
Metabolic Rewiring and the Characterization of Oncometabolites



Abstract
:Simple Summary
Abstract
1. Introduction
2. Metabolomics
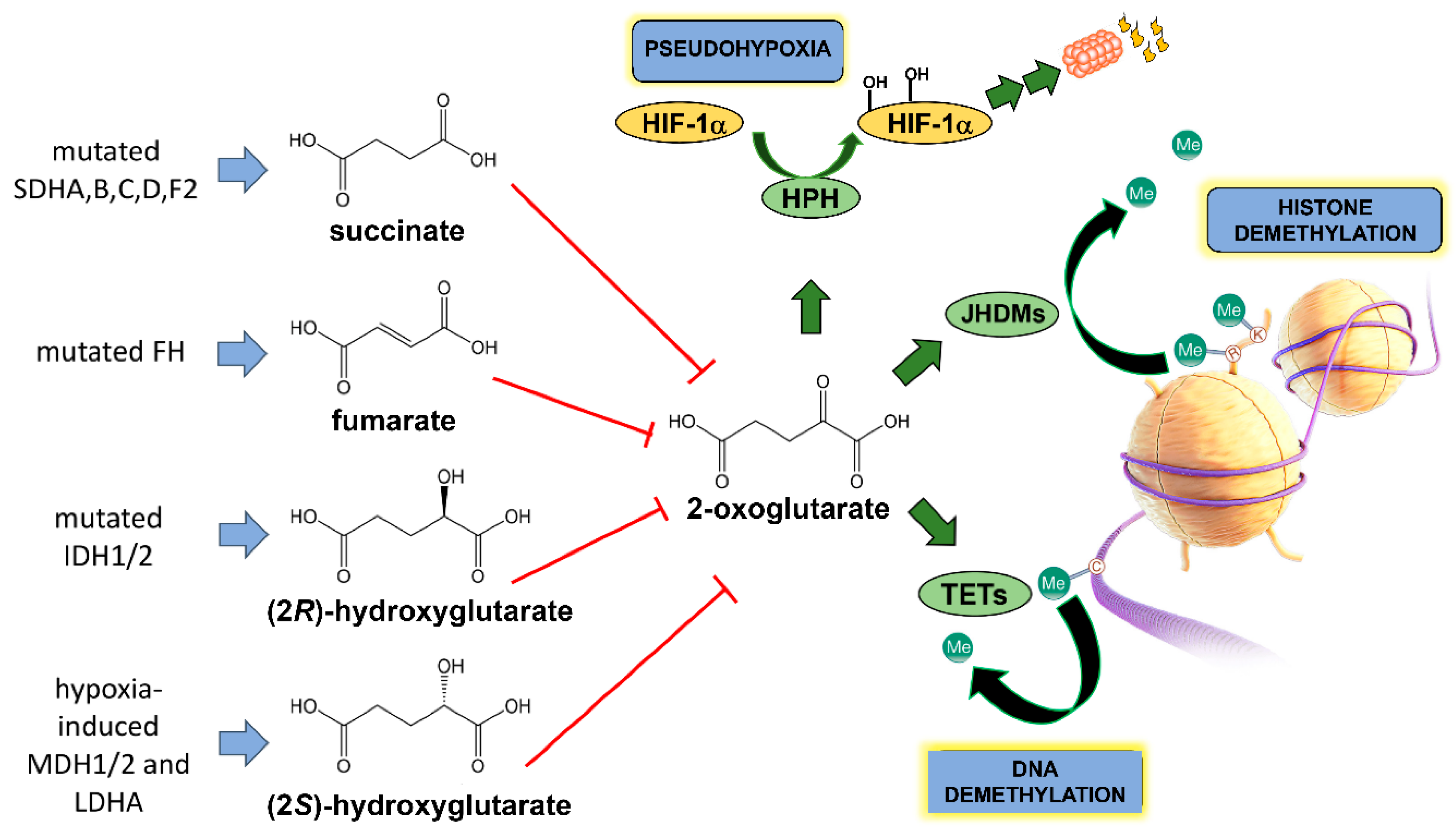
3. TCA Cycle Oncometabolites—Role of Metabolomics
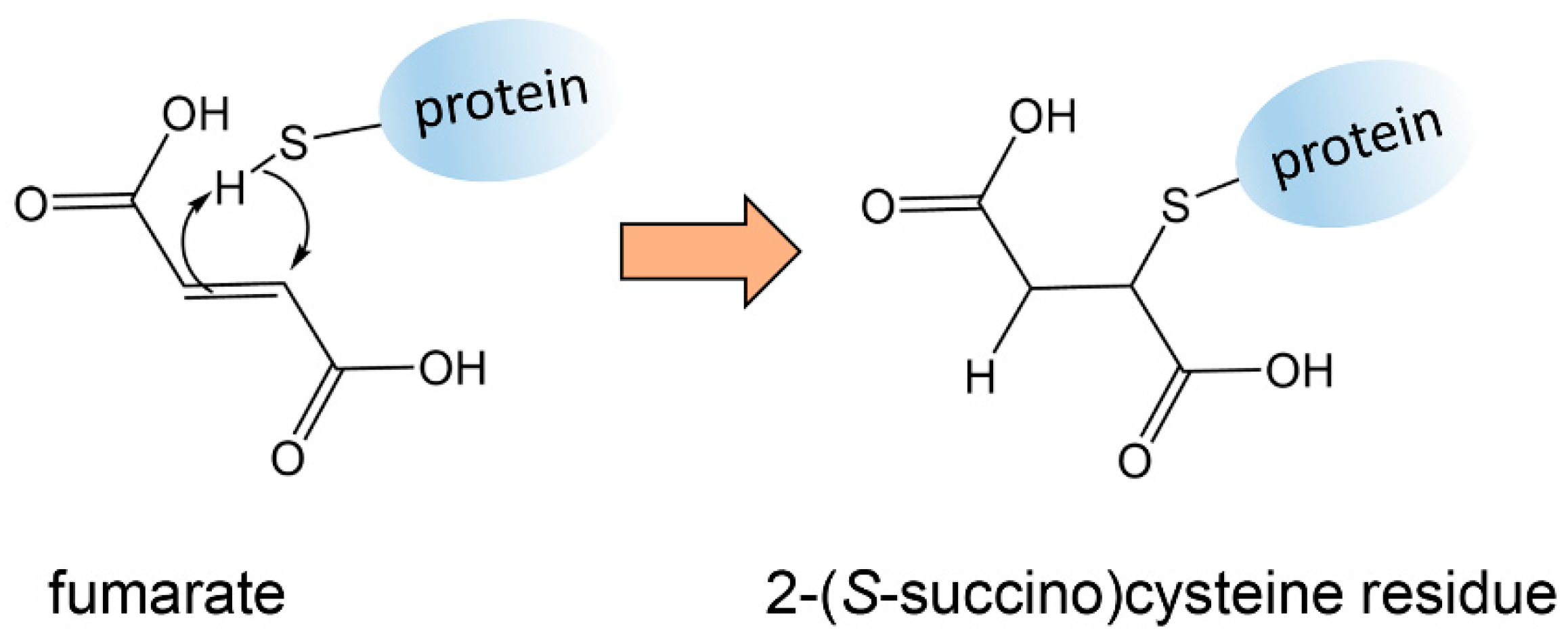
3.1. Fumarate
3.2. Succinate
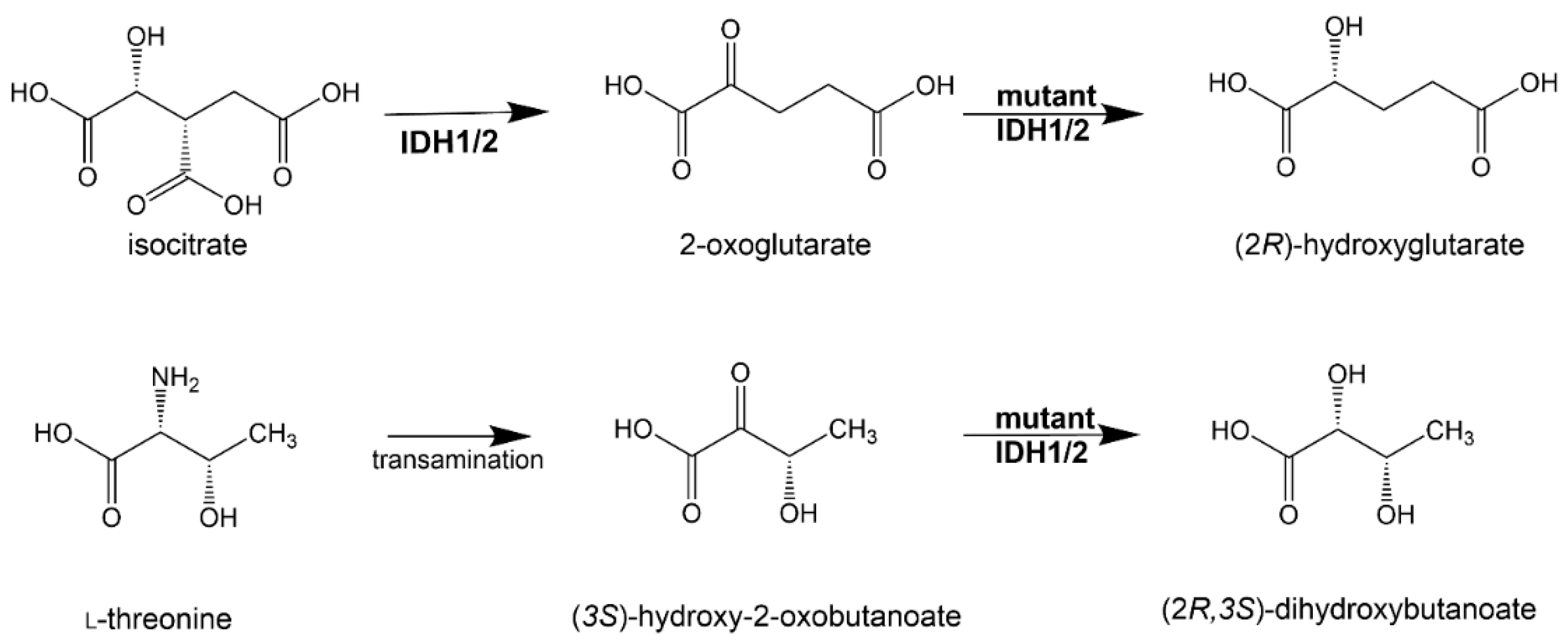
3.3. (2R)-Hydroxyglutarate
3.4. (2S)-Hydroxyglutarate
4. Nontraditional Oncometabolites—The Role of Metabolomics
4.1. Sarcosine
4.2. Glycine
4.3. Hypotaurine
4.4. Lactate
4.5. Kynurenine
4.6. Methylglyoxal
4.7. Miscellaneous
5. Rewiring Cancer Metabolism as a Therapeutic Strategy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Watson, J.D.; Crick, F.H. Genetical implications of the structure of deoxyribonucleic acid. Nature 1953, 171, 964–967. [Google Scholar] [CrossRef]
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Stehelin, D.; Varmus, H.E.; Bishop, J.M.; Vogt, P.K. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature 1976, 260, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Hoagland, M.B.; Stephenson, M.L.; Scott, J.F.; Hecht, L.I.; Zamecnik, P.C. A soluble ribonucleic acid intermediate in protein synthesis. J. Biol. Chem. 1958, 231, 241–257. [Google Scholar] [CrossRef]
- Volkin, E.; Astrachan, L. Phosphorus incorporation in Escherichia coli ribo-nucleic acid after infection with bacteriophage T2. Virology 1956, 2, 149–161. [Google Scholar] [CrossRef]
- Volkin, E. The Function of Rna in T2-Infected Bacteria. Proc. Natl. Acad. Sci. USA 1960, 46, 1336–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palade, G.E. A small particulate component of the cytoplasm. J. Biophys. Biochem. Cytol. 1955, 1, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, R.A. Racing to the Beginning of the Road. The Search for the Origin of Cancer; W.H. Freeman and Company: New York, NY, USA, 1996; p. 274. [Google Scholar]
- Wishart, D.S. Is Cancer a Genetic Disease or a Metabolic Disease? EBioMedicine 2015, 2, 478–479. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O.; Posener, K.; Negelein, E. Ueber den Stoffwechsel der Tumoren. Biochem. Z. 1924, 152, 319–344. [Google Scholar]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, K.M.; Dewhirst, M.W. Tumor metabolism of lactate: The influence and therapeutic potential for MCT and CD147 regulation. Future Oncol. 2010, 6, 127–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, R.R.J.; Patel, K.; Putnam, W.C.; Kapur, P.; Rakheja, D. Oncometabolites: A New Paradigm for Oncology, Metabolism, and the Clinical Laboratory. Clin. Chem. 2017, 63, 1812–1820. [Google Scholar] [CrossRef] [Green Version]
- Morin, A.; Letouze, E.; Gimenez-Roqueplo, A.P.; Favier, J. Oncometabolites-driven tumorigenesis: From genetics to targeted therapy. Int. J. Cancer 2014, 135, 2237–2248. [Google Scholar] [CrossRef]
- Xiang, K.; Jendrossek, V.; Matschke, J. Oncometabolites and the response to radiotherapy. Radiat. Oncol. 2020, 15, 197. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Frezza, C. Oncometabolites: Unconventional triggers of oncogenic signalling cascades. Free Radic. Biol. Med. 2016, 100, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Zdzisinska, B.; Zurek, A.; Kandefer-Szerszen, M. Alpha-Ketoglutarate as a Molecule with Pleiotropic Activity: Well-Known and Novel Possibilities of Therapeutic Use. Arch. Immunol. Ther. Exp. 2017, 65, 21–36. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Soga, T.; Pollard, P.J. Oncometabolites: Linking altered metabolism with cancer. J. Clin. Investig. 2013, 123, 3652–3658. [Google Scholar] [CrossRef] [Green Version]
- Beyoglu, D.; Zhou, Y.; Chen, C.; Idle, J.R. Mass isotopomer-guided decluttering of metabolomic data to visualize endogenous biomarkers of drug toxicity. Biochem. Pharmacol. 2018, 156, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, K.; Dudley, E. Metabolomics and Biomarker Discovery. Adv. Exp. Med. Biol. 2019, 1140, 613–633. [Google Scholar] [CrossRef]
- Beyoglu, D.; Idle, J.R. Metabolomic and Lipidomic Biomarkers for Premalignant Liver Disease Diagnosis and Therapy. Metabolites 2020, 10, 50. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tweeddale, H.; Notley-McRobb, L.; Ferenci, T. Effect of slow growth on metabolism of Escherichia coli, as revealed by global metabolite pool (“metabolome”) analysis. J. Bacteriol. 1998, 180, 5109–5116. [Google Scholar] [CrossRef] [Green Version]
- Fiehn, O. Metabolomics--the link between genotypes and phenotypes. Plant. Mol. Biol 2002, 48, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. ‘Metabonomics’ Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef]
- Fahrner, R.; Beyoglu, D.; Beldi, G.; Idle, J.R. Metabolomic markers for intestinal ischemia in a mouse model. J. Surg. Res. 2012, 178, 879–887. [Google Scholar] [CrossRef] [Green Version]
- Semmo, N.; Weber, T.; Idle, J.R.; Beyoglu, D. Metabolomics reveals that aldose reductase activity due to AKR1B10 is upregulated in hepatitis C virus infection. J. Viral. Hepat. 2015, 22, 617–624. [Google Scholar] [CrossRef]
- Patel, D.P.; Krausz, K.W.; Xie, C.; Beyoglu, D.; Gonzalez, F.J.; Idle, J.R. Metabolic profiling by gas chromatography-mass spectrometry of energy metabolism in high-fat diet-fed obese mice. PLoS ONE 2017, 12, e0177953. [Google Scholar] [CrossRef]
- Simillion, C.; Semmo, N.; Idle, J.R.; Beyoglu, D. Robust Regression Analysis of GCMS Data Reveals Differential Rewiring of Metabolic Networks in Hepatitis B and C Patients. Metabolites 2017, 7, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idle, J.R.; Seipel, K.; Bacher, U.; Pabst, T.; Beyoglu, D. (2R,3S)-Dihydroxybutanoic Acid Synthesis as a Novel Metabolic Function of Mutant Isocitrate Dehydrogenase 1 and 2 in Acute Myeloid Leukemia. Cancers 2020, 12, 2842. [Google Scholar] [CrossRef]
- Beyoglu, D.; Imbeaud, S.; Maurhofer, O.; Bioulac-Sage, P.; Zucman-Rossi, J.; Dufour, J.F.; Idle, J.R. Tissue metabolomics of hepatocellular carcinoma: Tumor energy metabolism and the role of transcriptomic classification. Hepatology 2013, 58, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Keogh, A.; Treves, S.; Idle, J.R.; Beyoglu, D. The metabolomic profile of gamma-irradiated human hepatoma and muscle cells reveals metabolic changes consistent with the Warburg effect. PeerJ 2016, 4, e1624. [Google Scholar] [CrossRef] [Green Version]
- Keogh, A.; Senkardes, S.; Idle, J.R.; Kucukguzel, S.G.; Beyoglu, D. A Novel Anti-Hepatitis C Virus and Antiproliferative Agent Alters Metabolic Networks in HepG2 and Hep3B Cells. Metabolites 2017, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Denkert, C.; Budczies, J.; Kind, T.; Weichert, W.; Tablack, P.; Sehouli, J.; Niesporek, S.; Konsgen, D.; Dietel, M.; Fiehn, O. Mass spectrometry-based metabolic profiling reveals different metabolite patterns in invasive ovarian carcinomas and ovarian borderline tumors. Cancer Res. 2006, 66, 10795–10804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiehn, O.; Kopka, J.; Dormann, P.; Altmann, T.; Trethewey, R.N.; Willmitzer, L. Metabolite profiling for plant functional genomics. Nat. Biotechnol. 2000, 18, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Zullig, T.; Zandl-Lang, M.; Trotzmuller, M.; Hartler, J.; Plecko, B.; Kofeler, H.C. A Metabolomics Workflow for Analyzing Complex Biological Samples Using a Combined Method of Untargeted and Target-List Based Approaches. Metabolites 2020, 10, 342. [Google Scholar] [CrossRef]
- Pezzatti, J.; Boccard, J.; Codesido, S.; Gagnebin, Y.; Joshi, A.; Picard, D.; Gonzalez-Ruiz, V.; Rudaz, S. Implementation of liquid chromatography-high resolution mass spectrometry methods for untargeted metabolomic analyses of biological samples: A tutorial. Anal. Chim. Acta 2020, 1105, 28–44. [Google Scholar] [CrossRef]
- Carvajal-Carmona, L.G.; Alam, N.A.; Pollard, P.J.; Jones, A.M.; Barclay, E.; Wortham, N.; Pignatelli, M.; Freeman, A.; Pomplun, S.; Ellis, I.; et al. Adult leydig cell tumors of the testis caused by germline fumarate hydratase mutations. J. Clin. Endocrinol. Metab. 2006, 91, 3071–3075. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, I.P.; Alam, N.A.; Rowan, A.J.; Barclay, E.; Jaeger, E.E.; Kelsell, D.; Leigh, I.; Gorman, P.; Lamlum, H.; Rahman, S.; et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 2002, 30, 406–410. [Google Scholar] [CrossRef]
- Isaacs, J.S.; Jung, Y.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.L.; Merino, M.; Trepel, J.; Zbar, B.; Toro, J.; et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 2005, 8, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Frezza, C.; Zheng, L.; Folger, O.; Rajagopalan, K.N.; MacKenzie, E.D.; Jerby, L.; Micaroni, M.; Chaneton, B.; Adam, J.; Hedley, A.; et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature 2011, 477, 225–228. [Google Scholar] [CrossRef]
- Pollard, P.J.; Briere, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 2005, 14, 2231–2239. [Google Scholar] [CrossRef]
- Yang, M.; Su, H.; Soga, T.; Kranc, K.R.; Pollard, P.J. Prolyl hydroxylase domain enzymes: Important regulators of cancer metabolism. Hypoxia 2014, 2, 127–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, P.J.; Wortham, N.C.; Tomlinson, I.P. The TCA cycle and tumorigenesis: The examples of fumarate hydratase and succinate dehydrogenase. Ann. Med. 2003, 35, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Zhang, J.; Huang, G.; Yan, J.; Xu, C.; Dou, Z.; Sun, C.; Zhang, H. The crosstalk between HIFs and mitochondrial dysfunctions in cancer development. Cell Death Dis. 2021, 12, 215. [Google Scholar] [CrossRef]
- Adam, J.; Yang, M.; Soga, T.; Pollard, P.J. Rare insights into cancer biology. Oncogene 2014, 33, 2547–2556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, C.; Karamurzin, Y.; Frizzell, N.; Garg, K.; Nonaka, D.; Chen, Y.B.; Soslow, R.A. Uterine smooth muscle tumors with features suggesting fumarate hydratase aberration: Detailed morphologic analysis and correlation with S-(2-succino)-cysteine immunohistochemistry. Mod. Pathol. 2014, 27, 1020–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinonen, H.R.; Mehine, M.; Makinen, N.; Pasanen, A.; Pitkanen, E.; Karhu, A.; Sarvilinna, N.S.; Sjoberg, J.; Heikinheimo, O.; Butzow, R.; et al. Global metabolomic profiling of uterine leiomyomas. Br. J. Cancer 2017, 117, 1855–1864. [Google Scholar] [CrossRef]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; van der Mey, A.; Taschner, P.E.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Niemann, S.; Muller, U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat. Genet. 2000, 26, 268–270. [Google Scholar] [CrossRef] [PubMed]
- Burnichon, N.; Briere, J.J.; Libe, R.; Vescovo, L.; Riviere, J.; Tissier, F.; Jouanno, E.; Jeunemaitre, X.; Benit, P.; Tzagoloff, A.; et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum. Mol. Genet. 2010, 19, 3011–3020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astuti, D.; Latif, F.; Dallol, A.; Dahia, P.L.; Douglas, F.; George, E.; Skoldberg, F.; Husebye, E.S.; Eng, C.; Maher, E.R. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am. J. Hum. Genet. 2001, 69, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Hao, H.X.; Khalimonchuk, O.; Schraders, M.; Dephoure, N.; Bayley, J.P.; Kunst, H.; Devilee, P.; Cremers, C.W.; Schiffman, J.D.; Bentz, B.G.; et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 2009, 325, 1139–1142. [Google Scholar] [CrossRef] [Green Version]
- Bayley, J.P.; Kunst, H.P.; Cascon, A.; Sampietro, M.L.; Gaal, J.; Korpershoek, E.; Hinojar-Gutierrez, A.; Timmers, H.J.; Hoefsloot, L.H.; Hermsen, M.A.; et al. SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol 2010, 11, 366–372. [Google Scholar] [CrossRef]
- Szeto, S.S.; Reinke, S.N.; Sykes, B.D.; Lemire, B.D. Mutations in the Saccharomyces cerevisiae succinate dehydrogenase result in distinct metabolic phenotypes revealed through (1)H NMR-based metabolic footprinting. J. Proteome Res. 2010, 9, 6729–6739. [Google Scholar] [CrossRef] [PubMed]
- Sjoblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [Green Version]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Gross, S.; Cairns, R.A.; Minden, M.D.; Driggers, E.M.; Bittinger, M.A.; Jang, H.G.; Sasaki, M.; Jin, S.; Schenkein, D.P.; Su, S.M.; et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 2010, 207, 339–344. [Google Scholar] [CrossRef]
- Dvorak, A.; Zelenka, J.; Smolkova, K.; Vitek, L.; JeZek, P. Background levels of neomorphic 2-hydroxyglutarate facilitate proliferation of primary fibroblasts. Physiol Res. 2017, 66, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Pollard, P.J.; Gottlieb, E. Inborn and acquired metabolic defects in cancer. J. Mol. Med. 2011, 89, 213–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, E.H.; Livi, C.B.; Rakheja, D.; Tan, J.; Benson, D.; Parekh, V.; Kho, E.Y.; Ghosh, A.P.; Kirkman, R.; Velu, S.; et al. L-2-Hydroxyglutarate: An epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov. 2014, 4, 1290–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rzem, R.; Veiga-da-Cunha, M.; Noel, G.; Goffette, S.; Nassogne, M.C.; Tabarki, B.; Scholler, C.; Marquardt, T.; Vikkula, M.; Van Schaftingen, E. A gene encoding a putative FAD-dependent L-2-hydroxyglutarate dehydrogenase is mutated in L-2-hydroxyglutaric aciduria. Proc. Natl. Acad. Sci. USA 2004, 101, 16849–16854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rzem, R.; Vincent, M.F.; Van Schaftingen, E.; Veiga-da-Cunha, M. L-2-hydroxyglutaric aciduria, a defect of metabolite repair. J. Inherit. Metab. Dis. 2007, 30, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Oldham, W.M.; Clish, C.B.; Yang, Y.; Loscalzo, J. Hypoxia-Mediated Increases in L-2-hydroxyglutarate Coordinate the Metabolic Response to Reductive Stress. Cell Metab. 2015, 22, 291–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C.V. c-Myc transactivation of LDH-A: Implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar] [CrossRef] [Green Version]
- Intlekofer, A.M.; Dematteo, R.G.; Venneti, S.; Finley, L.W.; Lu, C.; Judkins, A.R.; Rustenburg, A.S.; Grinaway, P.B.; Chodera, J.D.; Cross, J.R.; et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab. 2015, 22, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Sreekumar, A.; Poisson, L.M.; Rajendiran, T.M.; Khan, A.P.; Cao, Q.; Yu, J.; Laxman, B.; Mehra, R.; Lonigro, R.J.; Li, Y.; et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 2009, 457, 910–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, F.; Dugheri, S.; Musci, M.; Bonacchi, A.; Salvadori, E.; Arcangeli, G.; Cupelli, V.; Lanciotti, M.; Masieri, L.; Serni, S.; et al. Fully automated solid-phase microextraction-fast gas chromatography-mass spectrometry method using a new ionic liquid column for high-throughput analysis of sarcosine and N-ethylglycine in human urine and urinary sediments. Anal. Chim. Acta 2011, 707, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, G.; Fanelli, M.; Larocca, A.M.; Germinario, C.A.; Rutigliano, M.; Vavallo, A.; Selvaggi, F.P.; Bettocchi, C.; Battaglia, M.; Ditonno, P. Serum sarcosine increases the accuracy of prostate cancer detection in patients with total serum PSA less than 4.0 ng/mL. Prostate 2012, 72, 1611–1621. [Google Scholar] [CrossRef] [PubMed]
- Koutros, S.; Meyer, T.E.; Fox, S.D.; Issaq, H.J.; Veenstra, T.D.; Huang, W.Y.; Yu, K.; Albanes, D.; Chu, L.W.; Andriole, G.; et al. Prospective evaluation of serum sarcosine and risk of prostate cancer in the Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial. Carcinogenesis 2013, 34, 2281–2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jentzmik, F.; Stephan, C.; Miller, K.; Schrader, M.; Erbersdobler, A.; Kristiansen, G.; Lein, M.; Jung, K. Sarcosine in urine after digital rectal examination fails as a marker in prostate cancer detection and identification of aggressive tumours. Eur. Urol. 2010, 58, 12–18; discussion 20–11. [Google Scholar] [CrossRef] [PubMed]
- Jentzmik, F.; Stephan, C.; Lein, M.; Miller, K.; Kamlage, B.; Bethan, B.; Kristiansen, G.; Jung, K. Sarcosine in prostate cancer tissue is not a differential metabolite for prostate cancer aggressiveness and biochemical progression. J. Urol. 2011, 185, 706–711. [Google Scholar] [CrossRef]
- Bohm, L.; Serafin, A.M.; Fernandez, P.; Van der Watt, G.; Bouic, P.J.; Harvey, J. Plasma sarcosine does not distinguish early and advanced stages of prostate cancer. S. Afr. Med. J. 2012, 102, 677–679. [Google Scholar] [CrossRef] [Green Version]
- Struys, E.A.; Heijboer, A.C.; van Moorselaar, J.; Jakobs, C.; Blankenstein, M.A. Serum sarcosine is not a marker for prostate cancer. Ann. Clin. Biochem. 2010, 47, 282. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Liu, T.; Ma, C.; Xue, R.; Deng, C.; Zeng, H.; Shen, X. GC/MS-based metabolomic approach to validate the role of urinary sarcosine and target biomarkers for human prostate cancer by microwave-assisted derivatization. Anal. Bioanal. Chem. 2011, 401, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.L.; Ye, D.W.; Zhu, Y.; Zhang, H.L.; Wang, Y.X.; Yao, X.D. Efforts to resolve the contradictions in early diagnosis of prostate cancer: A comparison of different algorithms of sarcosine in urine. Prostate Cancer Prostatic. Dis. 2011, 14, 166–172. [Google Scholar] [CrossRef]
- de Vogel, S.; Ulvik, A.; Meyer, K.; Ueland, P.M.; Nygard, O.; Vollset, S.E.; Tell, G.S.; Gregory, J.F., 3rd; Tretli, S.; Bjorge, T. Sarcosine and other metabolites along the choline oxidation pathway in relation to prostate cancer--a large nested case-control study within the JANUS cohort in Norway. Int. J. Cancer 2014, 134, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markin, P.A.; Brito, A.; Moskaleva, N.; Fodor, M.; Lartsova, E.V.; Shpot, Y.V.; Lerner, Y.V.; Mikhajlov, V.Y.; Potoldykova, N.V.; Enikeev, D.V.; et al. Plasma Sarcosine Measured by Gas Chromatography-Mass Spectrometry Distinguishes Prostatic Intraepithelial Neoplasia and Prostate Cancer from Benign Prostate Hyperplasia. Lab. Med. 2020, 51, 566–573. [Google Scholar] [CrossRef]
- Strmiska, V.; Michalek, P.; Eckschlager, T.; Stiborova, M.; Adam, V.; Krizkova, S.; Heger, Z. Prostate cancer-specific hallmarks of amino acids metabolism: Towards a paradigm of precision medicine. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Strmiska, V.; Michalek, P.; Lackova, Z.; Guran, R.; Krizkova, S.; Vanickova, L.; Zitka, O.; Stiborova, M.; Eckschlager, T.; Klejdus, B.; et al. Sarcosine is a prostate epigenetic modifier that elicits aberrant methylation patterns through the SAMe-Dnmts axis. Mol. Oncol. 2019, 13, 1002–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [Green Version]
- Sniegowski, T.; Korac, K.; Bhutia, Y.D.; Ganapathy, V. SLC6A14 and SLC38A5 Drive the Glutaminolysis and Serine-Glycine-One-Carbon Pathways in Cancer. Pharmaceuticals 2021, 14, 216. [Google Scholar] [CrossRef]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef]
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E.; et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 2012, 148, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Yang, C.; Nesvick, C.L.; Feldman, M.J.; Sizdahkhani, S.; Liu, H.; Chu, H.; Yang, F.; Tang, L.; Tian, J.; et al. Hypotaurine evokes a malignant phenotype in glioma through aberrant hypoxic signaling. Oncotarget 2016, 7, 15200–15214. [Google Scholar] [CrossRef] [Green Version]
- Chinnaiyan, P.; Kensicki, E.; Bloom, G.; Prabhu, A.; Sarcar, B.; Kahali, S.; Eschrich, S.; Qu, X.; Forsyth, P.; Gillies, R. The metabolomic signature of malignant glioma reflects accelerated anabolic metabolism. Cancer Res. 2012, 72, 5878–5888. [Google Scholar] [CrossRef] [Green Version]
- Prabhu, A.; Sarcar, B.; Kahali, S.; Yuan, Z.; Johnson, J.J.; Adam, K.P.; Kensicki, E.; Chinnaiyan, P. Cysteine catabolism: A novel metabolic pathway contributing to glioblastoma growth. Cancer Res. 2014, 74, 787–796. [Google Scholar] [CrossRef] [Green Version]
- Ueki, I.; Stipanuk, M.H. 3T3-L1 adipocytes and rat adipose tissue have a high capacity for taurine synthesis by the cysteine dioxygenase/cysteinesulfinate decarboxylase and cysteamine dioxygenase pathways. J. Nutr 2009, 139, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, D.; Tian, L.; Yang, F.; Li, J.; Li, X.; Yao, Y.; Lam, E.W.; Gao, P.; Jin, B.; Wang, R. ADO/hypotaurine: A novel metabolic pathway contributing to glioblastoma development. Cell Death Discov. 2021, 7, 21. [Google Scholar] [CrossRef]
- Ippolito, L.; Morandi, A.; Giannoni, E.; Chiarugi, P. Lactate: A Metabolic Driver in the Tumour Landscape. Trends Biochem. Sci. 2019, 44, 153–166. [Google Scholar] [CrossRef]
- Pereira-Nunes, A.; Afonso, J.; Granja, S.; Baltazar, F. Lactate and Lactate Transporters as Key Players in the Maintenance of the Warburg Effect. Adv. Exp. Med. Biol. 2020, 1219, 51–74. [Google Scholar] [CrossRef]
- Brooks, G.A. The lactate shuttle during exercise and recovery. Med. Sci. Sports Exerc. 1986, 18, 360–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, G.A. Lactate production under fully aerobic conditions: The lactate shuttle during rest and exercise. Fed. Proc. 1986, 45, 2924–2929. [Google Scholar] [PubMed]
- Baltazar, F.; Afonso, J.; Costa, M.; Granja, S. Lactate Beyond a Waste Metabolite: Metabolic Affairs and Signaling in Malignancy. Front. Oncol. 2020, 10, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Zuo, H.; Xiong, H.; Kolar, M.J.; Chu, Q.; Saghatelian, A.; Siegwart, D.J.; Wan, Y. Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2017, 114, 580–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Goede, K.E.; Driessen, A.J.M.; Van den Bossche, J. Metabolic Cancer-Macrophage Crosstalk in the Tumor Microenvironment. Biology 2020, 9, 380. [Google Scholar] [CrossRef] [PubMed]
- Emir, U.E.; Larkin, S.J.; de Pennington, N.; Voets, N.; Plaha, P.; Stacey, R.; Al-Qahtani, K.; McCullagh, J.; Schofield, C.J.; Clare, S.; et al. Noninvasive Quantification of 2-Hydroxyglutarate in Human Gliomas with IDH1 and IDH2 Mutations. Cancer Res. 2016, 76, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S.; Mandal, R.; Stanislaus, A.; Ramirez-Gaona, M. Cancer Metabolomics and the Human Metabolome Database. Metabolites 2016, 6, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatami, F.; Payab, M.; Sarvari, M.; Gilany, K.; Larijani, B.; Arjmand, B.; Tavangar, S.M. Oncometabolites as biomarkers in thyroid cancer: A systematic review. Cancer Manag. Res. 2019, 11, 1829–1841. [Google Scholar] [CrossRef] [Green Version]
- Miccoli, P.; Torregrossa, L.; Shintu, L.; Magalhaes, A.; Chandran, J.; Tintaru, A.; Ugolini, C.; Minuto, M.N.; Miccoli, M.; Basolo, F.; et al. Metabolomics approach to thyroid nodules: A high-resolution magic-angle spinning nuclear magnetic resonance-based study. Surgery 2012, 152, 1118–1124. [Google Scholar] [CrossRef]
- Tessem, M.B.; Swanson, M.G.; Keshari, K.R.; Albers, M.J.; Joun, D.; Tabatabai, Z.L.; Simko, J.P.; Shinohara, K.; Nelson, S.J.; Vigneron, D.B.; et al. Evaluation of lactate and alanine as metabolic biomarkers of prostate cancer using 1H HR-MAS spectroscopy of biopsy tissues. Magn. Reson. Med. 2008, 60, 510–516. [Google Scholar] [CrossRef] [Green Version]
- Beadle, G.W.; Mitchell, H.K.; Nyc, J.F. Kynurenine as an Intermediate in the Formation of Nicotinic Acid from Tryptophane by Neurospora. Proc. Natl. Acad. Sci. USA 1947, 33, 155–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkateswaran, N.; Lafita-Navarro, M.C.; Hao, Y.H.; Kilgore, J.A.; Perez-Castro, L.; Braverman, J.; Borenstein-Auerbach, N.; Kim, M.; Lesner, N.P.; Mishra, P.; et al. MYC promotes tryptophan uptake and metabolism by the kynurenine pathway in colon cancer. Genes Dev. 2019, 33, 1236–1251. [Google Scholar] [CrossRef] [Green Version]
- Venkateswaran, N.; Conacci-Sorrell, M. Kynurenine: An oncometabolite in colon cancer. Cell Stress 2020, 4, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Sauve, A.A. NAD(+) metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim. Biophys. Acta 2016, 1864, 1787–1800. [Google Scholar] [CrossRef] [Green Version]
- Rather, G.M.; Pramono, A.A.; Szekely, Z.; Bertino, J.R.; Tedeschi, P.M. In cancer, all roads lead to NADPH. Pharmacol. Ther. 2021, 226, 107864. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.A.; Wu, C.H.; Lu, C.C.; Hsia, S.M.; Yen, G.C. Glycative stress from advanced glycation end products (AGEs) and dicarbonyls: An emerging biological factor in cancer onset and progression. Mol. Nutr. Food Res. 2016, 60, 1850–1864. [Google Scholar] [CrossRef]
- Wilker, S.C.; Chellan, P.; Arnold, B.M.; Nagaraj, R.H. Chromatographic quantification of argpyrimidine, a methylglyoxal-derived product in tissue proteins: Comparison with pentosidine. Anal. Biochem. 2001, 290, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Ashour, A.; Thornalley, P.J. Mass spectrometric determination of early and advanced glycation in biology. Glycoconj. J. 2016, 33, 553–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellier, J.; Nokin, M.J.; Larde, E.; Karoyan, P.; Peulen, O.; Castronovo, V.; Bellahcene, A. Methylglyoxal, a potent inducer of AGEs, connects between diabetes and cancer. Diabetes Res. Clin. Pract 2019, 148, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Leone, A.; Nigro, C.; Nicolo, A.; Prevenzano, I.; Formisano, P.; Beguinot, F.; Miele, C. The Dual-Role of Methylglyoxal in Tumor Progression—Novel Therapeutic Approaches. Front. Oncol 2021, 11, 645686. [Google Scholar] [CrossRef]
- Koo, J.H.; Guan, K.L. Interplay between YAP/TAZ and Metabolism. Cell Metab. 2018, 28, 196–206. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yuan, Y.; Ji, X.; Xia, X.; Li, Z.; Yi, X.; Zhu, Y.; Guo, T.; Wang, Y.; Chen, L.; et al. The Hippo-TAZ axis mediates vascular endothelial growth factor C in glioblastoma-derived exosomes to promote angiogenesis. Cancer Lett. 2021. [Google Scholar] [CrossRef]
- Nokin, M.J.; Durieux, F.; Peixoto, P.; Chiavarina, B.; Peulen, O.; Blomme, A.; Turtoi, A.; Costanza, B.; Smargiasso, N.; Baiwir, D.; et al. Methylglyoxal, a glycolysis side-product, induces Hsp90 glycation and YAP-mediated tumor growth and metastasis. eLife 2016, 5. [Google Scholar] [CrossRef]
- Roci, I.; Watrous, J.D.; Lagerborg, K.A.; Jain, M.; Nilsson, R. Mapping metabolic oscillations during cell cycle progression. Cell Cycle 2020, 19, 2676–2684. [Google Scholar] [CrossRef]
- Kani, S.; Nakayama, E.; Yoda, A.; Onishi, N.; Sougawa, N.; Hazaka, Y.; Umeda, T.; Takeda, K.; Ichijo, H.; Hamada, Y.; et al. Chk2 kinase is required for methylglyoxal-induced G2/M cell-cycle checkpoint arrest: Implication of cell-cycle checkpoint regulation in diabetic oxidative stress signaling. Genes Cells 2007, 12, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Bultman, S.J. Interplay between diet, gut microbiota, epigenetic events, and colorectal cancer. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- San-Millan, I.; Julian, C.G.; Matarazzo, C.; Martinez, J.; Brooks, G.A. Is Lactate an Oncometabolite? Evidence Supporting a Role for Lactate in the Regulation of Transcriptional Activity of Cancer-Related Genes in MCF7 Breast Cancer Cells. Front. Oncol. 2019, 9, 1536. [Google Scholar] [CrossRef] [PubMed]
- Comito, G.; Iscaro, A.; Bacci, M.; Morandi, A.; Ippolito, L.; Parri, M.; Montagnani, I.; Raspollini, M.R.; Serni, S.; Simeoni, L.; et al. Lactate modulates CD4(+) T-cell polarization and induces an immunosuppressive environment, which sustains prostate carcinoma progression via TLR8/miR21 axis. Oncogene 2019, 38, 3681–3695. [Google Scholar] [CrossRef]
- Bellier, J.; Nokin, M.J.; Caprasse, M.; Tiamiou, A.; Blomme, A.; Scheijen, J.L.; Koopmansch, B.; MacKay, G.M.; Chiavarina, B.; Costanza, B.; et al. Methylglyoxal Scavengers Resensitize KRAS-Mutated Colorectal Tumors to Cetuximab. Cell Rep. 2020, 30, 1400–1416 e1046. [Google Scholar] [CrossRef]
- Dang, L.; Jin, S.; Su, S.M. IDH mutations in glioma and acute myeloid leukemia. Trends Mol. Med. 2010, 16, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Yen, K.; Travins, J.; Wang, F.; David, M.D.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DeLaBarre, B.; Tobin, E.; et al. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov. 2017, 7, 478–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullard, A. Cancer metabolism pipeline breaks new ground. Nat. Rev. Drug Discov. 2016, 15, 735–737. [Google Scholar] [CrossRef] [PubMed]
- Rai, S.K.; Bril, F.; Hatch, H.M.; Xu, Y.; Shelton, L.; Kalavalapalli, S.; Click, A.; Lee, D.; Beecher, C.; Kirby, A.; et al. Targeting pheochromocytoma/paraganglioma with polyamine inhibitors. Metabolism 2020, 110, 154297. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Oncometabolite | Tumors | Metabolomic Contribution | Strength of Evidence | Reference |
|---|---|---|---|---|
| Fumarate | HLRCC | Linked FH mutation to TCA and glycolytic metabolites | +++ | [43,44,45] |
| Succinate | Hereditary paraganglioma Pheochromocytoma | Linked SDH mutations to succinate and other metabolites | +++ | [59] |
| (2R)-Hydroxyglutarate | Glioblastoma multiforme Acute myeloid leukemia | Linked IDH1 and IDH2 mutations to 2R-HG | +++ | [33,62,64,65] |
| (2S)-Hydroxyglutarate | Clear cell RCC | Linked L2HGDH activity in tumors to 2S-HG | +++ | [68] |
| Lactate | Several cancers | Signaling molecule contributing to proliferation, migration, invasion, angiogenesis, immune system escape and resistance to therapy | +++ | [97,101,125,126] |
| Kynurenine | Colon cancer | Activation of transcription factor AHR that regulates growth-promoting genes | + | [110,111] |
| Methylglyoxal | Breast and colorectal cancer | AKT activation through PI3K/mTORC2 and Hsp27 regulation | + | [118,127] |
| Sarcosine | Prostate cancer | Variable reports of linkage of sarcosine to prostate cancer; no clear mechanism; potential biomarker | ± | [74,75,76,77,78,79,80,81,82,83,84,85,86] |
| Glycine | NCI-60 cell lines | Linked glycine metabolism to rapidly proliferating cancer cells; postulated mechanism involving glycine decarboxylase (GLDC); cells overexpressing GLDC → ↑glycolytic intermediates and ↑pyrimidines | + | [88,91] |
| Hypotaurine | Glioblastoma multiforme | Established role for hypotaurine in glioblastoma multiforme; correlative and mechanistic data point to hypotaurine as an oncometabolite | ++ | [92] |
| (2R,3S)-Dihydroxybutanoate | Acute myeloid leukemia | Found in plasmas of mutant IDH1/2, 2,3-DHBA is greater than WT IDH1/2 and strongly correlated with 2R-HG; 2,3-DHBA is a better biomarker for mutated IDH than classical oncometabolite 2R-HG | ± | [33] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beyoğlu, D.; Idle, J.R. Metabolic Rewiring and the Characterization of Oncometabolites. Cancers 2021, 13, 2900. https://doi.org/10.3390/cancers13122900
Beyoğlu D, Idle JR. Metabolic Rewiring and the Characterization of Oncometabolites. Cancers. 2021; 13(12):2900. https://doi.org/10.3390/cancers13122900
Chicago/Turabian StyleBeyoğlu, Diren, and Jeffrey R. Idle. 2021. "Metabolic Rewiring and the Characterization of Oncometabolites" Cancers 13, no. 12: 2900. https://doi.org/10.3390/cancers13122900
APA StyleBeyoğlu, D., & Idle, J. R. (2021). Metabolic Rewiring and the Characterization of Oncometabolites. Cancers, 13(12), 2900. https://doi.org/10.3390/cancers13122900