
Advances in Chemokine Signaling Pathways as Therapeutic Targets in Glioblastoma



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
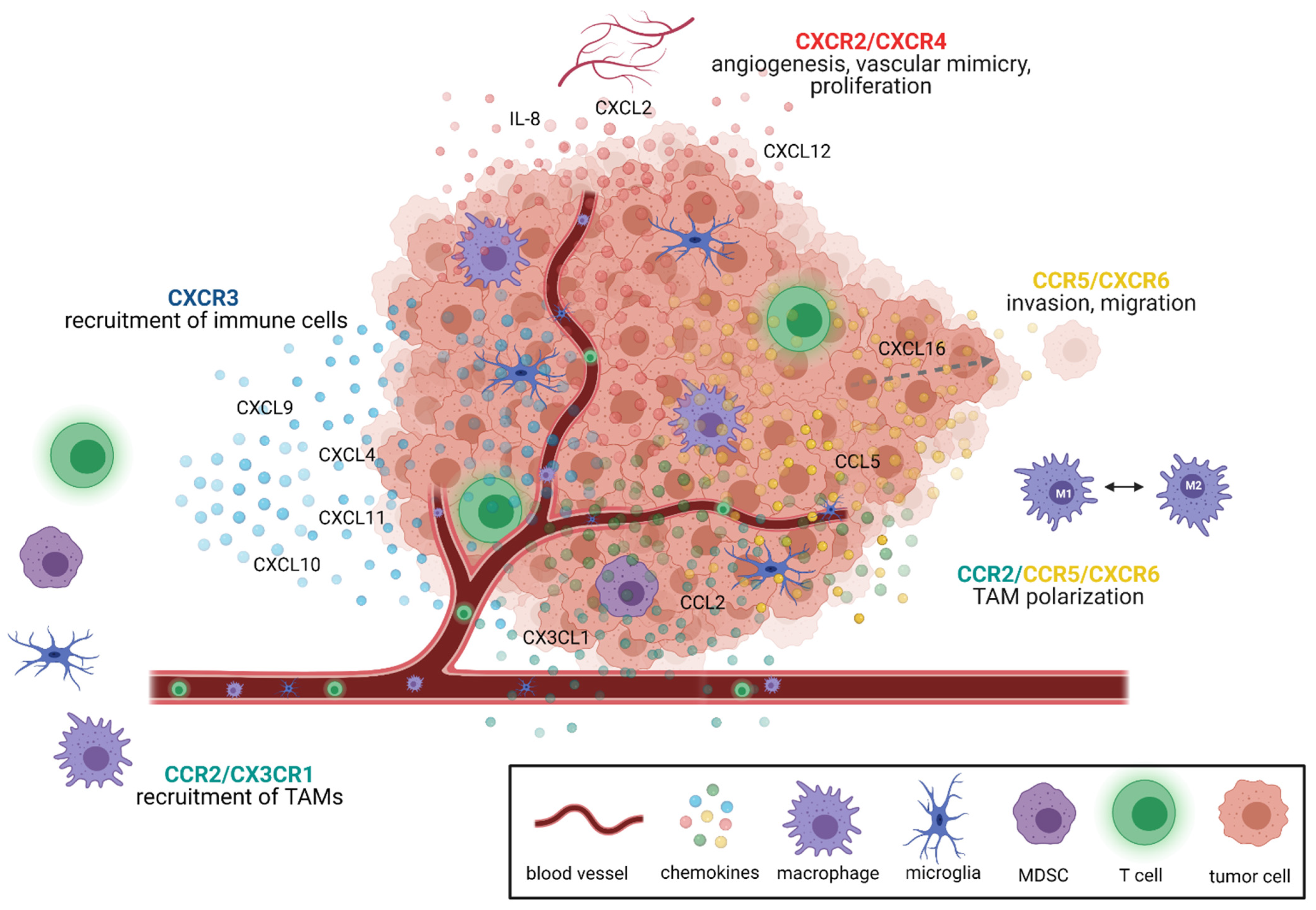
2. Chemokine Signaling Pathways as Therapeutic Targets in GBM
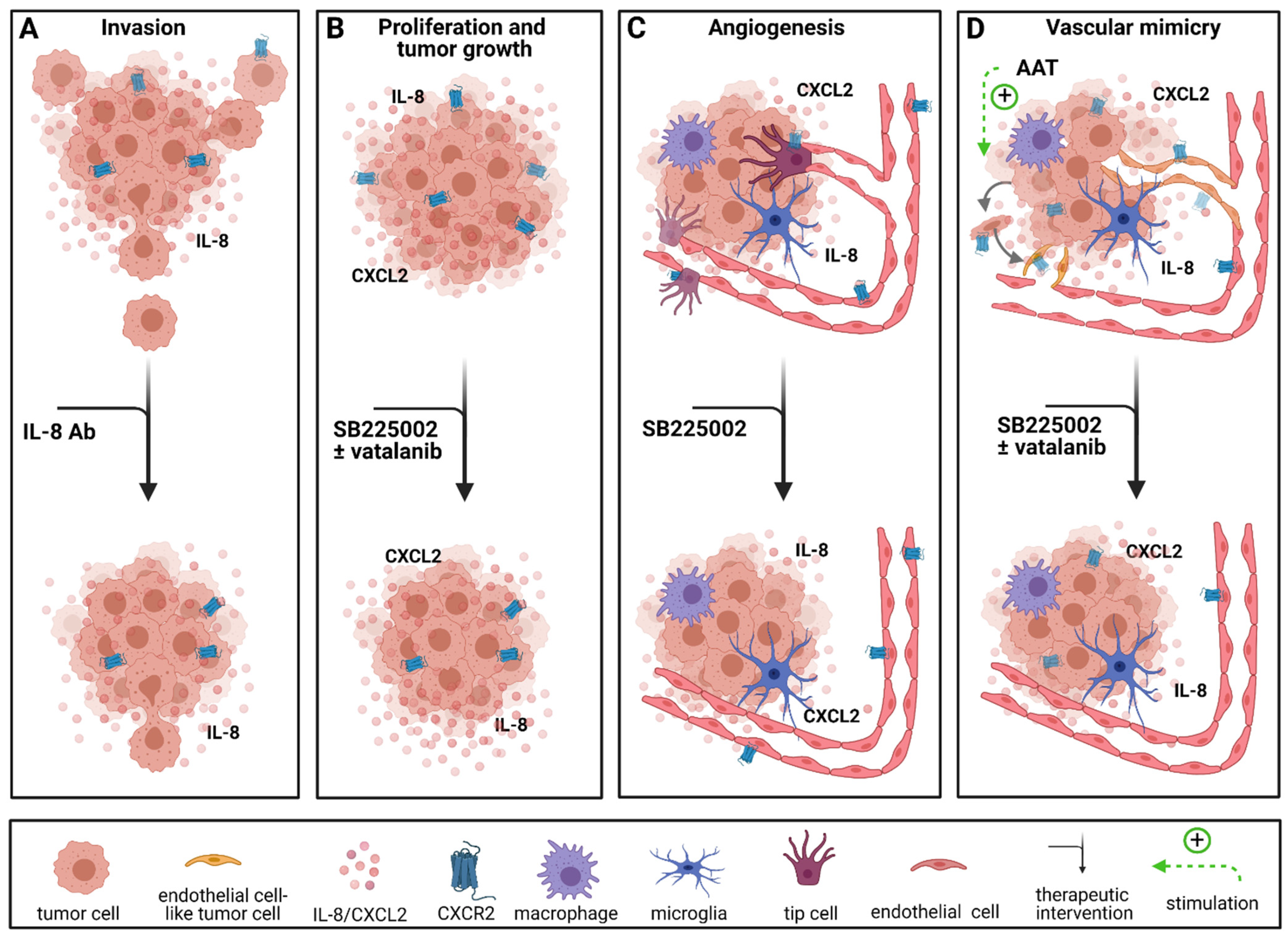
2.1. Targeted Therapies in Chemokine Signaling
2.2. Combination of Targeting Chemokine Receptors/Chemokines with the Standard-of-Care Therapy
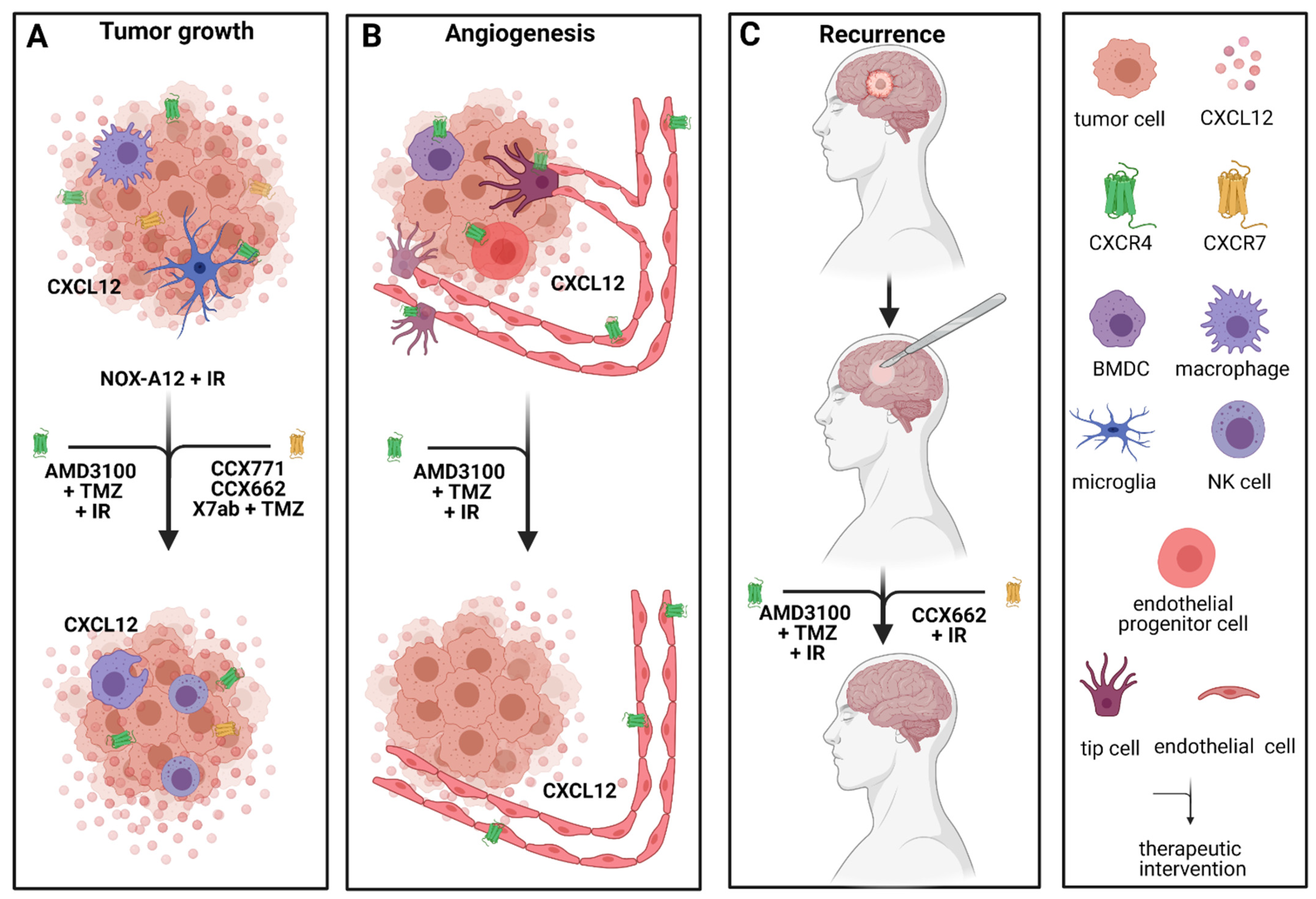
2.3. Antiangiogenic Approaches Targeting Chemokine Receptors/Chemokines
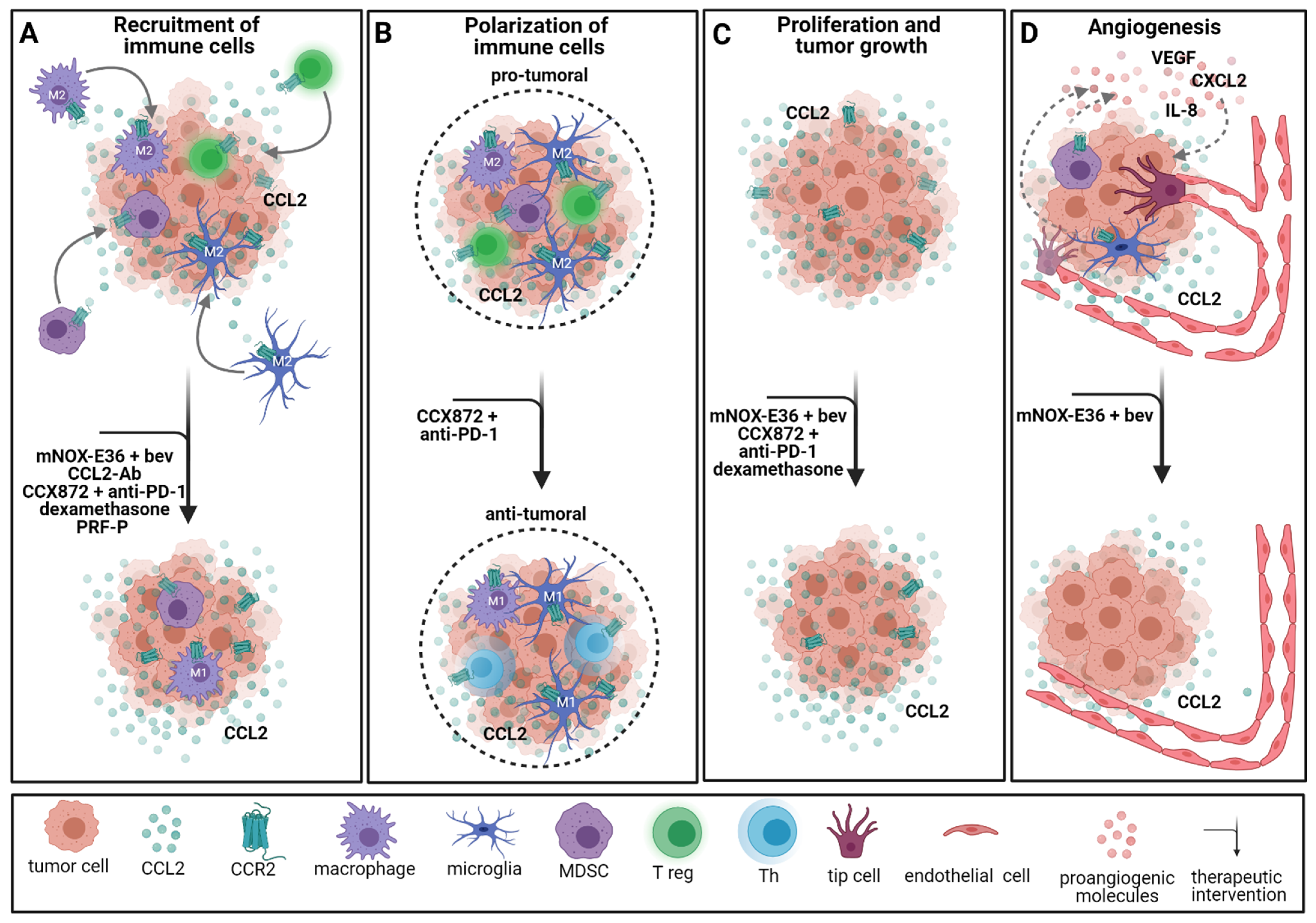
2.4. Immunotherapeutic Approaches Involving Chemokine Signaling
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro-Oncology 2019, 21, v1–v100. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Brodbelt, A.; Greenberg, D.; Winters, T.; Williams, M.; Vernon, S.; Collins, V.P.; National Cancer Information Network Brain Tumour, G. Glioblastoma in England: 2007–2011. Eur. J. Cancer 2015, 51, 533–542. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binabaj, M.M.; Bahrami, A.; ShahidSales, S.; Joodi, M.; Joudi Mashhad, M.; Hassanian, S.M.; Anvari, K.; Avan, A. The prognostic value of MGMT promoter methylation in glioblastoma: A meta-analysis of clinical trials. J. Cell. Physiol. 2018, 233, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.H.; Liu, W.L.; Cao, H.; Wen, C.; Chen, L.; Jiang, G. O6-methylguanine DNA methyltransferase as a promising target for the treatment of temozolomide-resistant gliomas. Cell Death Dis. 2013, 4, e876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohn, A.; Braley, A.; Rodriguez de la Vega, P.; Zevallos, J.C.; Barengo, N.C. The association between race and survival in glioblastoma patients in the US: A retrospective cohort study. PLoS ONE 2018, 13, e0198581. [Google Scholar] [CrossRef] [PubMed]
- Ahir, B.K.; Engelhard, H.H.; Lakka, S.S. Tumor Development and Angiogenesis in Adult Brain Tumor: Glioblastoma. Mol. Neurobiol. 2020, 57, 2461–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, S.; Yamashita, T.; Ohneda, O. Molecular therapeutic targets for glioma angiogenesis. J. Oncol. 2010, 2010, 351908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norden, A.D.; Drappatz, J.; Wen, P.Y. Antiangiogenic therapy in malignant gliomas. Curr. Opin. Oncol. 2008, 20, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Roggendorf, W.; Strupp, S.; Paulus, W. Distribution and characterization of microglia/macrophages in human brain tumors. Acta Neuropathol. 1996, 92, 288–293. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, H.; Heppner, F.L.; Tsirka, S.E. Microglia/macrophages promote glioma progression. Glia 2011, 59, 472–485. [Google Scholar] [CrossRef] [Green Version]
- Ellert-Miklaszewska, A.; Dabrowski, M.; Lipko, M.; Sliwa, M.; Maleszewska, M.; Kaminska, B. Molecular definition of the pro-tumorigenic phenotype of glioma-activated microglia. Glia 2013, 61, 1178–1190. [Google Scholar] [CrossRef] [PubMed]
- Galarneau, H.; Villeneuve, J.; Gowing, G.; Julien, J.P.; Vallieres, L. Increased glioma growth in mice depleted of macrophages. Cancer Res. 2007, 67, 8874–8881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szulzewsky, F.; Arora, S.; de Witte, L.; Ulas, T.; Markovic, D.; Schultze, J.L.; Holland, E.C.; Synowitz, M.; Wolf, S.A.; Kettenmann, H. Human glioblastoma-associated microglia/monocytes express a distinct RNA profile compared to human control and murine samples. Glia 2016, 64, 1416–1436. [Google Scholar] [CrossRef]
- Szulzewsky, F.; Pelz, A.; Feng, X.; Synowitz, M.; Markovic, D.; Langmann, T.; Holtman, I.R.; Wang, X.; Eggen, B.J.; Boddeke, H.W.; et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS ONE 2015, 10, e0116644. [Google Scholar] [CrossRef] [Green Version]
- Brandenburg, S.; Turkowski, K.; Mueller, A.; Radev, Y.T.; Seidlitz, S.; Vajkoczy, P. Myeloid cells expressing high level of CD45 are associated with a distinct activated phenotype in glioma. Immunol. Res. 2017, 65, 757–768. [Google Scholar] [CrossRef]
- Fanelli, G.N.; Grassini, D.; Ortenzi, V.; Pasqualetti, F.; Montemurro, N.; Perrini, P.; Naccarato, A.G.; Scatena, C. Decipher the Glioblastoma Microenvironment: The First Milestone for New Groundbreaking Therapeutic Strategies. Genes 2021, 12, 445. [Google Scholar] [CrossRef]
- Huang, B.; Li, X.; Li, Y.; Zhang, J.; Zong, Z.; Zhang, H. Current Immunotherapies for Glioblastoma Multiforme. Front. Immunol. 2020, 11, 603911. [Google Scholar] [CrossRef]
- Sampson, J.H.; Maus, M.V.; June, C.H. Immunotherapy for Brain Tumors. J. Clin. Oncol. 2017, 35, 2450–2456. [Google Scholar] [CrossRef] [Green Version]
- Pombo Antunes, A.R.; Scheyltjens, I.; Duerinck, J.; Neyns, B.; Movahedi, K.; Van Ginderachter, J.A. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. Elife 2020, 9. [Google Scholar] [CrossRef]
- Yi, Y.; Hsieh, I.Y.; Huang, X.; Li, J.; Zhao, W. Glioblastoma Stem-Like Cells: Characteristics, Microenvironment, and Therapy. Front. Pharmacol. 2016, 7, 477. [Google Scholar] [CrossRef] [Green Version]
- Iwadate, Y. Plasticity in Glioma Stem Cell Phenotype and Its Therapeutic Implication. Neurol. Med. Chir. 2018, 58, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Koso, H.; Takeda, H.; Yew, C.C.; Ward, J.M.; Nariai, N.; Ueno, K.; Nagasaki, M.; Watanabe, S.; Rust, A.G.; Adams, D.J.; et al. Transposon mutagenesis identifies genes that transform neural stem cells into glioma-initiating cells. Proc. Natl. Acad. Sci. USA 2012, 109, E2998–E3007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zhang, H.; Xu, S.; Liu, Z.; Cheng, Q. The adaptive transition of glioblastoma stem cells and its implications on treatments. Signal Transduct. Target. Ther. 2021, 6, 124. [Google Scholar] [CrossRef] [PubMed]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrlinger, U.; Schafer, N.; Steinbach, J.P.; Weyerbrock, A.; Hau, P.; Goldbrunner, R.; Friedrich, F.; Rohde, V.; Ringel, F.; Schlegel, U.; et al. Bevacizumab Plus Irinotecan Versus Temozolomide in Newly Diagnosed O6-Methylguanine-DNA Methyltransferase Nonmethylated Glioblastoma: The Randomized GLARIUS Trial. J. Clin. Oncol. 2016, 34, 1611–1619. [Google Scholar] [CrossRef]
- Soffietti, R.; Trevisan, E.; Bertero, L.; Bosa, C.; Ruda, R. Anti-angiogenic approaches to malignant gliomas. Curr. Cancer Drug Targets 2012, 12, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Zlotnik, A.; Yoshie, O.; Nomiyama, H. The chemokine and chemokine receptor superfamilies and their molecular evolution. Genome Biol. 2006, 7, 243. [Google Scholar] [CrossRef] [PubMed]
- Charo, I.F.; Ransohoff, R.M. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 2006, 354, 610–621. [Google Scholar] [CrossRef]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [Green Version]
- Groblewska, M.; Litman-Zawadzka, A.; Mroczko, B. The Role of Selected Chemokines and Their Receptors in the Development of Gliomas. Int. J. Mol. Sci 2020, 21, 704. [Google Scholar] [CrossRef]
- Huynh, C.; Dingemanse, J.; Meyer Zu Schwabedissen, H.E.; Sidharta, P.N. Relevance of the CXCR4/CXCR7-CXCL12 axis and its effect in pathophysiological conditions. Pharmacol. Res. 2020, 161, 105092. [Google Scholar] [CrossRef]
- Brandenburg, S.; Muller, A.; Turkowski, K.; Radev, Y.T.; Rot, S.; Schmidt, C.; Bungert, A.D.; Acker, G.; Schorr, A.; Hippe, A.; et al. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol. 2016, 131, 365–378. [Google Scholar] [CrossRef]
- Blank, A.; Kremenetskaia, I.; Urbantat, R.M.; Acker, G.; Turkowski, K.; Radke, J.; Schneider, U.C.; Vajkoczy, P.; Brandenburg, S. Microglia/macrophages express alternative proangiogenic factors depending on granulocyte content in human glioblastoma. J. Pathol. 2020, 253, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Hillyer, P.; Mordelet, E.; Flynn, G.; Male, D. Chemokines, chemokine receptors and adhesion molecules on different human endothelia: Discriminating the tissue-specific functions that affect leucocyte migration. Clin. Exp. Immunol. 2003, 134, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Subileau, E.A.; Rezaie, P.; Davies, H.A.; Colyer, F.M.; Greenwood, J.; Male, D.K.; Romero, I.A. Expression of chemokines and their receptors by human brain endothelium: Implications for multiple sclerosis. J. Neuropathol. Exp. Neurol. 2009, 68, 227–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, I.; Singh, A.; Siraj, F.; Saxena, S. IL-8/CXCR1/2 signalling promotes tumor cell proliferation, invasion and vascular mimicry in glioblastoma. J. Biomed. Sci. 2018, 25, 62. [Google Scholar] [CrossRef]
- Hasan, T.; Caragher, S.P.; Shireman, J.M.; Park, C.H.; Atashi, F.; Baisiwala, S.; Lee, G.; Guo, D.; Wang, J.Y.; Dey, M.; et al. Interleukin-8/CXCR2 signaling regulates therapy-induced plasticity and enhances tumorigenicity in glioblastoma. Cell Death Dis. 2019, 10, 292. [Google Scholar] [CrossRef] [Green Version]
- Bruyere, C.; Mijatovic, T.; Lonez, C.; Spiegl-Kreinecker, S.; Berger, W.; Kast, R.E.; Ruysschaert, J.M.; Kiss, R.; Lefranc, F. Temozolomide-induced modification of the CXC chemokine network in experimental gliomas. Int. J. Oncol. 2011, 38, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, M.; Blank, A.; Bungert, A.D.; Mueller, A.; Ghori, A.; Kremenetskaia, I.; Rung, O.; Broggini, T.; Turkowski, K.; Scherschinski, L.; et al. CCR2 of Tumor Microenvironmental Cells Is a Relevant Modulator of Glioma Biology. Cancers 2020, 12, 1882. [Google Scholar] [CrossRef]
- Urbantat, R.M.; Blank, A.; Kremenetskaia, I.; Vajkoczy, P.; Acker, G.; Brandenburg, S. The CXCL2/IL8/CXCR2 Pathway Is Relevant for Brain Tumor Malignancy and Endothelial Cell Function. Int. J. Mol. Sci. 2021, 22, 2634. [Google Scholar] [CrossRef]
- Angara, K.; Borin, T.F.; Rashid, M.H.; Lebedyeva, I.; Ara, R.; Lin, P.C.; Iskander, A.; Bollag, R.J.; Achyut, B.R.; Arbab, A.S. CXCR2-Expressing Tumor Cells Drive Vascular Mimicry in Antiangiogenic Therapy-Resistant Glioblastoma. Neoplasia 2018, 20, 1070–1082. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Li, S.; Zhang, C.; Bao, Z.; Yang, Z.; Sun, L. High expression of CXCR3 is an independent prognostic factor in glioblastoma patients that promotes an invasive phenotype. J. Neurooncol. 2015, 122, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Huizer, K.; Sacchetti, A.; Swagemakers, S.; van der Spek, P.J.; Dik, W.; Mustafa, D.A.; Kros, J.M. Circulating angiogenic cells in glioblastoma: Toward defining crucial functional differences in CAC-induced neoplastic versus reactive neovascularization. Neuro-Oncol. Adv. 2020, 2, vdaa040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, Y.C.; Chen, J.N.; Chen, Y.H.; Chou, R.H.; Lee, H.C.; Yu, Y.L. Epigenetic Silencing of miR-9 Promotes Migration and Invasion by EZH2 in Glioblastoma Cells. Cancers 2020, 12, 1781. [Google Scholar] [CrossRef] [PubMed]
- Hattermann, K.; Held-Feindt, J.; Ludwig, A.; Mentlein, R. The CXCL16-CXCR6 chemokine axis in glial tumors. J. Neuroimmunol. 2013, 260, 47–54. [Google Scholar] [CrossRef]
- Salazar, N.; Carlson, J.C.; Huang, K.; Zheng, Y.; Oderup, C.; Gross, J.; Jang, A.D.; Burke, T.M.; Lewen, S.; Scholz, A.; et al. A Chimeric Antibody against ACKR3/CXCR7 in Combination with TMZ Activates Immune Responses and Extends Survival in Mouse GBM Models. Mol. Ther. 2018, 26, 1354–1365. [Google Scholar] [CrossRef] [Green Version]
- Hattermann, K.; Held-Feindt, J.; Lucius, R.; Muerkoster, S.S.; Penfold, M.E.; Schall, T.J.; Mentlein, R. The chemokine receptor CXCR7 is highly expressed in human glioma cells and mediates antiapoptotic effects. Cancer Res. 2010, 70, 3299–3308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novak, M.; Koprivnikar Krajnc, M.; Hrastar, B.; Breznik, B.; Majc, B.; Mlinar, M.; Rotter, A.; Porcnik, A.; Mlakar, J.; Stare, K.; et al. CCR5-Mediated Signaling Is Involved in Invasion of Glioblastoma Cells in Its Microenvironment. Int. J. Mol. Sci. 2020, 21, 4199. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, Y.; Xue, Y.; Lv, W.; Zhang, Y.; He, S. Critical roles of chemokine receptor CCR5 in regulating glioblastoma proliferation and invasion. Acta Biochim. Biophys. Sin. 2015, 47, 890–898. [Google Scholar] [CrossRef] [Green Version]
- Erreni, M.; Solinas, G.; Brescia, P.; Osti, D.; Zunino, F.; Colombo, P.; Destro, A.; Roncalli, M.; Mantovani, A.; Draghi, R.; et al. Human glioblastoma tumours and neural cancer stem cells express the chemokine CX3CL1 and its receptor CX3CR1. Eur. J. Cancer 2010, 46, 3383–3392. [Google Scholar] [CrossRef]
- Sharma, I.; Singh, A.; Sharma, K.; Saxena, S. Gene Expression Profiling of Chemokines and Their Receptors in Low and High Grade Astrocytoma. Asian Pac. J. Cancer Prev. 2017, 18, 1307–1313. [Google Scholar] [CrossRef]
- Raychaudhuri, B.; Vogelbaum, M.A. IL-8 is a mediator of NF-kappaB induced invasion by gliomas. J. Neurooncol. 2011, 101, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Addison, C.L.; Daniel, T.O.; Burdick, M.D.; Liu, H.; Ehlert, J.E.; Xue, Y.Y.; Buechi, L.; Walz, A.; Richmond, A.; Strieter, R.M. The CXC chemokine receptor 2, CXCR2, is the putative receptor for ELR+ CXC chemokine-induced angiogenic activity. J. Immunol. 2000, 165, 5269–5277. [Google Scholar] [CrossRef] [Green Version]
- Murdoch, C.; Monk, P.N.; Finn, A. Cxc chemokine receptor expression on human endothelial cells. Cytokine 1999, 11, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Stafford, J.H.; Liu, S.C.; Chernikova, S.B.; Merchant, M.; Recht, L.; Martin Brown, J. SDF-1 Blockade Enhances Anti-VEGF Therapy of Glioblastoma and Can Be Monitored by MRI. Neoplasia 2017, 19, 1–7. [Google Scholar] [CrossRef]
- Laudati, E.; Curro, D.; Navarra, P.; Lisi, L. Blockade of CCR5 receptor prevents M2 microglia phenotype in a microglia-glioma paradigm. Neurochem. Int. 2017, 108, 100–108. [Google Scholar] [CrossRef]
- Lepore, F.; D’Alessandro, G.; Antonangeli, F.; Santoro, A.; Esposito, V.; Limatola, C.; Trettel, F. CXCL16/CXCR6 Axis Drives Microglia/Macrophages Phenotype in Physiological Conditions and Plays a Crucial Role in Glioma. Front. Immunol. 2018, 9, 2750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Liu, Z.; Wu, R.; Yao, Q.; Gu, Z.; Liu, M. Correlation of C-X-C chemokine receptor 2 upregulation with poor prognosis and recurrence in human glioma. Onco Targets Ther. 2015, 8, 3203–3209. [Google Scholar] [CrossRef] [Green Version]
- Acker, G.; Zollfrank, J.; Jelgersma, C.; Nieminen-Kelha, M.; Kremenetskaia, I.; Mueller, S.; Ghori, A.; Vajkoczy, P.; Brandenburg, S. The CXCR2/CXCL2 signalling pathway—An alternative therapeutic approach in high-grade glioma. Eur. J. Cancer 2020, 126, 106–115. [Google Scholar] [CrossRef]
- Brat, D.J.; Bellail, A.C.; Van Meir, E.G. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro-Oncology 2005, 7, 122–133. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.G.; Nyanyo, D.; Hung, C.K.; Goerger, J.P.; Zipfel, W.R.; Williams, R.M.; Nishimura, N.; Fischbach, C. Endothelial cells promote 3D invasion of GBM by IL-8-dependent induction of cancer stem cell properties. Sci. Rep. 2019, 9, 9069. [Google Scholar] [CrossRef]
- Ding, D.C.; Shyu, W.C.; Lin, S.Z. Mesenchymal stem cells. Cell Transplant. 2011, 20, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markov, A.; Thangavelu, L.; Aravindhan, S.; Zekiy, A.O.; Jarahian, M.; Chartrand, M.S.; Pathak, Y.; Marofi, F.; Shamlou, S.; Hassanzadeh, A. Mesenchymal stem/stromal cells as a valuable source for the treatment of immune-mediated disorders. Stem Cell Res. Ther. 2021, 12, 192. [Google Scholar] [CrossRef]
- Yagi, H.; Kitagawa, Y. The role of mesenchymal stem cells in cancer development. Front. Genet. 2013, 4, 261. [Google Scholar] [CrossRef] [Green Version]
- Klopp, A.H.; Gupta, A.; Spaeth, E.; Andreeff, M.; Marini, F., 3rd. Concise review: Dissecting a discrepancy in the literature: Do mesenchymal stem cells support or suppress tumor growth? Stem Cells 2011, 29, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Bajetto, A.; Pattarozzi, A.; Corsaro, A.; Barbieri, F.; Daga, A.; Bosio, A.; Gatti, M.; Pisaturo, V.; Sirito, R.; Florio, T. Different Effects of Human Umbilical Cord Mesenchymal Stem Cells on Glioblastoma Stem Cells by Direct Cell Interaction or Via Released Soluble Factors. Front. Cell Neurosci. 2017, 11, 312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, K.J.; Lee, J.I.; Eom, Y.W. Mesenchymal Stem Cell-Mediated Effects of Tumor Support or Suppression. Int. J. Mol. Sci. 2015, 16, 30015–30033. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Yang, S.J.; Wen, Q.; Zhong, J.F.; Chen, X.L.; Stucky, A.; Press, M.F.; Zhang, X. Human-derived normal mesenchymal stem/stromal cells in anticancer therapies. J. Cancer 2017, 8, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Hübner, M.; Moellhoff, N.; Effinger, D.; Hinske, C.L.; Hirschberger, S.; Wu, T.; Müller, M.B.; Strauß, G.; Kreth, F.W.; Kreth, S. MicroRNA-93 acts as an “anti-inflammatory tumor suppressor” in glioblastoma. Neuro-Oncol. Adv. 2020, 2, vdaa047. [Google Scholar] [CrossRef]
- Strieter, R.M.; Burdick, M.D.; Mestas, J.; Gomperts, B.; Keane, M.P.; Belperio, J.A. Cancer CXC chemokine networks and tumour angiogenesis. Eur. J. Cancer 2006, 42, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Datta, D.; Banerjee, P.; Gasser, M.; Waaga-Gasser, A.M.; Pal, S. CXCR3-B can mediate growth-inhibitory signals in human renal cancer cells by down-regulating the expression of heme oxygenase-1. J. Biol. Chem. 2010, 285, 36842–36848. [Google Scholar] [CrossRef] [Green Version]
- Datta, D.; Flaxenburg, J.A.; Laxmanan, S.; Geehan, C.; Grimm, M.; Waaga-Gasser, A.M.; Briscoe, D.M.; Pal, S. Ras-induced modulation of CXCL10 and its receptor splice variant CXCR3-B in MDA-MB-435 and MCF-7 cells: Relevance for the development of human breast cancer. Cancer Res. 2006, 66, 9509–9518. [Google Scholar] [CrossRef] [Green Version]
- Boye, K.; Pujol, N.; Alves, I.D.; Chen, Y.P.; Daubon, T.; Lee, Y.Z.; Dedieu, S.; Constantin, M.; Bello, L.; Rossi, M.; et al. The role of CXCR3/LRP1 cross-talk in the invasion of primary brain tumors. Nat. Commun. 2017, 8, 1571. [Google Scholar] [CrossRef]
- Wang, P.; Peng, X.; Zhang, J.; Wang, Z.; Meng, J.; Cen, B.; Ji, A.; He, S. LncRNA-135528 inhibits tumor progression by up-regulating CXCL10 through the JAK/STAT pathway. Apoptosis 2018, 23, 651–666. [Google Scholar] [CrossRef]
- Walters, M.J.; Ebsworth, K.; Berahovich, R.D.; Penfold, M.E.; Liu, S.C.; Al Omran, R.; Kioi, M.; Chernikova, S.B.; Tseng, D.; Mulkearns-Hubert, E.E.; et al. Inhibition of CXCR7 extends survival following irradiation of brain tumours in mice and rats. Br. J. Cancer 2014, 110, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Sharma, I.; Siraj, F.; Sharma, K.C.; Singh, A. Immunohistochemical expression of chemokine receptor CXCR3 and its ligand CXCL10 in low-grade astrocytomas and glioblastoma multiforme: A tissue microarray-based comparison. J. Cancer Res. Ther. 2016, 12, 793–797. [Google Scholar] [CrossRef]
- Gjorgjevski, M.; Hannen, R.; Carl, B.; Li, Y.; Landmann, E.; Buchholz, M.; Bartsch, J.W.; Nimsky, C. Molecular profiling of the tumor microenvironment in glioblastoma patients: Correlation of microglia/macrophage polarization state with metalloprotease expression profiles and survival. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Billottet, C.; Quemener, C.; Bikfalvi, A. CXCR3, a double-edged sword in tumor progression and angiogenesis. Biochim. Biophys. Acta 2013, 1836, 287–295. [Google Scholar] [CrossRef]
- Han, J.H.; Yoon, J.S.; Chang, D.Y.; Cho, K.G.; Lim, J.; Kim, S.S.; Suh-Kim, H. CXCR4-STAT3 Axis Plays a Role in Tumor Cell Infiltration in an Orthotopic Mouse Glioblastoma Model. Mol. Cells 2020, 43, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Dono, A.; Zhu, P.; Zorofchian, S.; Takayasu, T.; Quezado, M.M.; Rios, A.; Powers, A.; Esquenazi, Y.; Ballester, L.Y. Variable expression of CXCR4 in molecular subtypes of infiltrating gliomas. Clin. Neuropathol. 2020. [Google Scholar] [CrossRef]
- Goffart, N.; Kroonen, J.; Di Valentin, E.; Dedobbeleer, M.; Denne, A.; Martinive, P.; Rogister, B. Adult mouse subventricular zones stimulate glioblastoma stem cells specific invasion through CXCL12/CXCR4 signaling. Neuro-Oncology 2015, 17, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Truong, D.; Fiorelli, R.; Barrientos, E.S.; Melendez, E.L.; Sanai, N.; Mehta, S.; Nikkhah, M. A three-dimensional (3D) organotypic microfluidic model for glioma stem cells—Vascular interactions. Biomaterials 2019, 198, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sarkar, S.; Yong, V.W. The chemokine stromal cell derived factor-1 (CXCL12) promotes glioma invasiveness through MT2-matrix metalloproteinase. Carcinogenesis 2005, 26, 2069–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, L.; Zhou, X.; Li, T.; Liu, P.; Hai, L.; Tong, L.; Ma, H.; Tao, Z.; Xie, Y.; Zhang, C.; et al. Notch1 signaling pathway promotes invasion, self-renewal and growth of glioma initiating cells via modulating chemokine system CXCL12/CXCR4. J. Exp. Clin. Cancer Res. 2019, 38, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kioi, M.; Vogel, H.; Schultz, G.; Hoffman, R.M.; Harsh, G.R.; Brown, J.M. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J. Clin. Investig. 2010, 120, 694–705. [Google Scholar] [CrossRef]
- Kozin, S.V.; Kamoun, W.S.; Huang, Y.; Dawson, M.R.; Jain, R.K.; Duda, D.G. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res. 2010, 70, 5679–5685. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Chen, C.; Li, J.; Xu, X.; Chen, W.; Li, F. The CXCL12/CXCR4 axis confers temozolomide resistance to human glioblastoma cells via up-regulation of FOXM1. J. Neurol. Sci. 2020, 414, 116837. [Google Scholar] [CrossRef] [PubMed]
- Contento, R.L.; Molon, B.; Boularan, C.; Pozzan, T.; Manes, S.; Marullo, S.; Viola, A. CXCR4-CCR5: A couple modulating T cell functions. Proc. Natl. Acad. Sci. USA 2008, 105, 10101–10106. [Google Scholar] [CrossRef] [Green Version]
- Strasser, G.A.; Kaminker, J.S.; Tessier-Lavigne, M. Microarray analysis of retinal endothelial tip cells identifies CXCR4 as a mediator of tip cell morphology and branching. Blood 2010, 115, 5102–5110. [Google Scholar] [CrossRef] [Green Version]
- Schulte, A.; Günther, H.S.; Phillips, H.S.; Kemming, D.; Martens, T.; Kharbanda, S.; Soriano, R.H.; Modrusan, Z.; Zapf, S.; Westphal, M.; et al. A distinct subset of glioma cell lines with stem cell-like properties reflects the transcriptional phenotype of glioblastomas and overexpresses CXCR4 as therapeutic target. Glia 2011, 59, 590–602. [Google Scholar] [CrossRef]
- Zagzag, D.; Lukyanov, Y.; Lan, L.; Ali, M.A.; Esencay, M.; Mendez, O.; Yee, H.; Voura, E.B.; Newcomb, E.W. Hypoxia-inducible factor 1 and VEGF upregulate CXCR4 in glioblastoma: Implications for angiogenesis and glioma cell invasion. Lab. Investig. J. Tech. Methods Pathol. 2006, 86, 1221–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, J.B.; Kung, A.L.; Klein, R.S.; Chan, J.A.; Sun, Y.; Schmidt, K.; Kieran, M.W.; Luster, A.D.; Segal, R.A. A small-molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 13513–13518. [Google Scholar] [CrossRef] [Green Version]
- Calinescu, A.A.; Yadav, V.N.; Carballo, E.; Kadiyala, P.; Tran, D.; Zamler, D.B.; Doherty, R.; Srikanth, M.; Lowenstein, P.R.; Castro, M.G. Survival and Proliferation of Neural Progenitor-Derived Glioblastomas Under Hypoxic Stress is Controlled by a CXCL12/CXCR4 Autocrine-Positive Feedback Mechanism. Clin. Cancer Res. 2017, 23, 1250–1262. [Google Scholar] [CrossRef] [Green Version]
- Hira, V.V.; Verbovšek, U.; Breznik, B.; Srdič, M.; Novinec, M.; Kakar, H.; Wormer, J.; der Swaan, B.V.; Lenarčič, B.; Juliano, L.; et al. Cathepsin K cleavage of SDF-1α inhibits its chemotactic activity towards glioblastoma stem-like cells. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 594–603. [Google Scholar] [CrossRef]
- Redjal, N.; Chan, J.A.; Segal, R.A.; Kung, A.L. CXCR4 inhibition synergizes with cytotoxic chemotherapy in gliomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 6765–6771. [Google Scholar] [CrossRef] [Green Version]
- Cornelison, R.C.; Brennan, C.E.; Kingsmore, K.M.; Munson, J.M. Convective forces increase CXCR4-dependent glioblastoma cell invasion in GL261 murine model. Sci. Rep. 2018, 8, 17057. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Wang, B.; Chen, Y.; Liu, H.; Shi, L. Novel CXCR4 Inhibitor CPZ1344 Inhibits the Proliferation, Migration and Angiogenesis of Glioblastoma. Pathol. Oncol. Res. 2020, 26, 2597–2604. [Google Scholar] [CrossRef]
- Yang, L.; Jackson, E.; Woerner, B.M.; Perry, A.; Piwnica-Worms, D.; Rubin, J.B. Blocking CXCR4-mediated cyclic AMP suppression inhibits brain tumor growth in vivo. Cancer Res. 2007, 67, 651–658. [Google Scholar] [CrossRef] [Green Version]
- Mercurio, L.; Ajmone-Cat, M.A.; Cecchetti, S.; Ricci, A.; Bozzuto, G.; Molinari, A.; Manni, I.; Pollo, B.; Scala, S.; Carpinelli, G.; et al. Targeting CXCR4 by a selective peptide antagonist modulates tumor microenvironment and microglia reactivity in a human glioblastoma model. J. Exp. Clin. Cancer Res. CR 2016, 35, 55. [Google Scholar] [CrossRef]
- Deng, L.; Zheng, W.; Dong, X.; Liu, J.; Zhu, C.; Lu, D.; Zhang, J.; Song, L.; Wang, Y.; Deng, D. Chemokine receptor CXCR7 is an independent prognostic biomarker in glioblastoma. Cancer Biomark. 2017, 20, 1–6. [Google Scholar] [CrossRef]
- Liu, Y.; Carson-Walter, E.; Walter, K.A. Targeting chemokine receptor CXCR7 inhibits glioma cell proliferation and mobility. Anticancer Res. 2015, 35, 53–64. [Google Scholar]
- Calatozzolo, C.; Canazza, A.; Pollo, B.; Di Pierro, E.; Ciusani, E.; Maderna, E.; Salce, E.; Sponza, V.; Frigerio, S.; Di Meco, F.; et al. Expression of the new CXCL12 receptor, CXCR7, in gliomas. Cancer Biol. Ther. 2011, 11, 242–253. [Google Scholar] [CrossRef] [Green Version]
- Sehgal, A.; Keener, C.; Boynton, A.L.; Warrick, J.; Murphy, G.P. CXCR-4, a chemokine receptor, is overexpressed in and required for proliferation of glioblastoma tumor cells. J. Surg. Oncol. 1998, 69, 99–104. [Google Scholar] [CrossRef]
- Barbero, S.; Bonavia, R.; Bajetto, A.; Porcile, C.; Pirani, P.; Ravetti, J.L.; Zona, G.L.; Spaziante, R.; Florio, T.; Schettini, G. Stromal cell-derived factor 1alpha stimulates human glioblastoma cell growth through the activation of both extracellular signal-regulated kinases 1/2 and Akt. Cancer Res. 2003, 63, 1969–1974. [Google Scholar]
- Shi, Y.; Chen, C.; Yu, S.Z.; Liu, Q.; Rao, J.; Zhang, H.R.; Xiao, H.L.; Fu, T.W.; Long, H.; He, Z.C.; et al. miR-663 Suppresses Oncogenic Function of CXCR4 in Glioblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 4004–4013. [Google Scholar] [CrossRef] [Green Version]
- Fareh, M.; Turchi, L.; Virolle, V.; Debruyne, D.; Almairac, F.; de-la-Forest Divonne, S.; Paquis, P.; Preynat-Seauve, O.; Krause, K.H.; Chneiweiss, H.; et al. The miR 302–367 cluster drastically affects self-renewal and infiltration properties of glioma-initiating cells through CXCR4 repression and consequent disruption of the SHH-GLI-NANOG network. Cell Death Differ. 2012, 19, 232–244. [Google Scholar] [CrossRef]
- Fareh, M.; Almairac, F.; Turchi, L.; Burel-Vandenbos, F.; Paquis, P.; Fontaine, D.; Lacas-Gervais, S.; Junier, M.P.; Chneiweiss, H.; Virolle, T. Cell-based therapy using miR-302-367 expressing cells represses glioblastoma growth. Cell Death Dis. 2017, 8, e2713. [Google Scholar] [CrossRef] [PubMed]
- Gascon, S.; Giraldo Solano, A.; El Kheir, W.; Therriault, H.; Berthelin, P.; Cattier, B.; Marcos, B.; Virgilio, N.; Paquette, B.; Faucheux, N.; et al. Characterization and Mathematical Modeling of Alginate/Chitosan-Based Nanoparticles Releasing the Chemokine CXCL12 to Attract Glioblastoma Cells. Pharmaceutics 2020, 12, 356. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Wang, C.; Fitch, S.; Yang, F. Targeting Tumor Hypoxia Using Nanoparticle-engineered CXCR4-overexpressing Adipose-derived Stem Cells. Theranostics 2018, 8, 1350–1360. [Google Scholar] [CrossRef]
- Li, M.; Zeng, L.; Liu, S.; Dangelmajer, S.; Kahlert, U.D.; Huang, H.; Han, Y.; Chi, X.; Zhu, M.; Lei, T. Transforming Growth Factor-β Promotes Homing and Therapeutic Efficacy of Human Mesenchymal Stem Cells to Glioblastoma. J. Neuropathol. Exp. Neurol. 2019, 78, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Pavon, L.F.; Sibov, T.T.; de Souza, A.V.; da Cruz, E.F.; Malheiros, S.M.F.; Cabral, F.R.; de Souza, J.G.; Boufleur, P.; de Oliveira, D.M.; de Toledo, S.R.C.; et al. Tropism of mesenchymal stem cell toward CD133(+) stem cell of glioblastoma in vitro and promote tumor proliferation in vivo. Stem Cell Res. Ther. 2018, 9, 310. [Google Scholar] [CrossRef]
- Kranjc, M.K.; Novak, M.; Pestell, R.G.; Lah, T.T. Cytokine CCL5 and receptor CCR5 axis in glioblastoma multiforme. Radiol. Oncol. 2019, 53, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Pham, K.; Luo, D.; Liu, C.; Harrison, J.K. CCL5, CCR1 and CCR5 in murine glioblastoma: Immune cell infiltration and survival rates are not dependent on individual expression of either CCR1 or CCR5. J. Neuroimmunol. 2012, 246, 10–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.Y.; Wu, C.Y.; Fang, J.H.; Chen, H.C.; Feng, L.Y.; Huang, C.Y.; Wei, K.C.; Fang, J.Y.; Lin, C.Y. Functional Change of Effector Tumor-Infiltrating CCR5(+)CD38(+)HLA-DR(+)CD8(+) T Cells in Glioma Microenvironment. Front. Immunol. 2019, 10, 2395. [Google Scholar] [CrossRef] [PubMed]
- Sciume, G.; Soriani, A.; Piccoli, M.; Frati, L.; Santoni, A.; Bernardini, G. CX3CR1/CX3CL1 axis negatively controls glioma cell invasion and is modulated by transforming growth factor-beta1. Neuro-Oncology 2010, 12, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Zhao, Q.; Huang, L.; Zheng, Y.; Li, L.; He, Q.; Zhang, C.; Li, F.; Maimela, N.R.; Sun, Z.; et al. The R132H mutation in IDH1 promotes the recruitment of NK cells through CX3CL1/CX3CR1 chemotaxis and is correlated with a better prognosis in gliomas. Immunol. Cell Biol. 2019, 97, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Held-Feindt, J.; Hattermann, K.; Muerkoster, S.S.; Wedderkopp, H.; Knerlich-Lukoschus, F.; Ungefroren, H.; Mehdorn, H.M.; Mentlein, R. CX3CR1 promotes recruitment of human glioma-infiltrating microglia/macrophages (GIMs). Exp. Cell Res. 2010, 316, 1553–1566. [Google Scholar] [CrossRef] [PubMed]
- Morisse, M.C.; Jouannet, S.; Dominguez-Villar, M.; Sanson, M.; Idbaih, A. Interactions between tumor-associated macrophages and tumor cells in glioblastoma: Unraveling promising targeted therapies. Expert Rev. Neurother. 2018, 18, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Latha, K.; Manyam, G.; Yang, Y.; Rao, A.; Rao, G. Role of CX3CR1 signaling in malignant transformation of gliomas. Neuro-Oncology 2020, 22, 1463–1473. [Google Scholar] [CrossRef]
- Lacroix, M.; Abi-Said, D.; Fourney, D.R.; Gokaslan, Z.L.; Shi, W.; DeMonte, F.; Lang, F.F.; McCutcheon, I.E.; Hassenbusch, S.J.; Holland, E.; et al. A multivariate analysis of 416 patients with glioblastoma multiforme: Prognosis, extent of resection, and survival. J. Neurosurg. 2001, 95, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.M.; Suki, D.; Hess, K.; Sawaya, R. The influence of maximum safe resection of glioblastoma on survival in 1229 patients: Can we do better than gross-total resection? J. Neurosurg. 2016, 124, 977–988. [Google Scholar] [CrossRef] [Green Version]
- Sanai, N.; Polley, M.Y.; McDermott, M.W.; Parsa, A.T.; Berger, M.S. An extent of resection threshold for newly diagnosed glioblastomas. J. Neurosurg. 2011, 115, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.C.; Alomran, R.; Chernikova, S.B.; Lartey, F.; Stafford, J.; Jang, T.; Merchant, M.; Zboralski, D.; Zöllner, S.; Kruschinski, A.; et al. Blockade of SDF-1 after irradiation inhibits tumor recurrences of autochthonous brain tumors in rats. Neuro-Oncology 2014, 16, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Rios, A.; Hsu, S.H.; Blanco, A.; Buryanek, J.; Day, A.L.; McGuire, M.F.; Brown, R.E. Durable response of glioblastoma to adjuvant therapy consisting of temozolomide and a weekly dose of AMD3100 (plerixafor), a CXCR4 inhibitor, together with lapatinib, metformin and niacinamide. Oncoscience 2016, 3, 156–163. [Google Scholar] [CrossRef] [Green Version]
- Tseng, D.; Vasquez-Medrano, D.A.; Brown, J.M. Targeting SDF-1/CXCR4 to inhibit tumour vasculature for treatment of glioblastomas. Br. J. Cancer 2011, 104, 1805–1809. [Google Scholar] [CrossRef] [Green Version]
- Thomas, R.P.; Nagpal, S.; Iv, M.; Soltys, S.G.; Bertrand, S.; Pelpola, J.S.; Ball, R.; Yang, J.; Sundaram, V.; Lavezo, J.; et al. Macrophage Exclusion after Radiation Therapy (MERT): A First in Human Phase I/II Trial using a CXCR4 Inhibitor in Glioblastoma. Clin. Cancer Res. 2019, 25, 6948–6957. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, M.C. Radiographic patterns of relapse in glioblastoma. J. Neurooncol. 2011, 101, 319–323. [Google Scholar] [CrossRef]
- Jin, U.H.; Michelhaugh, S.K.; Polin, L.A.; Shrestha, R.; Mittal, S.; Safe, S. Omeprazole Inhibits Glioblastoma Cell Invasion and Tumor Growth. Cancers 2020, 12, 2097. [Google Scholar] [CrossRef] [PubMed]
- Séhédic, D.; Chourpa, I.; Tétaud, C.; Griveau, A.; Loussouarn, C.; Avril, S.; Legendre, C.; Lepareur, N.; Wion, D.; Hindré, F.; et al. Locoregional Confinement and Major Clinical Benefit of (188)Re-Loaded CXCR4-Targeted Nanocarriers in an Orthotopic Human to Mouse Model of Glioblastoma. Theranostics 2017, 7, 4517–4536. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, I.H.; O’Donovan, D.G.; Brenchley, P.E.; Reid, H.; Roberts, I.S. Vascular endothelial growth factor expression correlates with tumour grade and vascularity in gliomas. Histopathology 2001, 39, 409–415. [Google Scholar] [CrossRef]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef]
- Piao, Y.; Liang, J.; Holmes, L.; Zurita, A.J.; Henry, V.; Heymach, J.V.; de Groot, J.F. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro-Oncology 2012, 14, 1379–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soda, Y.; Myskiw, C.; Rommel, A.; Verma, I.M. Mechanisms of neovascularization and resistance to anti-angiogenic therapies in glioblastoma multiforme. J. Mol. Med. 2013, 91, 439–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folberg, R.; Maniotis, A.J. Vasculogenic mimicry. APMIS 2004, 112, 508–525. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef] [Green Version]
- Hong, X.; Jiang, F.; Kalkanis, S.N.; Zhang, Z.G.; Zhang, X.P.; DeCarvalho, A.C.; Katakowski, M.; Bobbitt, K.; Mikkelsen, T.; Chopp, M. SDF-1 and CXCR4 are up-regulated by VEGF and contribute to glioma cell invasion. Cancer Lett. 2006, 236, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Ping, Y.F.; Yao, X.H.; Jiang, J.Y.; Zhao, L.T.; Yu, S.C.; Jiang, T.; Lin, M.C.; Chen, J.H.; Wang, B.; Zhang, R.; et al. The chemokine CXCL12 and its receptor CXCR4 promote glioma stem cell-mediated VEGF production and tumour angiogenesis via PI3K/AKT signalling. J. Pathol. 2011, 224, 344–354. [Google Scholar] [CrossRef]
- Pham, K.; Luo, D.; Siemann, D.W.; Law, B.K.; Reynolds, B.A.; Hothi, P.; Foltz, G.; Harrison, J.K. VEGFR inhibitors upregulate CXCR4 in VEGF receptor-expressing glioblastoma in a TGFβR signaling-dependent manner. Cancer Lett. 2015, 360, 60–67. [Google Scholar] [CrossRef]
- Ping, Y.F.; Yao, X.H.; Chen, J.H.; Liu, H.; Chen, D.L.; Zhou, X.D.; Wang, J.M.; Bian, X.W. The anti-cancer compound Nordy inhibits CXCR4-mediated production of IL-8 and VEGF by malignant human glioma cells. J. Neurooncol. 2007, 84, 21–29. [Google Scholar] [CrossRef]
- Ricard, C.; Stanchi, F.; Rodriguez, T.; Amoureux, M.C.; Rougon, G.; Debarbieux, F. Dynamic quantitative intravital imaging of glioblastoma progression reveals a lack of correlation between tumor growth and blood vessel density. PLoS ONE 2013, 8, e72655. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.M.; Kumar, S.; Shankar, A.; Varma, N.R.; Iskander, A.S.; Janic, B.; Chwang, W.B.; Jain, R.; Babajeni-Feremi, A.; Borin, T.F.; et al. Effects of tyrosine kinase inhibitors and CXCR4 antagonist on tumor growth and angiogenesis in rat glioma model: MRI and protein analysis study. Transl. Oncol. 2013, 6, 660–669. [Google Scholar] [CrossRef] [Green Version]
- Shaaban, S.; Alsulami, M.; Arbab, S.A.; Ara, R.; Shankar, A.; Iskander, A.; Angara, K.; Jain, M.; Bagher-Ebadian, H.; Achyut, B.R.; et al. Targeting Bone Marrow to Potentiate the Anti-Tumor Effect of Tyrosine Kinase Inhibitor in Preclinical Rat Model of Human Glioblastoma. Int. J. Cancer Res. 2016, 12, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Achyut, B.R.; Shankar, A.; Iskander, A.S.; Ara, R.; Knight, R.A.; Scicli, A.G.; Arbab, A.S. Chimeric Mouse model to track the migration of bone marrow derived cells in glioblastoma following anti-angiogenic treatments. Cancer Biol. Ther. 2016, 17, 280–290. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.Q.; Duda, D.G.; Muzikansky, A.; Gerstner, E.R.; Kuhn, J.G.; Reardon, D.A.; Nayak, L.; Norden, A.D.; Doherty, L.; LaFrankie, D.; et al. Phase I and Biomarker Study of Plerixafor and Bevacizumab in Recurrent High-Grade Glioma. Clin. Cancer Res. 2018, 24, 4643–4649. [Google Scholar] [CrossRef] [Green Version]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef]
- Gravina, G.L.; Mancini, A.; Colapietro, A.; Vitale, F.; Vetuschi, A.; Pompili, S.; Rossi, G.; Marampon, F.; Richardson, P.J.; Patient, L.; et al. The novel CXCR4 antagonist, PRX177561, reduces tumor cell proliferation and accelerates cancer stem cell differentiation in glioblastoma preclinical models. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2017, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravina, G.L.; Mancini, A.; Marampon, F.; Colapietro, A.; Delle Monache, S.; Sferra, R.; Vitale, F.; Richardson, P.J.; Patient, L.; Burbidge, S.; et al. The brain-penetrating CXCR4 antagonist, PRX177561, increases the antitumor effects of bevacizumab and sunitinib in preclinical models of human glioblastoma. J. Hematol. Oncol. 2017, 10, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagner, J.P.; Sarfraz, Y.; Ortenzi, V.; Alotaibi, F.M.; Chiriboga, L.A.; Tayyib, A.T.; Douglas, G.J.; Chevalier, E.; Romagnoli, B.; Tuffin, G.; et al. Multifaceted C-X-C Chemokine Receptor 4 (CXCR4) Inhibition Interferes with Anti-Vascular Endothelial Growth Factor Therapy-Induced Glioma Dissemination. Am. J. Pathol. 2017, 187, 2080–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egorova, A.; Shubina, A.; Sokolov, D.; Selkov, S.; Baranov, V.; Kiselev, A. CXCR4-targeted modular peptide carriers for efficient anti-VEGF siRNA delivery. Int. J. Pharm. 2016, 515, 431–440. [Google Scholar] [CrossRef]
- da Fonseca, A.C.; Badie, B. Microglia and macrophages in malignant gliomas: Recent discoveries and implications for promising therapies. Clin. Dev. Immunol. 2013, 2013, 264124. [Google Scholar] [CrossRef]
- Cho, H.R.; Kumari, N.; Thi Vu, H.; Kim, H.; Park, C.K.; Choi, S.H. Increased Antiangiogenic Effect by Blocking CCL2-dependent Macrophages in a Rodent Glioblastoma Model: Correlation Study with Dynamic Susceptibility Contrast Perfusion MRI. Sci. Rep. 2019, 9, 11085. [Google Scholar] [CrossRef]
- Chan, H.Y.; Choi, J.; Jackson, C.; Lim, M. Combination immunotherapy strategies for glioblastoma. J. Neurooncol. 2021, 151, 375–391. [Google Scholar] [CrossRef] [PubMed]
- Medikonda, R.; Dunn, G.; Rahman, M.; Fecci, P.; Lim, M. A review of glioblastoma immunotherapy. J. Neurooncol. 2021, 151, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhou, H.; Xu, J.; Lu, Y.; Ji, X.; Yao, Y.; Chao, H.; Zhang, J.; Zhang, X.; Yao, S.; et al. Different T-cell subsets in glioblastoma multiforme and targeted immunotherapy. Cancer Lett. 2021, 496, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Mangani, D.; Weller, M.; Roth, P. The network of immunosuppressive pathways in glioblastoma. Biochem. Pharmacol. 2017, 130, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Larson, R.C.; Maus, M.V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef]
- Jin, L.; Tao, H.; Karachi, A.; Long, Y.; Hou, A.Y.; Na, M.; Dyson, K.A.; Grippin, A.J.; Deleyrolle, L.P.; Zhang, W.; et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat. Commun. 2019, 10, 4016. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.Q.; Liu, D.Q.; Chen, S.P.; Sun, J.; Zhou, X.R.; Xing, C.; Ye, D.W.; Tian, Y.K. The Role of CXCR3 in Neurological Diseases. Curr. Neuropharmacol. 2019, 17, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Enderlin, M.; Kleinmann, E.V.; Struyf, S.; Buracchi, C.; Vecchi, A.; Kinscherf, R.; Kiessling, F.; Paschek, S.; Sozzani, S.; Rommelaere, J.; et al. TNF-alpha and the IFN-gamma-inducible protein 10 (IP-10/CXCL-10) delivered by parvoviral vectors act in synergy to induce antitumor effects in mouse glioblastoma. Cancer Gene Ther. 2009, 16, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Luo, D.; Reynolds, B.A.; Meher, G.; Katritzky, A.R.; Lu, B.; Gerard, C.J.; Bhadha, C.P.; Harrison, J.K. Chemokine receptor CXCR3 promotes growth of glioma. Carcinogenesis 2011, 32, 129–137. [Google Scholar] [CrossRef]
- Fujita, M.; Kohanbash, G.; Fellows-Mayle, W.; Hamilton, R.L.; Komohara, Y.; Decker, S.A.; Ohlfest, J.R.; Okada, H. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res. 2011, 71, 2664–2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosaka, A.; Ohkuri, T.; Okada, H. Combination of an agonistic anti-CD40 monoclonal antibody and the COX-2 inhibitor celecoxib induces anti-glioma effects by promotion of type-1 immunity in myeloid cells and T-cells. Cancer Immunol. Immunother. 2014, 63, 847–857. [Google Scholar] [CrossRef] [Green Version]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Shono, K.; Yamaguchi, I.; Mizobuchi, Y.; Kagusa, H.; Sumi, A.; Fujihara, T.; Nakajima, K.; Kitazato, K.T.; Matsuzaki, K.; Saya, H.; et al. Downregulation of the CCL2/CCR2 and CXCL10/CXCR3 axes contributes to antitumor effects in a mouse model of malignant glioma. Sci. Rep. 2020, 10, 15286. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Zhu, X.; Ueda, R.; Sasaki, K.; Kohanbash, G.; Kastenhuber, E.R.; McDonald, H.A.; Gibson, G.A.; Watkins, S.C.; Muthuswamy, R.; et al. Effective immunotherapy against murine gliomas using type 1 polarizing dendritic cells—Significant roles of CXCL10. Cancer Res. 2009, 69, 1587–1595. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.B.; Lu, X.L.; Hu, P.; Liu, R.E. Improved therapeutic efficacy using vaccination with glioma lysate-pulsed dendritic cells combined with IP-10 in murine glioma. Vaccine 2009, 27, 6210–6216. [Google Scholar] [CrossRef]
- Srivastava, S.; Jackson, C.; Kim, T.; Choi, J.; Lim, M. A Characterization of Dendritic Cells and Their Role in Immunotherapy in Glioblastoma: From Preclinical Studies to Clinical Trials. Cancers 2019, 11, 537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Zeng, S.; Gong, Z.; Xu, Z. Clinical implication of cellular vaccine in glioma: Current advances and future prospects. J. Exp. Clin. Cancer Res. 2020, 39, 257. [Google Scholar] [CrossRef]
- De Waele, J.; Marcq, E.; Van Audenaerde, J.R.; Van Loenhout, J.; Deben, C.; Zwaenepoel, K.; Van de Kelft, E.; Van der Planken, D.; Menovsky, T.; Van den Bergh, J.M.; et al. Poly(I:C) primes primary human glioblastoma cells for an immune response invigorated by PD-L1 blockade. Oncoimmunology 2018, 7, e1407899. [Google Scholar] [CrossRef]
- Ai, L.; Xu, A.; Xu, J. Roles of PD-1/PD-L1 Pathway: Signaling, Cancer, and Beyond. Adv. Exp. Med. Biol. 2020, 1248, 33–59. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bahr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Kmiecik, J.; Zimmer, J.; Chekenya, M. Natural killer cells in intracranial neoplasms: Presence and therapeutic efficacy against brain tumours. J. Neurooncol. 2014, 116, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Müller, N.; Michen, S.; Tietze, S.; Töpfer, K.; Schulte, A.; Lamszus, K.; Schmitz, M.; Schackert, G.; Pastan, I.; Temme, A. Engineering NK Cells Modified With an EGFRvIII-specific Chimeric Antigen Receptor to Overexpress CXCR4 Improves Immunotherapy of CXCL12/SDF-1α-secreting Glioblastoma. J. Immunother. 2015, 38, 197–210. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.; Maxwell, R.; Xia, Y.; Cardarelli, P.; Oyasu, M.; Belcaid, Z.; Kim, E.; Hung, A.; Luksik, A.S.; Garzon-Muvdi, T.; et al. Combination anti-CXCR4 and anti-PD-1 immunotherapy provides survival benefit in glioblastoma through immune cell modulation of tumor microenvironment. J. Neurooncol. 2019, 143, 241–249. [Google Scholar] [CrossRef]
- Jordan, J.T.; Sun, W.; Hussain, S.F.; DeAngulo, G.; Prabhu, S.S.; Heimberger, A.B. Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol. Immunother. 2008, 57, 123–131. [Google Scholar] [CrossRef]
- Panek, W.K.; Pituch, K.C.; Miska, J.; Kim, J.W.; Rashidi, A.; Kanojia, D.; Lopez-Rosas, A.; Han, Y.; Yu, D.; Chang, C.L.; et al. Local Application of Autologous Platelet-Rich Fibrin Patch (PRF-P) Suppresses Regulatory T Cell Recruitment in a Murine Glioma Model. Mol. Neurobiol. 2019, 56, 5032–5040. [Google Scholar] [CrossRef]
- Kim, C.H.; Johnston, B.; Butcher, E.C. Trafficking machinery of NKT cells: Shared and differential chemokine receptor expression among V alpha 24(+)V beta 11(+) NKT cell subsets with distinct cytokine-producing capacity. Blood 2002, 100, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Bollen, A.W.; Gupta, N. CC chemokine receptor-2A is frequently overexpressed in glioblastoma. J. Neurooncol. 2008, 86, 153–163. [Google Scholar] [CrossRef]
- Ye, X.Z.; Xu, S.L.; Xin, Y.H.; Yu, S.C.; Ping, Y.F.; Chen, L.; Xiao, H.L.; Wang, B.; Yi, L.; Wang, Q.L.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-beta1 signaling pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Crane, C.A.; Ahn, B.J.; Han, S.J.; Parsa, A.T. Soluble factors secreted by glioblastoma cell lines facilitate recruitment, survival, and expansion of regulatory T cells: Implications for immunotherapy. Neuro-Oncology 2012, 14, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Vasco, C.; Canazza, A.; Rizzo, A.; Mossa, A.; Corsini, E.; Silvani, A.; Fariselli, L.; Salmaggi, A.; Ciusani, E. Circulating T regulatory cells migration and phenotype in glioblastoma patients: An in vitro study. J. Neurooncol. 2013, 115, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, M.P.; Lin, Y.; New, K.C.; Bulur, P.A.; O’Neill, B.P.; Gastineau, D.A.; Dietz, A.B. Systemic immune suppression in glioblastoma: The interplay between CD14+HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro-Oncology 2010, 12, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Alieva, M.; Margarido, A.S.; Wieles, T.; Abels, E.R.; Colak, B.; Boquetale, C.; Jan Noordmans, H.; Snijders, T.J.; Broekman, M.L.; van Rheenen, J. Preventing inflammation inhibits biopsy-mediated changes in tumor cell behavior. Sci. Rep. 2017, 7, 7529. [Google Scholar] [CrossRef] [Green Version]
- Flores-Toro, J.A.; Luo, D.; Gopinath, A.; Sarkisian, M.R.; Campbell, J.J.; Charo, I.F.; Singh, R.; Schall, T.J.; Datta, M.; Jain, R.K.; et al. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc. Natl. Acad. Sci. USA 2020, 117, 1129–1138. [Google Scholar] [CrossRef]
- Hattermann, K.; Bartsch, K.; Gebhardt, H.H.; Mehdorn, H.M.; Synowitz, M.; Schmitt, A.D.; Mentlein, R.; Held-Feindt, J. “Inverse signaling” of the transmembrane chemokine CXCL16 contributes to proliferative and anti-apoptotic effects in cultured human meningioma cells. Cell Commun. Signal 2016, 14, 26. [Google Scholar] [CrossRef] [Green Version]
- Hattermann, K.; Ludwig, A.; Gieselmann, V.; Held-Feindt, J.; Mentlein, R. The chemokine CXCL16 induces migration and invasion of glial precursor cells via its receptor CXCR6. Mol. Cell Neurosci. 2008, 39, 133–141. [Google Scholar] [CrossRef]
- Hattermann, K.; Sebens, S.; Helm, O.; Schmitt, A.D.; Mentlein, R.; Mehdorn, H.M.; Held-Feindt, J. Chemokine expression profile of freshly isolated human glioblastoma-associated macrophages/microglia. Oncol. Rep. 2014, 32, 270–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urbantat, R.M.; Vajkoczy, P.; Brandenburg, S. Advances in Chemokine Signaling Pathways as Therapeutic Targets in Glioblastoma. Cancers 2021, 13, 2983. https://doi.org/10.3390/cancers13122983
Urbantat RM, Vajkoczy P, Brandenburg S. Advances in Chemokine Signaling Pathways as Therapeutic Targets in Glioblastoma. Cancers. 2021; 13(12):2983. https://doi.org/10.3390/cancers13122983
Chicago/Turabian StyleUrbantat, Ruth M., Peter Vajkoczy, and Susan Brandenburg. 2021. "Advances in Chemokine Signaling Pathways as Therapeutic Targets in Glioblastoma" Cancers 13, no. 12: 2983. https://doi.org/10.3390/cancers13122983
APA StyleUrbantat, R. M., Vajkoczy, P., & Brandenburg, S. (2021). Advances in Chemokine Signaling Pathways as Therapeutic Targets in Glioblastoma. Cancers, 13(12), 2983. https://doi.org/10.3390/cancers13122983