of Circulating-Tumor DNA for Tracking Minimal Residual Disease in Localized Colon Cancer. Ann. Oncol. 2019, 30, 1804–1812. [Google Scholar] [CrossRef] [Green Version]
- Schøler, L.V.; Reinert, T.; Ørntoft, M.-B.W.; Kassentoft, C.G.; Árnadóttir, S.S.; Vang, S.; Nordentoft, I.; Knudsen, M.; Lamy, P.; Andreasen, D.; et al. Clinical Implications of Monitoring Circulating Tumor DNA in Patients with Colorectal Cancer. Clin. Cancer Res. 2017, 23, 5437–5445. [Google Scholar] [CrossRef] [Green Version]
- Maas, M.; Nelemans, P.J.; Valentini, V.; Das, P.; Rödel, C.; Kuo, L.-J.; Calvo, F.A.; García-Aguilar, J.; Glynne-Jones, R.; Haustermans, K.; et al. Long-Term Outcome in Patients with a Pathological Complete Response after Chemoradiation for Rectal Cancer: A Pooled Analysis of Individual Patient Data. Lancet Oncol. 2010, 11, 835–844. [Google Scholar] [CrossRef]
- Janjan, N.A.; Khoo, V.S.; Abbruzzese, J.; Pazdur, R.; Dubrow, R.; Cleary, K.R.; Allen, P.K.; Lynch, P.M.; Glober, G.; Wolff, R.; et al. Tumor Downstaging and Sphincter Preservation with Preoperative Chemoradiation in Locally Advanced Rectal Cancer: The M. D. Anderson Cancer Center Experience. Int. J. Radiat. Oncol. 1999, 44, 1027–1038. [Google Scholar] [CrossRef]
- Hendren, S.K.; O’Connor, B.I.; Liu, M.; Asano, T.; Cohen, Z.; Swallow, C.J.; MacRae, H.M.; Gryfe, R.; McLeod, R.S. Prevalence of Male and Female Sexual Dysfunction Is High Following Surgery for Rectal Cancer. Ann. Surg. 2005, 242, 212–223. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Wang, Y.; Li, L.; Christie, M.; Simons, K.; Elsaleh, H.; Kosmider, S.; Wong, R.; Yip, D.; et al. Serial Circulating Tumour DNA Analysis during Multimodality Treatment of Locally Advanced Rectal Cancer: A Prospective Biomarker Study. Gut 2019, 68, 663–671. [Google Scholar] [CrossRef]
- Khakoo, S.; Carter, P.D.; Brown, G.; Valeri, N.; Picchia, S.; Bali, M.A.; Shaikh, R.; Jones, T.; Begum, R.; Rana, I.; et al. MRI Tumor Regression Grade and Circulating Tumor DNA as Complementary Tools to Assess Response and Guide Therapy Adaptation in Rectal Cancer. Clin. Cancer Res. 2020, 26, 183–192. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, C.; Lin, G.; Xiao, Y.; Jia, W.; Xiao, G.; Liu, Q.; Wu, B.; Wu, A.; Qiu, H.; et al. Serial Circulating Tumor DNA in Predicting and Monitoring the Effect of Neoadjuvant Chemoradiotherapy in Patients with Rectal Cancer: A Prospective Multicenter Study. Clin. Cancer Res. 2021, 27, 301–310. [Google Scholar] [CrossRef]
- Murahashi, S.; Akiyoshi, T.; Sano, T.; Fukunaga, Y.; Noda, T.; Ueno, M.; Zembutsu, H. Serial Circulating Tumour DNA Analysis for Locally Advanced Rectal Cancer Treated with Preoperative Therapy: Prediction of Pathological Response and Postoperative Recurrence. Br. J. Cancer 2020, 123, 803–810. [Google Scholar] [CrossRef]
- Pazdirek, F.; Minarik, M.; Benesova, L.; Halkova, T.; Belsanova, B.; Macek, M.; Stepanek, L.; Hoch, J. Monitoring of Early Changes of Circulating Tumor DNA in the Plasma of Rectal Cancer Patients Receiving Neoadjuvant Concomitant Chemoradiotherapy: Evaluation for Prognosis and Prediction of Therapeutic Response. Front. Oncol. 2020, 10, 1028. [Google Scholar] [CrossRef]
- Vidal, J.; Casadevall, D.; Bellosillo, B.; Pericay, C.; Garcia-Carbonero, R.; Losa, F.; Layos, L.; Alonso, V.; Capdevila, J.; Gallego, J.; et al. Clinical Impact of Presurgery Circulating Tumor DNA after Total Neoadjuvant Treatment in Locally Advanced Rectal Cancer: A Biomarker Study from the GEMCAD 1402 Trial. Clin. Cancer Res. 2021, 27. [Google Scholar] [CrossRef]
- Snyder, R.A.; Hu, C.-Y.; Cuddy, A.; Francescatti, A.B.; Schumacher, J.R.; Van Loon, K.; You, Y.N.; Kozower, B.D.; Greenberg, C.C.; Schrag, D.; et al. Association Between Intensity of Posttreatment Surveillance Testing and Detection of Recurrence in Patients with Colorectal Cancer. JAMA 2018, 319, 2104. [Google Scholar] [CrossRef]
- Wille-Jørgensen, P.; Syk, I.; Smedh, K.; Laurberg, S.; Nielsen, D.T.; Petersen, S.H.; Renehan, A.G.; Horváth-Puhó, E.; Påhlman, L.; Sørensen, H.T.; et al. Effect of More vs Less Frequent Follow-up Testing on Overall and Colorectal Cancer–Specific Mortality in Patients With Stage II or III Colorectal Cancer: The COLOFOL Randomized Clinical Trial. JAMA 2018, 319, 2095. [Google Scholar] [CrossRef] [PubMed]
- Coakley, M.; Garcia-Murillas, I.; Turner, N.C. Molecular Residual Disease and Adjuvant Trial Design in Solid Tumors. Clin. Cancer Res. 2019, 25, 6026–6034. [Google Scholar] [CrossRef] [PubMed]
- Merker, J.D.; Oxnard, G.R.; Compton, C.; Diehn, M.; Hurley, P.; Lazar, A.J.; Lindeman, N.; Lockwood, C.M.; Rai, A.J.; Schilsky, R.L.; et al. Circulating Tumor DNA Analysis in Patients with Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J. Clin. Oncol. 2018, 36, 1631–1641. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Fowler, J.C.; Wabik, A.; Lawson, A.R.J.; Abascal, F.; Hall, M.W.J.; Cagan, A.; Murai, K.; Mahbubani, K.; Stratton, M.R.; et al. Somatic Mutant Clones Colonize the Human Esophagus with Age. Science 2018, 362, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Reinert, T.; Schøler, L.V.; Thomsen, R.; Tobiasen, H.; Vang, S.; Nordentoft, I.; Lamy, P.; Kannerup, A.-S.; Mortensen, F.V.; Stribolt, K.; et al. Analysis of Circulating Tumour DNA to Monitor Disease Burden Following Colorectal Cancer Surgery. Gut 2016, 65, 625–634. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Sample Size | Study Population | Timepoint of ctDNA Collection | ctDNA Detection Assay | Post-op ctDNA Detection Rate | RFS Post-op ctDNA+ vs. ctDNA− |
|---|---|---|---|---|---|---|
| Tie et al. [56] | 230 | Stage II CC | Weeks 4–10 post-op | Safe-SeqS (1 variant; 15 genes) | 8.7% | 18 (95% CI 7.9–40) p < 0.001 |
| Taieb et al. [46] | 805 | Stage III CC | NA | ddPCR (2 methylated markers) | 13.5% | 1.85 (95% CI 1.31–2.61) p < 0.001 |
| Wang et al. [59] | 58 | Stage I–III CRC | Week 4 post-op | Safe-SeqS (1 variant; 15 genes) | 22.4% | Recurrence-free at 49 months: 33% vs. 100% (non-compared) |
| Reinert et al. [76] | 130 | Stage I–III CRC | Week 4 post-op | Multiplex PCR based NGS assay (SignateraTM) | 10.6% | 7.2 (95% CI 2.7–19.0) p < 0.001 |
| Tie et al. [77] | 96 | Stage III CC (all chemo) | Weeks 4–10 post-op | Safe SeqS (1 variant; 15 genes) | 21% | 3.8 (95% CI 2.4–21.0) p < 0.001 |
| Tie et al. [78] | 485 | Stage II–III CRC and LARC | Weeks 4–10 post-op | Safe-SeqS (1 variant; 15 genes) | 12% | Recurrence-free at 5 years: 38.6% vs. 85.5% p < 0.001 |
| Tarazona et al. [79] | 69 | Stage I–III CC | Weeks 6–8 post-op | ddPCR (2 variants; 29 genes) | 20.3% | 6.96 (95% CI 2.57–18.91) p < 0.001 |
| Scholer et al. [80] | 21 | Stage I–III CRC | Weeks 1–4 post-op | ddPCR (NA) | 28.5% | 37.7 (95% CI 4.2–335.5) p < 0.001 |
| Study | Sample Size | Study Population | Timepoint of ctDNA Collection | ctDNA Detection Assay | Baseline ctDNA Detection Rate | Pre-op ctDNA Detection Rate | Post-op ctDNA Detection Rate | Main Results for ctDNA+ vs. ctDNA− |
|---|---|---|---|---|---|---|---|---|
| Tie et al. [84] | 159 | LARC | Pre-treatment (CRT), weeks 4–6 post-CRT, and weeks 4–10 post-op | Safe-SeqS (1 variant; 15 genes) | 77% | 8.3% | 12% | Post-CRT RFS: HR 6.6 (95% CI 2.6–17) p < 0.001 Post-op RFS: HR 13 (95% CI 5.5–31) p < 0.001 |
| Khakoo et al. [85] | 47 | Localized rectal cancer | Pre-treatment (CRT), mid-CRT, post-CRT, and weeks 4–12 post-op | ddPCR (up to 3 variants; 6 genes) | 74% | 21% | 13% | Post-CRT MFS: 7.1 (95% CI 2.4–21.5) p < 0.001 Post-op DFS: 39.9 (95% CI 4.0–399.5) p = 0.002 |
| Zhou et al. [86] | 104 | LARC | Pre-treatment (CRT), 1 week from the start of treatment, post-CRT, and 4 weeks post-op | HiSeq 3000 Sequencing System (IlluminaTM). Panel of 1021 genes | 75% | 10.5% | 6.7% | Post-CRT MFS:19.82 (95% CI 2.029–193.7) p < 0.001 Post-op MFS: 25.30 (95% CI, 1.475–434) p < 0.001 |
| Murahashi et al. [87] | 85 | LARC | Pre-treatment (CRT), post-CRT, and 12 weeks post-op | Oncomine CRC (14 genes) | 57.6% | 22.3% | NA | Post-op RFS: 17.1 (95% CI, 1.0–282) p < 0.001 |
| Pazdirek et al. [88] | 36 | LARC | Pre-treatment (CRT), 1 week from the start of treatment | Denaturing capillary electrophoresis (DCE) and High sensitivity Beaming assay | 21.2% | NA | NA | Prior CRT: reduction DFS by 1.47 years (p = 0.015) and OS by 1.41 years (p = 0.010) |
| Vidal et al. [89] | 62 | LARC | Pre-treatment (TNT) and post-CRT (48 h pre-op) | LUNAR-1 | 83% | 15% | NA | Post-CRT RFS: HR 4.029 (95% CI, 1.004–16.16) p = 0.033 Post-CRT OS: HR 23(95% CI, 2.4–212) p < 0.0001 |
| Trial Name | Sample Size | Study Design | Study Population | Timepoint of ctDNA Collection | ctDNA Detection Assay | Design | ctDNA Treatment Intervention |
|---|---|---|---|---|---|---|---|
| DYNAMIC (ACTRN12615000381583) Australia/NZ | 450 | NA | Stage II CRC | Week 4 post-op | Safe-SeqS (1 variant; 15 genes) | Randomization 1:1 SOC- vs. ctDNA− guided treatment | ctDNA+: 5FU-based regimen ± oxaliplatin for 3–6 months; ctDNA− no chemotherapy |
| DYNAMIC III (ACTRN12617001566325) Australia/NZ | 1000 | Phase II/III | Stage III CC | Weeks 5–6 post-op | Safe-SeqS (1 variant; 15 genes) | Randomization 1:1 SOC- vs. ctDNA− guided treatment | ctDNA+: escalated chemotherapy regimen from pre-planned treatment (increase duration or number of agents); ctDNA−: de-escalated chemotherapy regimen from pre-planned treatment (reduction in duration or number of agents) |
| DYNAMIC RECTAL (ACTRN12617001560381) Australia/NZ | 408 | NA | LARC | Week 4 post-op | Safe-SeqS (1 variant; 15 genes) | Randomization 1:1 SOC vs. ctDNA guided | ctDNA+: adjuvant chemotherapy ctDNA− and ypN0: surveillance ctDNA− and ypN+ surveillance or adjuvant chemotherapy at clinician’s choice |
| CIRCULATE (NCT04089631) Germany | 4812 | Phase III | Stage II CRC | Week 5 post-op | NGS (NA) | ctDNA+ randomization 2:1 chemotherapy vs. follow-up | Capecitabine × 6 months vs. follow-up |
| CIRCULATE-PRODIGE 70 (NCT04120701) France | 198 | Phase III | Stage II (pT3-pT4aN0) CRC | Week 2 post-op | ddPCR (2 methylated markers) | ctDNA+ randomization 2:1 chemotherapy vs. follow-up | mFOLFOX (× 6 months) vs. follow-up |
| COBRA (NCT04068103) US | 1408 | Phase II/III | Stage II (low risk CC) | NA | Guardant LUNAR-1 (NA) | Randomization 1:1 surveillance- vs. ctDNA− guided treatment | ctDNA+: CAPOX or FOLFOX vs. ctDNA−: no chemotherapy |
| TRACC (NCT04050345) UK | 1000 | Observational study | Stage II/III CRC | Weeks 4–8 post-op | Customized NGS panel (NA) | Randomization 1:1 SOC- vs. ctDNA− guided treatment | ctDNA+: SOC ctDNA−: de-escalate treatment (from 3 months CAPOX to 3 months Capecitabine and from 6 months capecitabine to no chemotherapy) but re-escalate if ctDNA becomes positive at 3 months |
| (UMIN000039205) Japan | 1240 | NA | High-risk stage II/low-risk III CRC | Week 4 post-op | Signatera-PCR NGS assay (16 specific somatic variants) | ctDNA− randomization SOC vs. no treatment | ctDNA− Randomization SOC vs. no treatment |
| MEDOCC-CrEATE (NL6281/NTR6455) Netherlands | 1320 | NA | Stage II (low risk CC) | Weeks 1–3 post-op | PGDx elio (panel of more than 30 genes) | Randomization 1:1 SOC- vs. ctDNA− guided treatment | ctDNA+: 6 months CAPOX or FOLFOX; ctDNA−: no chemotherapy |
| PEGASUS (NCT04259944) Italy and Spain | 140 | Phase II | High-risk stage II/III CC | Weeks 2–4 post-op | Guardant LUNAR-1 (NA) | ctDNA− guided treatment | ctDNA+: CAPOX × 3 months → 2nd ctDNA+ switch FOLFIRI; secind ctDNA− capecitabine × 3 month ctDNA−: capecitabine × 6 months. 2nd ctDNA+ → CAPOX × 6 months |
| ACT-3 trial (NCT04259944) | 500 | NA | Stage III CRC | Weeks 3–6 post-op and 3–6 months after adjuvant treatment | Guardant LUNAR-1 (NA) | ctDNA− guided treatment | ctDNA+ after adjuvant treatment: randomized to follow-up or molecular target-directed therapy ctDNA−: follow-up |
| ALTAIR trial (UMIN000039205)Japan | 240 | NA | Stage II/III CRC or stage IV with resectable metastases | 1 month after surgery and 3 months after standard adjuvant treatment | Signatera-PCR NGS assay | Signatera-PCR NGS assay | ctDNA+ after adjuvant treatment: randomized to follow-up or second-line trifluridine/Tipiracil ctDNA−: follow-up |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masfarré, L.; Vidal, J.; Fernández-Rodríguez, C.; Montagut, C. ctDNA to Guide Adjuvant Therapy in Localized Colorectal Cancer (CRC). Cancers 2021, 13, 2869. https://doi.org/10.3390/cancers13122869
Masfarré L, Vidal J, Fernández-Rodríguez C, Montagut C. ctDNA to Guide Adjuvant Therapy in Localized Colorectal Cancer (CRC). Cancers. 2021; 13(12):2869. https://doi.org/10.3390/cancers13122869
Chicago/Turabian StyleMasfarré, Laura, Joana Vidal, Concepción Fernández-Rodríguez, and Clara Montagut. 2021. "ctDNA to Guide Adjuvant Therapy in Localized Colorectal Cancer (CRC)" Cancers 13, no. 12: 2869. https://doi.org/10.3390/cancers13122869
APA StyleMasfarré, L., Vidal, J., Fernández-Rodríguez, C., & Montagut, C. (2021). ctDNA to Guide Adjuvant Therapy in Localized Colorectal Cancer (CRC). Cancers, 13(12), 2869. https://doi.org/10.3390/cancers13122869