
Upper Aerodigestive Tract Squamous Cell Carcinomas Show Distinct Overall DNA Methylation Profiles and Different Molecular Mechanisms behind WNT Signaling Disruption

 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Methylome Analysis
2.3. Cellular Component Analysis
2.4. RNA-Seq Analysis
2.5. TCGA Data
3. Results
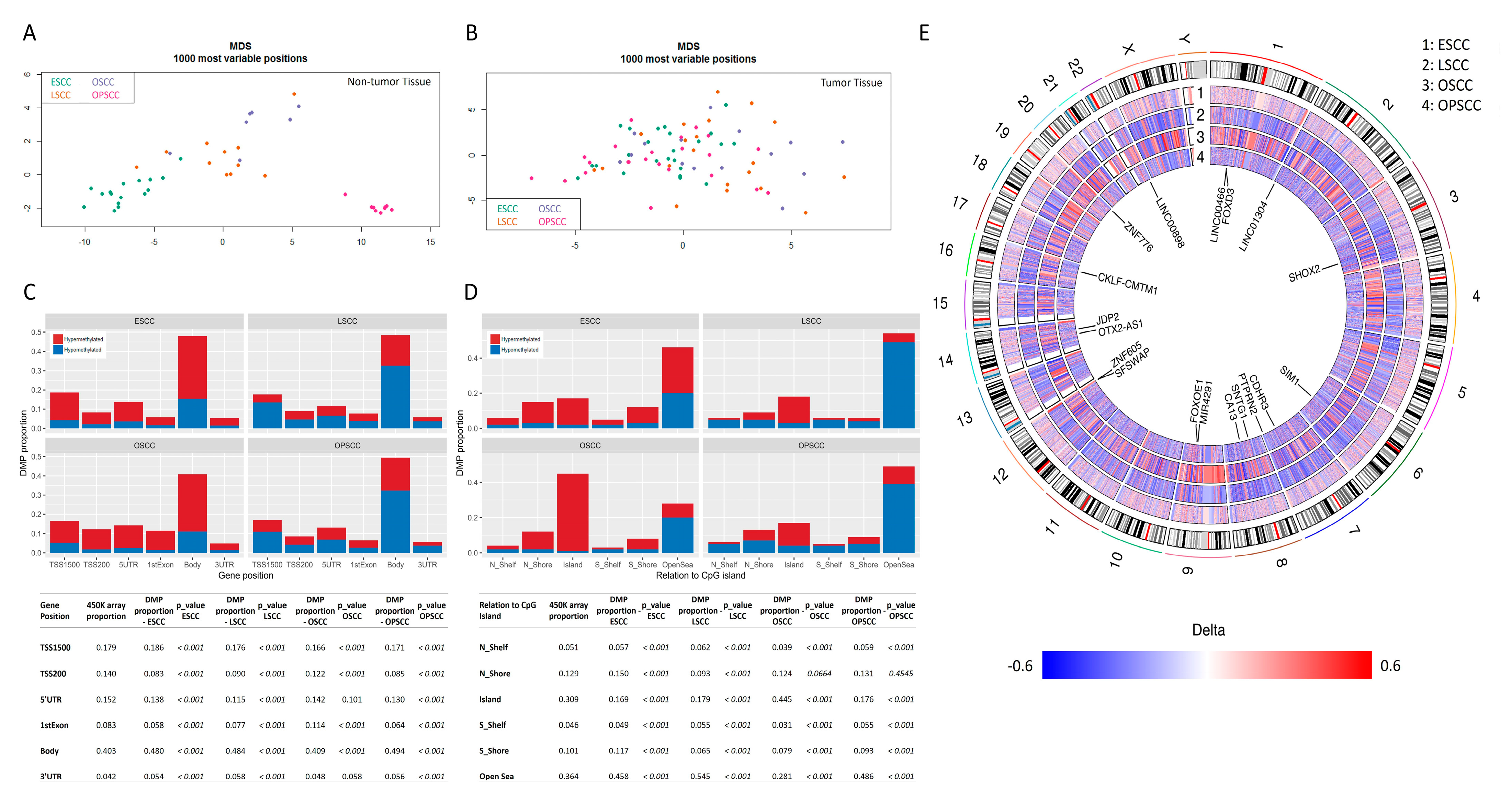
3.1. Global DNA Methylation Profile Is Quite Distinct among UADT Squamous Cell Carcinomas
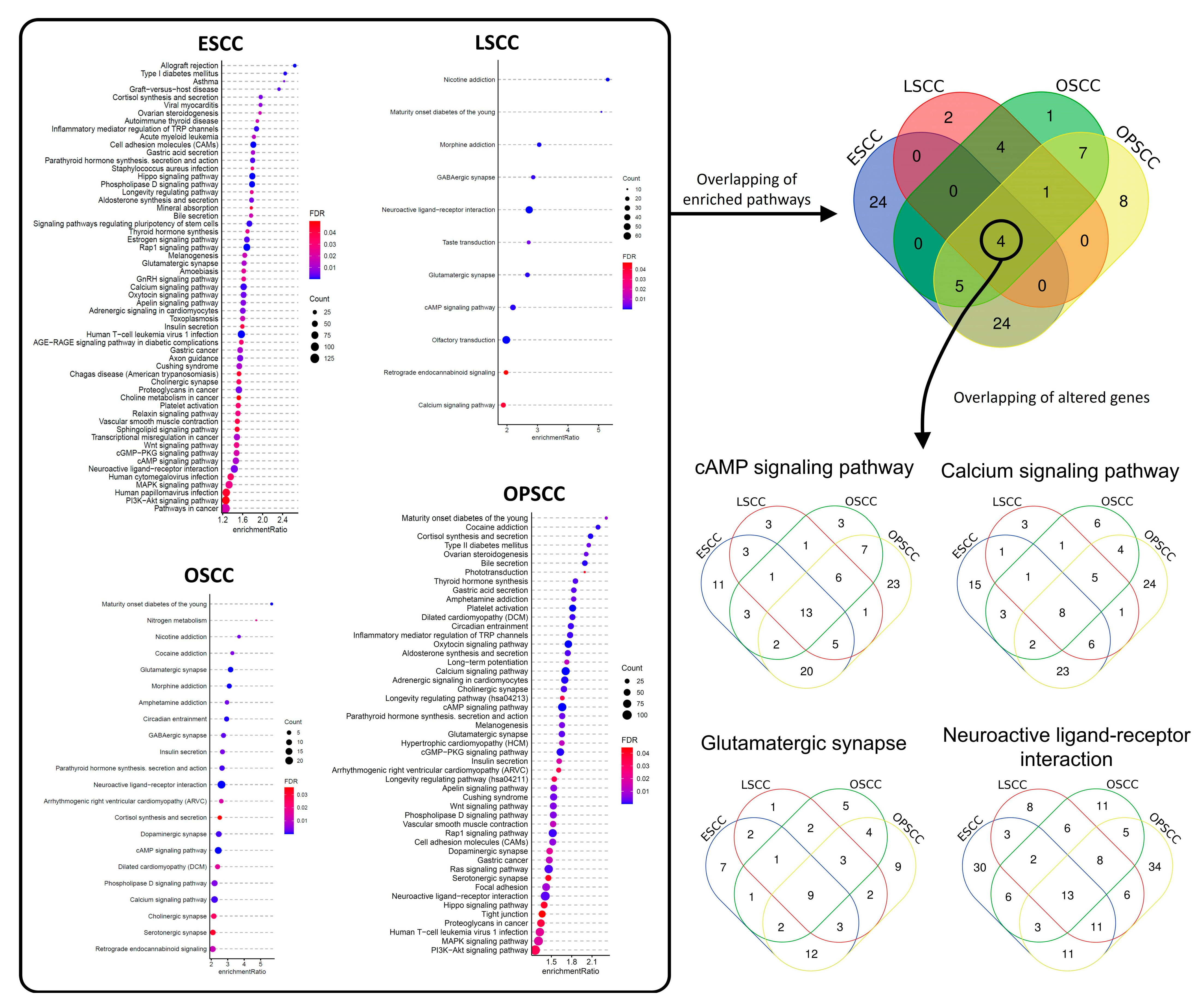
3.2. Differentially Methylated Regions Affect Different Cell Pathways and Different Genes in the Same Cell Signaling Pathways, According to Tumor Subsite
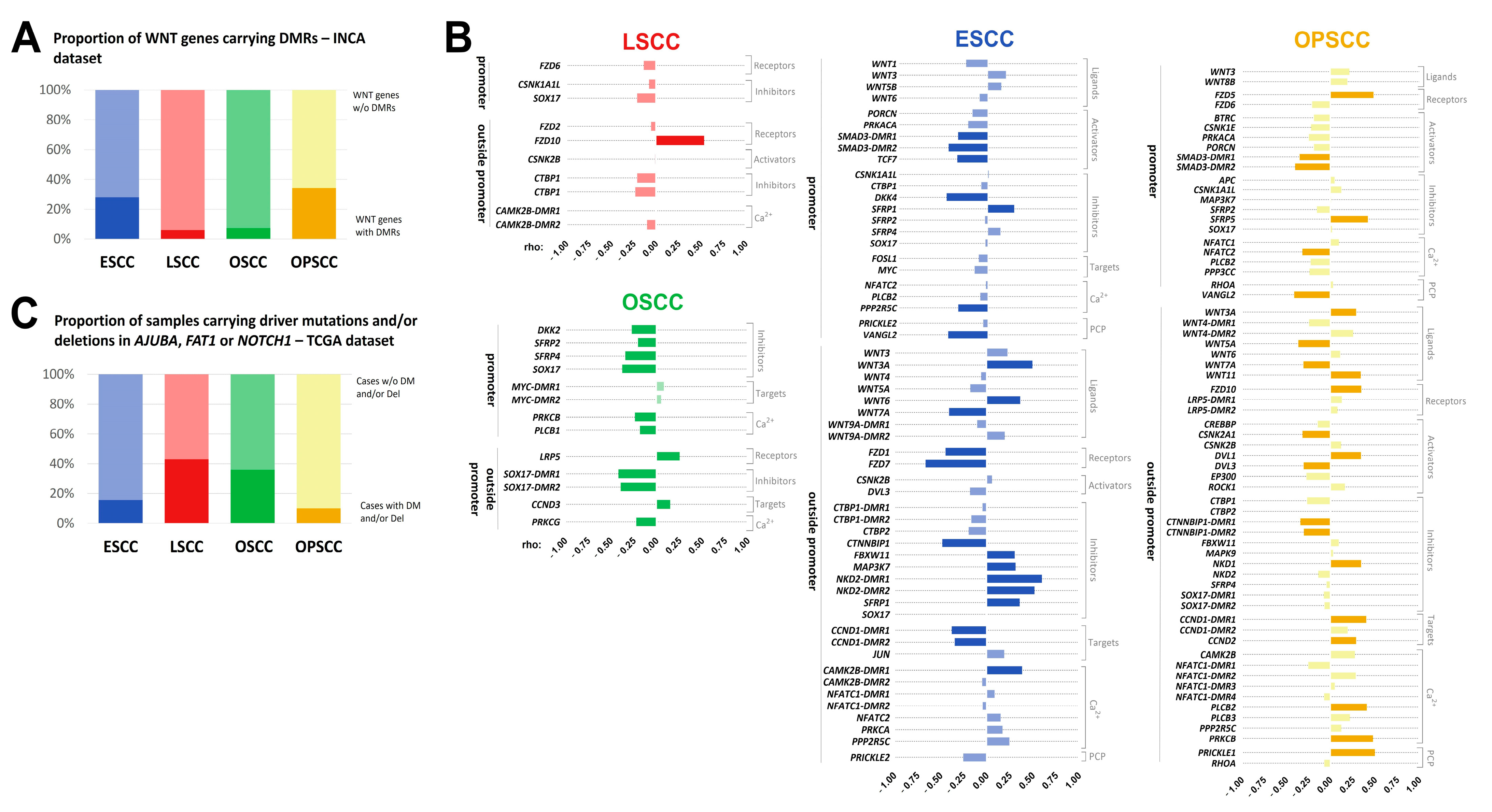
3.3. Methylation Levels of DMRs in WNT Signaling Genes Are Potentially Associated with Gene Expression
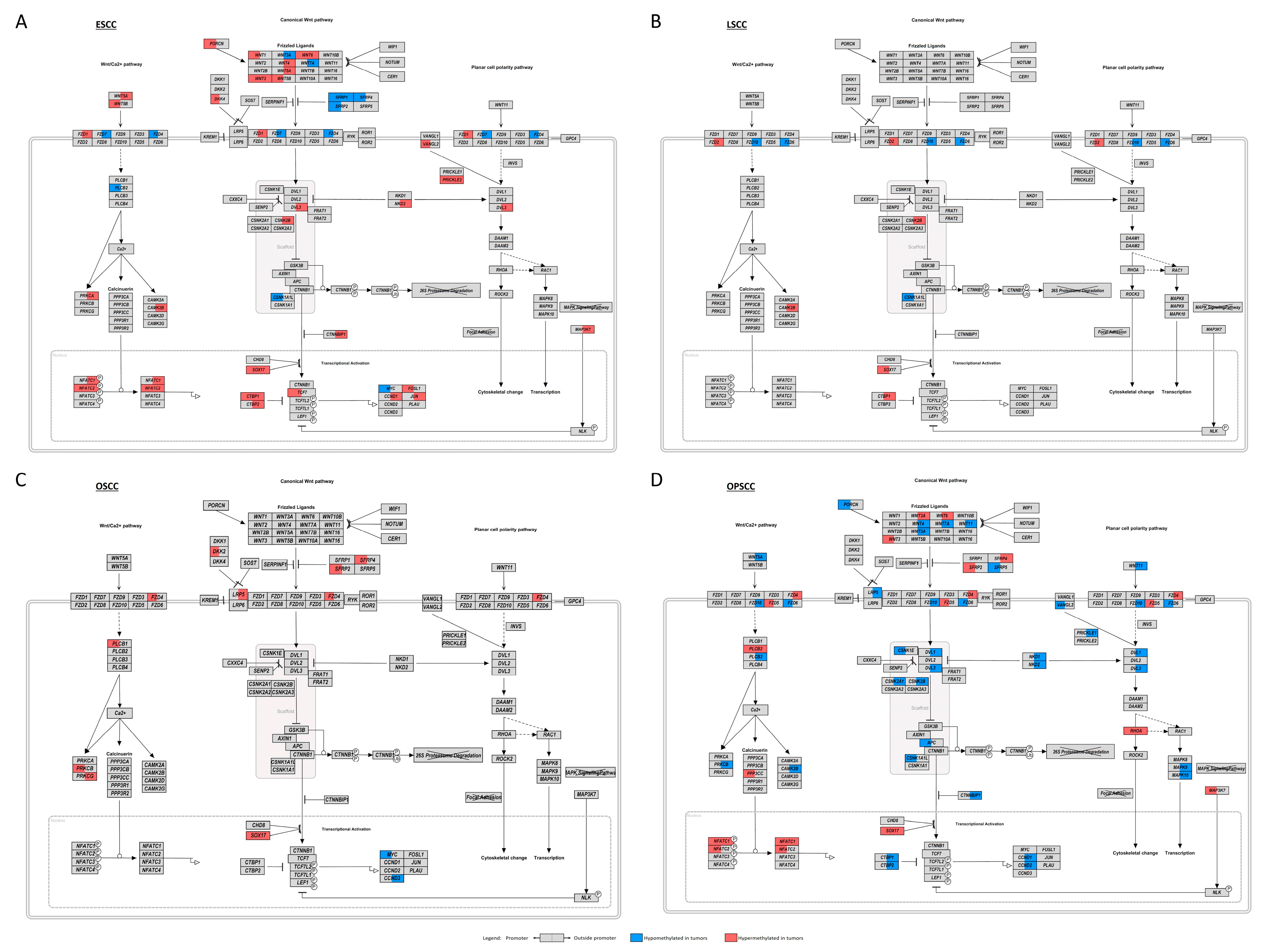
3.4. WNT Signaling Pathway May Be Affected by Different Molecular Mechanisms in UADT Subsites
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Rettig, E.M.; D’Souza, G. Epidemiology of head and neck cancer. Surg. Oncol. Clin. N. Am. 2015, 24, 379–396. [Google Scholar] [CrossRef]
- Prabhu, A.; Obi, K.O.; Rubenstein, J.H. The synergistic effects of alcohol and tobacco consumption on the risk of esophageal squamous cell carcinoma: A meta-analysis. Am. J. Gastroenterol. 2014, 109, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Priante, A.V.; Castilho, E.C.; Kowalski, L.P. Second primary tumors in patients with head and neck cancer. Curr. Oncol. Rep. 2011, 13, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Lampri, E.S.; Chondrogiannis, G.; Ioachim, E.; Varouktsi, A.; Mitselou, A.; Galani, A.; Briassoulis, E.; Kanavaros, P.; Galani, V. Biomarkers of head and neck cancer, tools or a gordian knot? Int. J. Clin. Exp. Med. 2015, 8, 10340–10357. [Google Scholar] [PubMed]
- Lim, H.; Kim, D.H.; Jung, H.Y.; Gong, E.J.; Na, H.K.; Ahn, J.Y.; Kim, M.Y.; Lee, J.H.; Choi, K.S.; Choi, K.D.; et al. Clinical significance of early detection of esophageal cancer in patients with head and neck cancer. Gut Liver 2015, 9, 159–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef]
- Amin, M.B.; Greene, F.L.; Edge, S.B.; Compton, C.C.; Gershenwald, J.E.; Brookland, R.K.; Meyer, L.; Gress, D.M.; Byrd, D.R.; Winchester, D.P. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J. Clin. 2017, 67, 93–99. [Google Scholar] [CrossRef]
- Peri, S.; Izumchenko, E.; Schubert, A.D.; Slifker, M.J.; Ruth, K.; Serebriiskii, I.G.; Guo, T.; Burtness, B.A.; Mehra, R.; Ross, E.A.; et al. NSD1- and NSD2-damaging mutations define a subset of laryngeal tumors with favorable prognosis. Nat. Commun. 2017, 8, 1772. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Izreig, S.; Yarbrough, W.G.; Issaeva, N. NSD1 mutations by HPV status in head and neck cancer: Differences in survival and response to DNA-damaging agents. Cancers Head Neck 2019, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Sawada, G.; Niida, A.; Uchi, R.; Hirata, H.; Shimamura, T.; Suzuki, Y.; Shiraishi, Y.; Chiba, K.; Imoto, S.; Takahashi, Y.; et al. Genomic Landscape of Esophageal Squamous Cell Carcinoma in a Japanese Population. Gastroenterology 2016, 150, 1171–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Network, C.G.A. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Network, C.G.A.R. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175. [Google Scholar]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Koch, A.; Joosten, S.C.; Feng, Z.; de Ruijter, T.C.; Draht, M.X.; Melotte, V.; Smits, K.M.; Veeck, J.; Herman, J.G.; Van Neste, L.; et al. Analysis of DNA methylation in cancer: Location revisited. Nat. Rev. Clin. Oncol. 2018, 15, 459–466. [Google Scholar] [CrossRef]
- Zhou, C.; Ye, M.; Ni, S.; Li, Q.; Ye, D.; Li, J.; Shen, Z.; Deng, H. DNA methylation biomarkers for head and neck squamous cell carcinoma. Epigenetics 2018, 13, 398–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, G.; Song, J.; Yuan, Y.; Chen, Z.; Tian, Y.; Yin, X.; Niu, Y.; Liu, J. Systematic analysis of differentially methylated expressed genes and site-specific methylation as potential prognostic markers in head and neck cancer. J. Cell Physiol. 2019, 234, 22687–22702. [Google Scholar] [CrossRef] [Green Version]
- Taciak, B.; Pruszynska, I.; Kiraga, L.; Bialasek, M.; Krol, M. Wnt signaling pathway in development and cancer. J. Physiol. Pharmacol. 2018, 69. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Tan, S.H.; Barker, N. Wnt Signaling in Adult Epithelial Stem Cells and Cancer. Prog. Mol. Biol. Transl. Sci. 2018, 153, 21–79. [Google Scholar] [CrossRef] [PubMed]
- Souza-Santos, P.T.; Soares Lima, S.C.; Nicolau-Neto, P.; Boroni, M.; Meireles Da Costa, N.; Brewer, L.; Menezes, A.N.; Furtado, C.; Moreira, M.A.M.; Seuanez, H.N.; et al. Mutations, Differential Gene Expression, and Chimeric Transcripts in Esophageal Squamous Cell Carcinoma Show High Heterogeneity. Transl. Oncol. 2018, 11, 1283–1291. [Google Scholar] [CrossRef]
- Leemans, C.R.; Snijders, P.J.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269. [Google Scholar] [CrossRef]
- Han, Q.; Wang, X.; Liao, X.; Han, C.; Yu, T.; Yang, C.; Li, G.; Han, B.; Huang, K.; Zhu, G.; et al. Diagnostic and prognostic value of WNT family gene expression in hepatitis B virus-related hepatocellular carcinoma. Oncol. Rep. 2019, 42, 895–910. [Google Scholar] [CrossRef]
- Davis, S.; Du, P.; Bilke, S.; Triche, T.; Bootwalla, M. methylumi: Handle Illumina Methylation Data. R Package Version. 2015. Available online: https://www.bioconductor.org/packages/release/bioc/html/methylumi.html (accessed on 9 April 2021).
- Chen, Y.A.; Lemire, M.; Choufani, S.; Butcher, D.T.; Grafodatskaya, D.; Zanke, B.W.; Gallinger, S.; Hudson, T.J.; Weksberg, R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013, 8, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Du, P.; Kibbe, W.A.; Lin, S.M. lumi: A pipeline for processing Illumina microarray. Bioinformatics 2008, 24, 1547–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pidsley, R.; Y Wong, C.C.; Volta, M.; Lunnon, K.; Mill, J.; Schalkwyk, L.C. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genom. 2013, 14, 293. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Bourgon, R. methyAnalysis: DNA Methylation Data Analysis and Visualization. R Package Version. 2014, pp. 1–33. Available online: http://bioconductor.org/packages/release/bioc/html/methyAnalysis.html (accessed on 9 April 2021).
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [Green Version]
- Kutmon, M.; van Iersel, M.P.; Bohler, A.; Kelder, T.; Nunes, N.; Pico, A.R.; Evelo, C.T. PathVisio 3: An extendable pathway analysis toolbox. PLoS Comput. Biol. 2015, 11, e1004085. [Google Scholar] [CrossRef] [Green Version]
- Chakravarthy, A.; Furness, A.; Joshi, K.; Ghorani, E.; Ford, K.; Ward, M.J.; King, E.V.; Lechner, M.; Marafioti, T.; Quezada, S.A.; et al. Pan-cancer deconvolution of tumour composition using DNA methylation. Nat. Commun. 2018, 9, 3220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díez-Villanueva, A.; Mallona, I.; Peinado, M.A. Wanderer, an interactive viewer to explore DNA methylation and gene expression data in human cancer. Epigenetics Chromatin 2015, 8, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nusse, R. The WNT Homepage. Available online: https://web.stanford.edu/group/nusselab/cgi-bin/wnt/target_genes (accessed on 24 July 2020).
- Poage, G.M.; Houseman, E.A.; Christensen, B.C.; Butler, R.A.; Avissar-Whiting, M.; McClean, M.D.; Waterboer, T.; Pawlita, M.; Marsit, C.J.; Kelsey, K.T. Global hypomethylation identifies Loci targeted for hypermethylation in head and neck cancer. Clin. Cancer Res. 2011, 17, 3579–3589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lleras, R.A.; Smith, R.V.; Adrien, L.R.; Schlecht, N.F.; Burk, R.D.; Harris, T.M.; Childs, G.; Prystowsky, M.B.; Belbin, T.J. Unique DNA methylation loci distinguish anatomic site and HPV status in head and neck squamous cell carcinoma. Clin. Cancer Res. 2013, 19, 5444–5455. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.D.; Yau, C.; Bowlby, R.; Liu, Y.; Brennan, K.; Fan, H.; Taylor, A.M.; Wang, C.; Walter, V.; Akbani, R.; et al. Genomic, Pathway Network, and Immunologic Features Distinguishing Squamous Carcinomas. Cell Rep. 2018, 23, 194–212.e196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talukdar, F.R.; Soares Lima, S.C.; Khoueiry, R.; Laskar, R.S.; Cuenin, C.; Sorroche, B.P.; Boisson, A.C.; Abedi-Ardekani, B.; Carreira, C.; Menya, D.; et al. Genome-wide DNA methylation profiling of esophageal squamous cell carcinoma from global high-incidence regions identifies crucial genes and potential cancer markers. Cancer Res. 2021, 81, 2612–2624. [Google Scholar] [CrossRef]
- Pyeon, D.; Newton, M.A.; Lambert, P.F.; den Boon, J.A.; Sengupta, S.; Marsit, C.J.; Woodworth, C.D.; Connor, J.P.; Haugen, T.H.; Smith, E.M.; et al. Fundamental differences in cell cycle deregulation in human papillomavirus-positive and human papillomavirus-negative head/neck and cervical cancers. Cancer Res. 2007, 67, 4605–4619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, T.; Sakai, A.; Afsari, B.; Considine, M.; Danilova, L.; Favorov, A.V.; Yegnasubramanian, S.; Kelley, D.Z.; Flam, E.; Ha, P.K.; et al. A Novel Functional Splice Variant of. Cancer Res. 2017, 77, 5248–5258. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Gaykalova, D.A.; Considine, M.; Wheelan, S.; Pallavajjala, A.; Bishop, J.A.; Westra, W.H.; Ideker, T.; Koch, W.M.; Khan, Z.; et al. Characterization of functionally active gene fusions in human papillomavirus related oropharyngeal squamous cell carcinoma. Int. J. Cancer 2016, 139, 373–382. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Gaykalova, D.; Wang, J.; Guo, T.; Danilova, L.; Favorov, A.; Fertig, E.; Bishop, J.; Khan, Z.; Flam, E.; et al. Discovery and development of differentially methylated regions in human papillomavirus-related oropharyngeal squamous cell carcinoma. Int. J. Cancer 2018, 143, 2425–2436. [Google Scholar] [CrossRef] [Green Version]
- Elhalawani, H.; Mohamed, A.S.R.; Elgohari, B.; Lin, T.A.; Sikora, A.G.; Lai, S.Y.; Abusaif, A.; Phan, J.; Morrison, W.H.; Gunn, G.B.; et al. Tobacco exposure as a major modifier of oncologic outcomes in human papillomavirus (HPV) associated oropharyngeal squamous cell carcinoma. BMC Cancer 2020, 20, 912. [Google Scholar] [CrossRef]
- Baliga, S.; Klamer, B.; Jhawar, S.; Gamez, M.; Mitchell, D.; Blakaj, A.; Grecula, J.; Gardner, U.; Dibs, K.; Old, M.; et al. Identification of Clinical and Socioeconomic Predictors of Adjuvant Therapy after Trans-Oral Robotic Surgery in Patients with Oropharyngeal Squamous Cell Carcinoma. Cancers 2020, 12, 2474. [Google Scholar] [CrossRef] [PubMed]
- Buexm, L.A.; Soares-Lima, S.C.; Brennan, P.; Fernandes, P.V.; de Souza Almeida Lopes, M.; Nascimento de Carvalho, F.; Santos, I.C.; Dias, L.F.; de Queiroz Chaves Lourenço, S.; Ribeiro Pinto, L.F. Hpv impact on oropharyngeal cancer patients treated at the largest cancer center from Brazil. Cancer Lett. 2020, 477, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Vossen, D.M.; Verhagen, C.V.M.; Verheij, M.; Wessels, L.F.A.; Vens, C.; van den Brekel, M.W.M. Comparative genomic analysis of oral versus laryngeal and pharyngeal cancer. Oral Oncol. 2018, 81, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.; Koenig, J.L.; Gentles, A.J.; Sunwoo, J.B.; Gevaert, O. Identification of an atypical etiological head and neck squamous carcinoma subtype featuring the CpG island methylator phenotype. EBioMedicine 2017, 17, 223–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraldi, L.; Leoncini, E.; Pastorino, R.; Wünsch-Filho, V.; de Carvalho, M.; Lopez, R.; Cadoni, G.; Arzani, D.; Petrelli, L.; Matsuo, K.; et al. Alcohol and cigarette consumption predict mortality in patients with head and neck cancer: A pooled analysis within the International Head and Neck Cancer Epidemiology (INHANCE) Consortium. Ann. Oncol. 2017, 28, 2843–2851. [Google Scholar] [CrossRef]
- Mochizuki, D.; Misawa, Y.; Kawasaki, H.; Imai, A.; Endo, S.; Mima, M.; Yamada, S.; Nakagawa, T.; Kanazawa, T.; Misawa, K. Aberrant Epigenetic Regulation in Head and Neck Cancer Due to Distinct EZH2 Overexpression and DNA Hypermethylation. Int. J. Mol. Sci. 2018, 19, 3707. [Google Scholar] [CrossRef] [Green Version]
- Hussein, Y.R.; Sood, A.K.; Bandyopadhyay, S.; Albashiti, B.; Semaan, A.; Nahleh, Z.; Roh, J.; Han, H.D.; Lopez-Berestein, G.; Ali-Fehmi, R. Clinical and biological relevance of enhancer of zeste homolog 2 in triple-negative breast cancer. Hum. Pathol. 2012, 43, 1638–1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.J.; Engers, R.; Florl, A.R.; Otte, A.P.; Muller, M.; Schulz, W.A. Expression changes in EZH2, but not in BMI-1, SIRT1, DNMT1 or DNMT3B are associated with DNA methylation changes in prostate cancer. Cancer Biol. Ther. 2007, 6, 1403–1412. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, J.; Valind, A.; Jansson, C.; O’Sullivan, M.J.; Holmquist Mengelbier, L.; Gisselsson, D. Aberrant epigenetic regulation in clear cell sarcoma of the kidney featuring distinct DNA hypermethylation and EZH2 overexpression. Oncotarget 2016, 7, 11127–11136. [Google Scholar] [CrossRef] [PubMed]
- Poirier, J.T.; Gardner, E.E.; Connis, N.; Moreira, A.L.; de Stanchina, E.; Hann, C.L.; Rudin, C.M. DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2. Oncogene 2015, 34, 5869–5878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, W.; Younis, R.H.; Li, J.; Chen, H.; Xia, R.; Mao, L.; Chen, W.; Ren, H. EZH2 promotes malignant phenotypes and is a predictor of oral cancer development in patients with oral leukoplakia. Cancer Prev. Res. 2011, 4, 1816–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.Y.; Hung, S.K.; Lee, M.S.; Chiou, W.Y.; Huang, T.T.; Tseng, C.E.; Shih, L.Y.; Lin, R.I.; Lin, J.M.; Lai, Y.H.; et al. DNA methylome analysis identifies epigenetic silencing of FHIT as a determining factor for radiosensitivity in oral cancer: An outcome-predicting and treatment-implicating study. Oncotarget 2015, 6, 915–934. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liu, L.; Mims, J.; Punska, E.C.; Williams, K.E.; Zhao, W.; Arcaro, K.F.; Tsang, A.W.; Zhou, X.; Furdui, C.M. Analysis of DNA methylation and gene expression in radiation-resistant head and neck tumors. Epigenetics 2015, 10, 545–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koumangoye, R.B.; Andl, T.; Taubenslag, K.J.; Zilberman, S.T.; Taylor, C.J.; Loomans, H.A.; Andl, C.D. SOX4 interacts with EZH2 and HDAC3 to suppress microRNA-31 in invasive esophageal cancer cells. Mol. Cancer 2015, 14, 24. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Gu, L.; Cao, Y.; Fan, X.; Zhang, F.; Sang, M. Aberrant overexpression of EZH2 and H3K27me3 serves as poor prognostic biomarker for esophageal squamous cell carcinoma patients. Biomarkers 2016, 21, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.; Schultz, B.; Coutifaris, C.; Sapienza, C. Highly variant DNA methylation in normal tissues identifies a distinct subclass of cancer patients. Adv. Cancer Res. 2019, 142, 1–22. [Google Scholar] [CrossRef]
- Kim, S.Y.; Han, Y.K.; Song, J.M.; Lee, C.H.; Kang, K.; Yi, J.M.; Park, H.R. Aberrantly hypermethylated tumor suppressor genes were identified in oral squamous cell carcinoma (OSCC). Clin. Epigenetics 2019, 11, 116. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Li, T.F.; Han, X.W.; Yuan, H.F. Downregulated MEG3 contributes to tumour progression and poor prognosis in oesophagal squamous cell carcinoma by interacting with miR-4261, downregulating DKK2 and activating the Wnt/β-catenin signalling. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1513–1523. [Google Scholar] [CrossRef]
- Singh, V.; Singh, A.P.; Sharma, I.; Singh, L.C.; Sharma, J.; Borthakar, B.B.; Rai, A.K.; Kataki, A.C.; Kapur, S.; Saxena, S. Epigenetic deregulations of Wnt/β-catenin and transforming growth factor beta-Smad pathways in esophageal cancer: Outcome of DNA methylation. J. Cancer Res. Ther. 2019, 15, 192–203. [Google Scholar] [CrossRef]
- Paluszczak, J.; Kiwerska, K.; Mielcarek-Kuchta, D. Frequent methylation of DAB2, a Wnt pathway antagonist, in oral and oropharyngeal squamous cell carcinomas. Pathol. Res. Pract. 2018, 214, 314–317. [Google Scholar] [CrossRef]
- Paluszczak, J. The Significance of the Dysregulation of Canonical Wnt Signaling in Head and Neck Squamous Cell Carcinomas. Cells 2020, 9, 723. [Google Scholar] [CrossRef] [Green Version]
- Papagerakis, P.; Pannone, G.; Shabana, A.H.; Depondt, J.; Santoro, A.; Ghirtis, K.; Berdal, A.; Papagerakis, S. Aberrant beta-catenin and LEF1 expression may predict the clinical outcome for patients with oropharyngeal cancer. Int. J. Immunopathol. Pharmacol. 2012, 25, 135–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, M.; Peña-Oyarzun, D.; Maturana, A.; Torres, V.A. Nuclear localization of β-catenin and expression of target genes are associated with increased Wnt secretion in oral dysplasia. Oral Oncol. 2019, 94, 58–67. [Google Scholar] [CrossRef]
- Silva, B.S.; Castro, C.A.; Von Zeidler, S.L.; Sousa, S.C.; Batista, A.C.; Yamamoto-Silva, F.P. Altered β-catenin expression in oral mucosal dysplasia: A comparative study. J. Appl. Oral Sci. 2015, 23, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Ishida, K.; Ito, S.; Wada, N.; Deguchi, H.; Hata, T.; Hosoda, M.; Nohno, T. Nuclear localization of beta-catenin involved in precancerous change in oral leukoplakia. Mol. Cancer 2007, 6, 62. [Google Scholar] [CrossRef] [Green Version]
- Kaur, J.; Sawhney, M.; DattaGupta, S.; Shukla, N.K.; Srivastava, A.; Walfish, P.G.; Ralhan, R. Clinical significance of altered expression of β-catenin and E-cadherin in oral dysplasia and cancer: Potential link with ALCAM expression. PLoS ONE 2013, 8, e67361. [Google Scholar] [CrossRef] [PubMed]
- Rampias, T.; Boutati, E.; Pectasides, E.; Sasaki, C.; Kountourakis, P.; Weinberger, P.; Psyrri, A. Activation of Wnt signaling pathway by human papillomavirus E6 and E7 oncogenes in HPV16-positive oropharyngeal squamous carcinoma cells. Mol. Cancer Res. 2010, 8, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Al Moustafa, A.E.; Foulkes, W.D.; Benlimame, N.; Wong, A.; Yen, L.; Bergeron, J.; Batist, G.; Alpert, L.; Alaoui-Jamali, M.A. E6/E7 proteins of HPV type 16 and ErbB-2 cooperate to induce neoplastic transformation of primary normal oral epithelial cells. Oncogene 2004, 23, 350–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osei-Sarfo, K.; Tang, X.H.; Urvalek, A.M.; Scognamiglio, T.; Gudas, L.J. The molecular features of tongue epithelium treated with the carcinogen 4-nitroquinoline-1-oxide and alcohol as a model for HNSCC. Carcinogenesis 2013, 34, 2673–2681. [Google Scholar] [CrossRef] [PubMed]
- Kanojia, D.; Vaidya, M.M. 4-nitroquinoline-1-oxide induced experimental oral carcinogenesis. Oral Oncol. 2006, 42, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Pannone, G.; Bufo, P.; Santoro, A.; Franco, R.; Aquino, G.; Longo, F.; Botti, G.; Serpico, R.; Cafarelli, B.; Abbruzzese, A.; et al. WNT pathway in oral cancer: Epigenetic inactivation of WNT-inhibitors. Oncol. Rep. 2010, 24, 1035–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, C.; Wang, L.; Zhu, L.; Zhang, C.; Zhou, J. Secreted frizzled-related protein 2 is epigenetically silenced and functions as a tumor suppressor in oral squamous cell carcinoma. Mol. Med. Rep. 2014, 10, 2293–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paluszczak, J.; Sarbak, J.; Kostrzewska-Poczekaj, M.; Kiwerska, K.; Jarmuż-Szymczak, M.; Grenman, R.; Mielcarek-Kuchta, D.; Baer-Dubowska, W. The negative regulators of Wnt pathway-DACH1, DKK1, and WIF1 are methylated in oral and oropharyngeal cancer and WIF1 methylation predicts shorter survival. Tumour Biol. 2015, 36, 2855–2861. [Google Scholar] [CrossRef] [Green Version]
- Paluszczak, J.; Wiśniewska, D.; Kostrzewska-Poczekaj, M.; Kiwerska, K.; Grénman, R.; Mielcarek-Kuchta, D.; Jarmuż-Szymczak, M. Prognostic significance of the methylation of Wnt pathway antagonists-CXXC4, DACT2, and the inhibitors of sonic hedgehog signaling-ZIC1, ZIC4, and HHIP in head and neck squamous cell carcinomas. Clin. Oral Investig. 2017, 21, 1777–1788. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Linghu, E.; Zhan, Q.; He, T.; Cao, B.; Brock, M.V.; Herman, J.G.; Xiang, R.; Guo, M. Methylation of DACT2 accelerates esophageal cancer development by activating Wnt signaling. Oncotarget 2016, 7, 17957–17969. [Google Scholar] [CrossRef]
- Cao, B.; Yang, W.; Jin, Y.; Zhang, M.; He, T.; Zhan, Q.; Herman, J.G.; Zhong, G.; Guo, M. Silencing NKD2 by Promoter Region Hypermethylation Promotes Esophageal Cancer Progression by Activating Wnt Signaling. J. Thorac. Oncol. 2016, 11, 1912–1926. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.; Wang, H.B.; Li, Y.H.; Li, H.F.; Li, T.T.; Zhang, W.X.; Xiang, S.S.; Sun, Z.Q. Correlations of Promoter Methylation in WIF-1, RASSF1A, and CDH13 Genes with the Risk and Prognosis of Esophageal Cancer. Med. Sci. Monit. 2016, 22, 2816–2824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Y.; Wang, Q.G.; Wang, J.X.; Zhu, S.T.; Jiao, Y.; Li, P.; Zhang, S.T. Epigenetic inactivation of the SFRP1 gene in esophageal squamous cell carcinoma. Dig. Dis. Sci. 2011, 56, 3195–3203. [Google Scholar] [CrossRef] [PubMed]
- Kishino, T.; Niwa, T.; Yamashita, S.; Takahashi, T.; Nakazato, H.; Nakajima, T.; Igaki, H.; Tachimori, Y.; Suzuki, Y.; Ushijima, T. Integrated analysis of DNA methylation and mutations in esophageal squamous cell carcinoma. Mol. Carcinog. 2016, 55, 2077–2088. [Google Scholar] [CrossRef]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutsik, P.; Slawski, M.; Gasparoni, G.; Vedeneev, N.; Hein, M.; Walter, J. MeDeCom: Discovery and quantification of latent components of heterogeneous methylomes. Genome Biol. 2017, 18, 55. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Shi, M.; Chen, T.; Zhang, B. Characterization of the Immune Cell Infiltration Landscape in Head and Neck Squamous Cell Carcinoma to Aid Immunotherapy. Mol. Ther. Nucleic Acids 2020, 22, 298–309. [Google Scholar] [CrossRef]
- Wood, O.; Clarke, J.; Woo, J.; Mirza, A.H.; Woelk, C.H.; Thomas, G.J.; Vijayanand, P.; King, E.; Ottensmeier, C.H. Head and Neck Squamous Cell Carcinomas Are Characterized by a Stable Immune Signature Within the Primary Tumor Over Time and Space. Clin. Cancer Res. 2017, 23, 7641–7649. [Google Scholar] [CrossRef] [Green Version]
- Barros, L.R.C.; Souza-Santos, P.T.; Pretti, M.A.M.; Vieira, G.F.; Bragatte, M.A.S.; Mendes, M.F.A.; De Freitas, M.V.; Scherer, N.M.; De Oliveira, I.M.; Rapozo, D.C.M.; et al. High infiltration of B cells in tertiary lymphoid structures, TCR oligoclonality, and neoantigens are part of esophageal squamous cell carcinoma microenvironment. J. Leukoc. Biol. 2020, 108, 1307–1318. [Google Scholar] [CrossRef]
- Seijas-Tamayo, R.; Fernández-Mateos, J.; Adansa Klain, J.C.; Mesía, R.; Pastor Borgoñón, M.; Pérez-Ruiz, E.; Vázquez Fernández, S.; Salvador Coloma, C.; Rueda Domínguez, A.; Taberna, M.; et al. Epidemiological characteristics of a Spanish cohort of patients diagnosed with squamous cell carcinoma of head and neck: Distribution of risk factors by tumor location. Clin. Transl. Oncol. 2016, 18, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Arnal, M.J.D.; Arenas, Á.F.; Arbeloa, Á.L. Esophageal cancer: Risk factors, screening and endoscopic treatment in Western and Eastern countries. World J. Gastroenterol. 2015, 21, 7933. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soares-Lima, S.C.; Mehanna, H.; Camuzi, D.; de Souza-Santos, P.T.; Simão, T.d.A.; Nicolau-Neto, P.; Almeida Lopes, M.d.S.; Cuenin, C.; Talukdar, F.R.; Batis, N.; et al. Upper Aerodigestive Tract Squamous Cell Carcinomas Show Distinct Overall DNA Methylation Profiles and Different Molecular Mechanisms behind WNT Signaling Disruption. Cancers 2021, 13, 3014. https://doi.org/10.3390/cancers13123014
Soares-Lima SC, Mehanna H, Camuzi D, de Souza-Santos PT, Simão TdA, Nicolau-Neto P, Almeida Lopes MdS, Cuenin C, Talukdar FR, Batis N, et al. Upper Aerodigestive Tract Squamous Cell Carcinomas Show Distinct Overall DNA Methylation Profiles and Different Molecular Mechanisms behind WNT Signaling Disruption. Cancers. 2021; 13(12):3014. https://doi.org/10.3390/cancers13123014
Chicago/Turabian StyleSoares-Lima, Sheila Coelho, Hisham Mehanna, Diego Camuzi, Paulo Thiago de Souza-Santos, Tatiana de Almeida Simão, Pedro Nicolau-Neto, Monique de Souza Almeida Lopes, Cyrille Cuenin, Fazlur Rahman Talukdar, Nikolaos Batis, and et al. 2021. "Upper Aerodigestive Tract Squamous Cell Carcinomas Show Distinct Overall DNA Methylation Profiles and Different Molecular Mechanisms behind WNT Signaling Disruption" Cancers 13, no. 12: 3014. https://doi.org/10.3390/cancers13123014
APA StyleSoares-Lima, S. C., Mehanna, H., Camuzi, D., de Souza-Santos, P. T., Simão, T. d. A., Nicolau-Neto, P., Almeida Lopes, M. d. S., Cuenin, C., Talukdar, F. R., Batis, N., Costa, I., Dias, F., Degli Esposti, D., Boroni, M., Herceg, Z., & Ribeiro Pinto, L. F. (2021). Upper Aerodigestive Tract Squamous Cell Carcinomas Show Distinct Overall DNA Methylation Profiles and Different Molecular Mechanisms behind WNT Signaling Disruption. Cancers, 13(12), 3014. https://doi.org/10.3390/cancers13123014