
A Retrospective 5-Year Single Center Study Highlighting the Risk of Cancer Predisposition in Adolescents and Young Adults

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
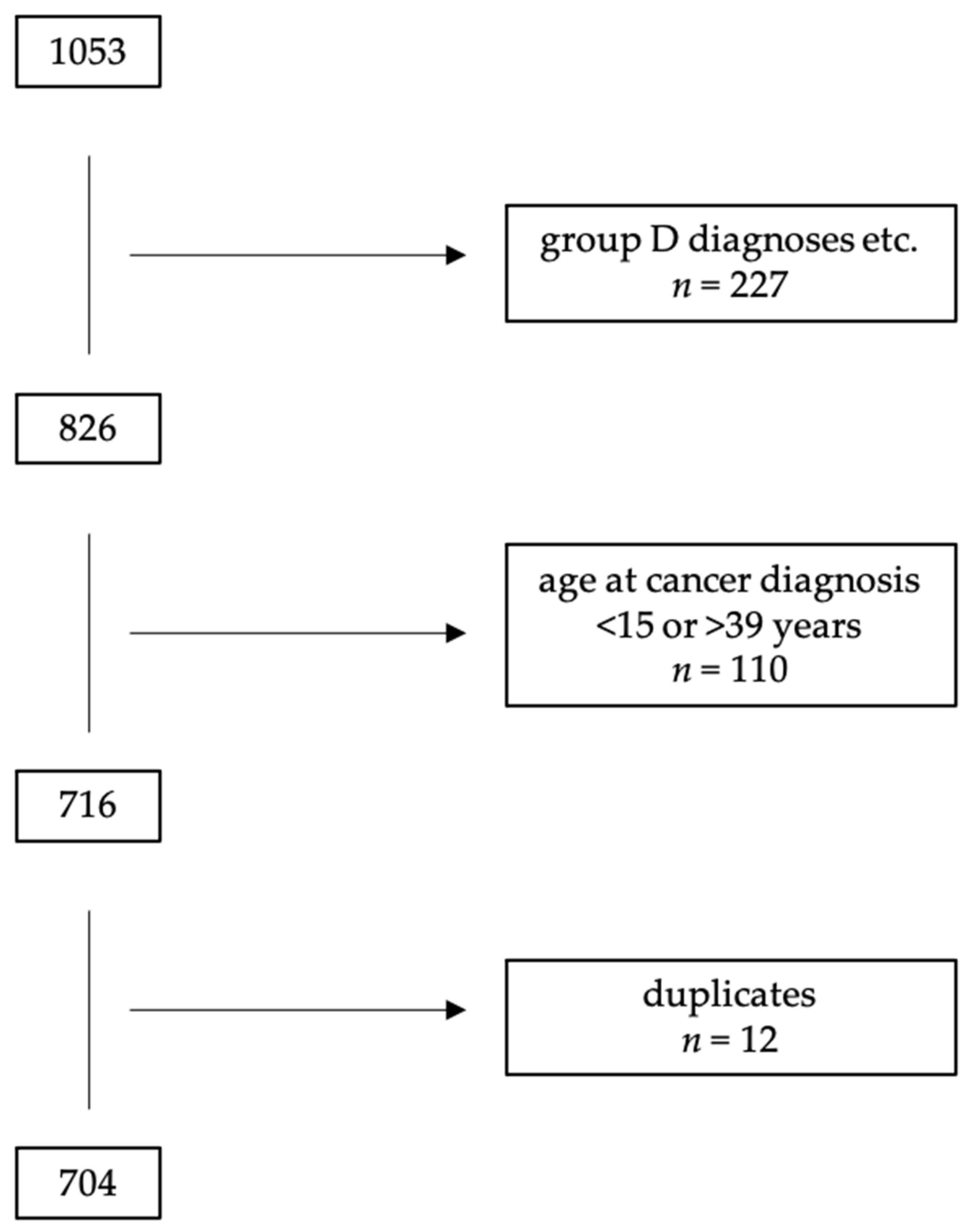
2. Materials and Methods
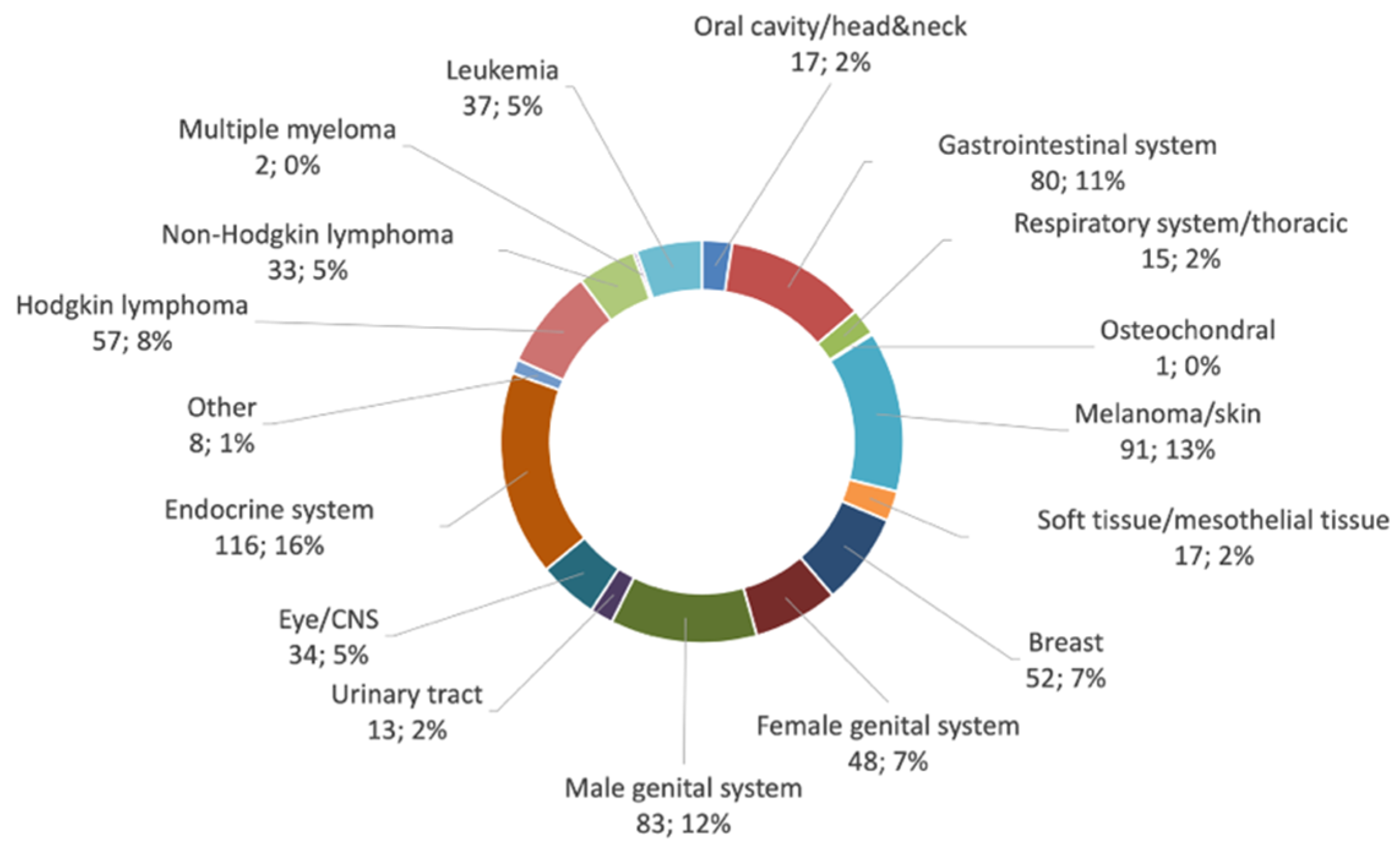
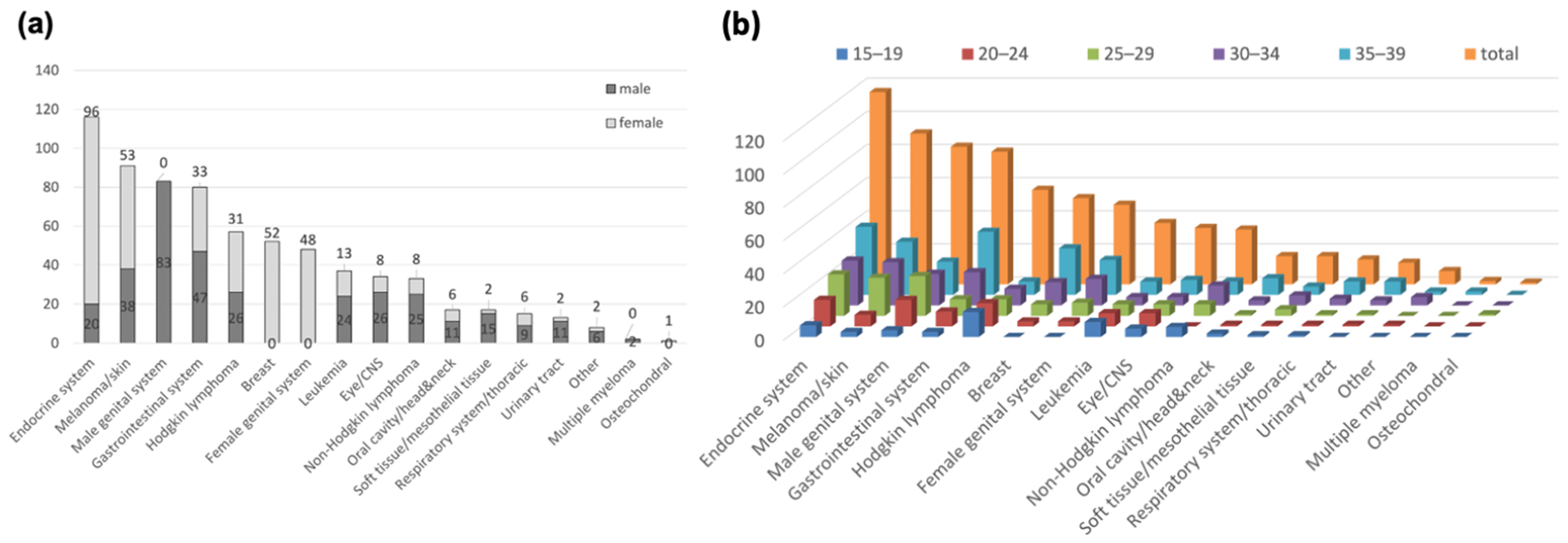
3. Results
3.1. Comparing Cancer Diagnosis in CCCA (ATDM) Patients and the GCR Database
3.2. Hereditary Cancer Risk Assessment in AYA Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.; Anagnostou, V.; Lytle, K.; Parpart-Li, S.; Nesselbush, M.; Riley, D.R.; Shukla, M.; Chesnick, B.; Kadan, M.; Papp, E.; et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci. Transl. Med. 2015, 7, 283ra253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef]
- Schrader, K.A.; Cheng, D.T.; Joseph, V.; Prasad, M.; Walsh, M.; Zehir, A.; Ni, A.; Thomas, T.; Benayed, R.; Ashraf, A.; et al. Germline Variants in Targeted Tumor Sequencing Using Matched Normal DNA. JAMA Oncol. 2016, 2, 104–111. [Google Scholar] [CrossRef]
- Seifert, B.A.; O’Daniel, J.M.; Amin, K.; Marchuk, D.S.; Patel, N.M.; Parker, J.S.; Hoyle, A.P.; Mose, L.E.; Marron, A.; Hayward, M.C.; et al. Germline Analysis from Tumor-Germline Sequencing Dyads to Identify Clinically Actionable Secondary Findings. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 4087–4094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robson, M.E.; Bradbury, A.R.; Arun, B.; Domchek, S.M.; Ford, J.M.; Hampel, H.L.; Lipkin, S.M.; Syngal, S.; Wollins, D.S.; Lindor, N.M. American Society of Clinical Oncology Policy Statement Update: Genetic and Genomic Testing for Cancer Susceptibility. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 3660–3667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampel, H.; Bennett, R.L.; Buchanan, A.; Pearlman, R.; Wiesner, G.L. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: Referral indications for cancer predisposition assessment. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 70–87. [Google Scholar] [CrossRef] [Green Version]
- Brodeur, G.M.; Nichols, K.E.; Plon, S.E.; Schiffman, J.D.; Malkin, D. Pediatric Cancer Predisposition and Surveillance: An Overview, and a Tribute to Alfred G. Knudson Jr. Clin. cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, e1–e5. [Google Scholar] [CrossRef] [Green Version]
- Mandelker, D.; Zhang, L.; Kemel, Y.; Stadler, Z.K.; Joseph, V.; Zehir, A.; Pradhan, N.; Arnold, A.; Walsh, M.F.; Li, Y.; et al. Mutation Detection in Patients With Advanced Cancer by Universal Sequencing of Cancer-Related Genes in Tumor and Normal DNA vs. Guideline-Based Germline Testing. JAMA J. Am. Med. Assoc. 2017, 318, 825–835. [Google Scholar] [CrossRef]
- Samadder, N.J.; Riegert-Johnson, D.; Boardman, L.; Rhodes, D.; Wick, M.; Okuno, S.; Kunze, K.L.; Golafshar, M.; Uson, P.L.S., Jr.; Mountjoy, L.; et al. Comparison of Universal Genetic Testing vs Guideline-Directed Targeted Testing for Patients with Hereditary Cancer Syndrome. JAMA Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Uson, P.L.S., Jr.; Riegert-Johnson, D.; Boardman, L.; Kisiel, J.; Mountjoy, L.; Patel, N.; Lizaola-Mayo, B.; Borad, M.J.; Ahn, D.; Sonbol, M.B.; et al. Germline Cancer Susceptibility Gene Testing in Unselected Patients with Colorectal Adenocarcinoma: A Multicenter Prospective Study. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2021. [Google Scholar] [CrossRef]
- Stadler, Z.K.; Maio, A.; Padunan, A.; Kemel, Y.; Salo-Mullen, E.; Sheehan, M.; Belanfanti, K.; Tejada, P.R.; Birsoy, O.; Mandelker, D.; et al. Abstract 1122: Germline mutation prevalence in young adults with cancer. Cancer Res. 2020, 80, 1122. [Google Scholar] [CrossRef]
- Kensler, T.W.; Spira, A.; Garber, J.E.; Szabo, E.; Lee, J.J.; Dong, Z.; Dannenberg, A.J.; Hait, W.N.; Blackburn, E.; Davidson, N.E.; et al. Transforming Cancer Prevention through Precision Medicine and Immune-oncology. Cancer Prev. Res. 2016, 9, 2–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spira, A.; Yurgelun, M.B.; Alexandrov, L.; Rao, A.; Bejar, R.; Polyak, K.; Giannakis, M.; Shilatifard, A.; Finn, O.J.; Dhodapkar, M.; et al. Precancer Atlas to Drive Precision Prevention Trials. Cancer Res. 2017, 77, 1510–1541. [Google Scholar] [CrossRef] [Green Version]
- Caswell-Jin, J.L.; Zimmer, A.D.; Stedden, W.; Kingham, K.E.; Zhou, A.Y.; Kurian, A.W. Cascade Genetic Testing of Relatives for Hereditary Cancer Risk: Results of an Online Initiative. J. Natl. Cancer Inst. 2019, 111, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Frey, M.K.; Kahn, R.M.; Chapman-Davis, E.; Tubito, F.; Pires, M.; Christos, P.; Anderson, S.; Mukherjee, S.; Jordan, B.; Blank, S.V.; et al. Prospective Feasibility Trial of a Novel Strategy of Facilitated Cascade Genetic Testing Using Telephone Counseling. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 1389–1397. [Google Scholar] [CrossRef] [PubMed]
- Close, A.G.; Dreyzin, A.; Miller, K.D.; Seynnaeve, B.K.N.; Rapkin, L.B. Adolescent and young adult oncology-past, present, and future. CA Cancer J. Clin. 2019, 69, 485–496. [Google Scholar] [CrossRef]
- Barr, R.D.; Ries, L.A.; Lewis, D.R.; Harlan, L.C.; Keegan, T.H.; Pollock, B.H.; Bleyer, W.A.; US National Cancer Institute Science of Adolescent; Young Adult Oncology Epidemiology Working Group. Incidence and incidence trends of the most frequent cancers in adolescent and young adult Americans, including “nonmalignant/noninvasive” tumors. Cancer 2016, 122, 1000–1008. [Google Scholar] [CrossRef]
- Fidler, M.M.; Gupta, S.; Soerjomataram, I.; Ferlay, J.; Steliarova-Foucher, E.; Bray, F. Cancer incidence and mortality among young adults aged 20-39 years worldwide in 2012: A population-based study. Lancet. Oncol. 2017, 18, 1579–1589. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.W.; Keegan, T.; Hamilton, A.; Lynch, C.; Wu, X.C.; Schwartz, S.M.; Kato, I.; Cress, R.; Harlan, L.; Group, A.H.S.C. Understanding care and outcomes in adolescents and young adult with Cancer: A review of the AYA HOPE study. Pediatric Blood Cancer 2019, 66, e27486. [Google Scholar] [CrossRef] [PubMed]
- Stiller, C.A.; Desandes, E.; Danon, S.E.; Izarzugaza, I.; Ratiu, A.; Vassileva-Valerianova, Z.; Steliarova-Foucher, E. Cancer incidence and survival in European adolescents (1978-1997). Report from the Automated Childhood Cancer Information System project. Eur. J. Cancer 2006, 42, 2006–2018. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef]
- Quante, A.S.; Ming, C.; Rottmann, M.; Engel, J.; Boeck, S.; Heinemann, V.; Westphalen, C.B.; Strauch, K. Projections of cancer incidence and cancer-related deaths in Germany by 2020 and 2030. Cancer Med. 2016, 5, 2649–2656. [Google Scholar] [CrossRef] [PubMed]
- Bleyer, A.; O’Leary, M.; Barr, R.; Ries, L.A.G. Cancer Epidemiology in Older Adolescents and Young Adults 15 to 29 Years of Age, Including SEER Incidence and Survival: 1975–2000; NIH Pub. No. 06-5767; NationalCancer Institute: Bethesda, MD, USA, 2006. [Google Scholar]
- Cancer Research UK. Available online: http://www.cancerresearchuk.org (accessed on 11 November 2020).
- Rummel, S.K.; Lovejoy, L.; Shriver, C.D.; Ellsworth, R.E. Contribution of germline mutations in cancer predisposition genes to tumor etiology in young women diagnosed with invasive breast cancer. Breast Cancer Res. Treat. 2017, 164, 593–601. [Google Scholar] [CrossRef]
- Mork, M.E.; You, Y.N.; Ying, J.; Bannon, S.A.; Lynch, P.M.; Rodriguez-Bigas, M.A.; Vilar, E. High Prevalence of Hereditary Cancer Syndromes in Adolescents and Young Adults with Colorectal Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 3544–3549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.H.; Lim, W.K.; Ishak, N.D.B.; Li, S.T.; Goh, W.L.; Tan, G.S.; Lim, K.H.; Teo, M.; Young, C.N.C.; Malik, S.; et al. Germline Mutations in Cancer Predisposition Genes are Frequent in Sporadic Sarcomas. Sci. Rep. 2017, 7, 10660. [Google Scholar] [CrossRef] [Green Version]
- Frebourg, T.; Bajalica Lagercrantz, S.; Oliveira, C.; Magenheim, R.; Evans, D.G.; The European Reference Network GENTURIS. Guidelines for the Li-Fraumeni and heritable TP53-related cancer syndromes. Eur. J. Hum. Genet. EJHG 2020, 28, 1379–1386. [Google Scholar] [CrossRef]
- Tutt, A.; Tovey, H.; Cheang, M.C.U.; Kernaghan, S.; Kilburn, L.; Gazinska, P.; Owen, J.; Abraham, J.; Barrett, S.; Barrett-Lee, P.; et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: The TNT Trial. Nat. Med. 2018, 24, 628–637. [Google Scholar] [CrossRef] [Green Version]
- Chopra, N.; Tovey, H.; Pearson, A.; Cutts, R.; Toms, C.; Proszek, P.; Hubank, M.; Dowsett, M.; Dodson, A.; Daley, F.; et al. Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple negative breast cancer. Nat. Commun. 2020, 11, 2662. [Google Scholar] [CrossRef]
- Villani, A.; Shore, A.; Wasserman, J.D.; Stephens, D.; Kim, R.H.; Druker, H.; Gallinger, B.; Naumer, A.; Kohlmann, W.; Novokmet, A.; et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. Lancet Oncol. 2016, 17, 1295–1305. [Google Scholar] [CrossRef]
- Tischkowitz, M.; Colas, C.; Pouwels, S.; Hoogerbrugge, N.; Group, P.G.D.; European Reference Network, G. Cancer Surveillance Guideline for individuals with PTEN hamartoma tumour syndrome. Eur. J. Hum. Genet. EJHG 2020, 28, 1387–1393. [Google Scholar] [CrossRef]
- Dean, M.; Rauscher, E.A. “It was an Emotional Baby”: Previvors’ Family Planning Decision-Making Styles about Hereditary Breast and Ovarian Cancer Risk. J. Genet. Couns. 2017, 26, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- German Centre for Cancer Registry Data. Cancer in Germany in 2015/2016, 12th ed.; Robert Koch Institute: Berlin, Germany, 2020. [Google Scholar]
- Alter, B.P. Cancer in Fanconi anemia, 1927–2001. Cancer 2003, 97, 425–440. [Google Scholar] [CrossRef] [PubMed]
- Kuhlen, M.; Wieczorek, D.; Siebert, R.; Fruhwald, M.C. How I approach hereditary cancer predisposition in a child with cancer. Pediatric Blood Cancer 2019, 66, e27916. [Google Scholar] [CrossRef]
- Vogt, A.; Schmid, S.; Heinimann, K.; Frick, H.; Herrmann, C.; Cerny, T.; Omlin, A. Multiple primary tumours: Challenges and approaches, a review. ESMO Open 2017, 2, e000172. [Google Scholar] [CrossRef] [Green Version]
- Mai, P.L.; Best, A.F.; Peters, J.A.; DeCastro, R.M.; Khincha, P.P.; Loud, J.T.; Bremer, R.C.; Rosenberg, P.S.; Savage, S.A. Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer 2016, 122, 3673–3681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bougeard, G.; Renaux-Petel, M.; Flaman, J.M.; Charbonnier, C.; Fermey, P.; Belotti, M.; Gauthier-Villars, M.; Stoppa-Lyonnet, D.; Consolino, E.; Brugieres, L.; et al. Revisiting Li-Fraumeni Syndrome from TP53 Mutation Carriers. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 2345–2352. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Ballinger, M.L.; Goode, D.L.; Ray-Coquard, I.; James, P.A.; Mitchell, G.; Niedermayr, E.; Puri, A.; Schiffman, J.D.; Dite, G.S.; Cipponi, A.; et al. Monogenic and polygenic determinants of sarcoma risk: An international genetic study. Lancet Oncol. 2016, 17, 1261–1271. [Google Scholar] [CrossRef]
- Mirabello, L.; Zhu, B.; Koster, R.; Karlins, E.; Dean, M.; Yeager, M.; Gianferante, M.; Spector, L.G.; Morton, L.M.; Karyadi, D.; et al. Frequency of Pathogenic Germline Variants in Cancer-Susceptibility Genes in Patients with Osteosarcoma. JAMA Oncol. 2020. [Google Scholar] [CrossRef]
- Waszak, S.M.; Tiao, G.; Zhu, B.; Rausch, T.; Muyas, F.; Rodríguez-Martín, B.; Rabionet, R.; Yakneen, S.; Escaramis, G.; Li, Y.; et al. Germline determinants of the somatic mutation landscape in 2,642 cancer genomes. bioRxiv 2017. [Google Scholar] [CrossRef] [Green Version]
- Copson, E.R.; Maishman, T.C.; Tapper, W.J.; Cutress, R.I.; Greville-Heygate, S.; Altman, D.G.; Eccles, B.; Gerty, S.; Durcan, L.T.; Jones, L.; et al. Germline BRCA mutation and outcome in young-onset breast cancer (POSH): A prospective cohort study. Lancet Oncol. 2018, 19, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Stoffel, E.M.; Koeppe, E.; Everett, J.; Ulintz, P.; Kiel, M.; Osborne, J.; Williams, L.; Hanson, K.; Gruber, S.B.; Rozek, L.S. Germline Genetic Features of Young Individuals with Colorectal Cancer. Gastroenterology 2018, 154, 897–905.e891. [Google Scholar] [CrossRef]
- Idos, G.E.; Kurian, A.W.; Ricker, C.; Sturgeon, D.; Culver, J.O.; Kingham, K.E.; Koff, R.; Chun, N.M.; Rowe-Teeter, C.; Lebensohn, A.P.; et al. Multicenter Prospective Cohort Study of the Diagnostic Yield and Patient Experience of Multiplex Gene Panel Testing For Hereditary Cancer Risk. JCO Precis. Oncol. 2019, 1–12. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ICD Groups | Specific Types of Cancer |
|---|---|
| Endocrine system | Thyroid cancer, medullary |
| Adrenocortical carcinoma | |
| Pheochromocytoma, paraganglioma | |
| Melanoma/skin | Basal cell carcinoma, >5 |
| Basal cell carcinoma, <30 years of age | |
| Sebaceous neoplasm | |
| Gastrointestinal system | Gastric cancer, diffuse |
| Small intestine cancer | |
| Colon cancer, <50 years of age | |
| Pancreatobiliary carcinoma | |
| Breast cancer | Breast cancer, unilateral (<36 years of age) |
| Breast cancer, bilateral (<51 years of age) | |
| Breast cancer, male | |
| Female genital system | Ovarian cancer |
| Tubal cancer | |
| Primary peritoneal carcinoma | |
| Leukemia | Acute lymphoblastic leukemia, low hypodiploid |
| Central nervous system/eye | Choroid plexus carcinoma |
| Brain tumor, <46 years of age | |
| Gangliocytoma, dysplastic cerebellar (Lhermitte-Duclos disease) | |
| Meningioma, clear-cell | |
| Schwannoma, ≥2 (non-dermal) | |
| Endolymphatic sac tumor | |
| Retinoblastoma | |
| Soft tissue/mesothelial tissue | Rhabdomyosarcoma, embryonal |
| Rhabdomyosarcoma, anaplastic | |
| Soft tissue sarcoma, <46 years of age | |
| Leiomyoma, cutaneous | |
| Urinary tract | Renal cell carcinoma (<47 years of age) |
| Renal cell carcinoma, papillary | |
| Renal cell carcinoma, clear cell tubulo-papillary | |
| Collecting-duct carcinoma | |
| Ureteric cancer | |
| Osteochondral | Osteosarcoma, <46 years of age |
| Other | Rhabdoid tumor |
| Hemangioblastoma | |
| Pineoblastoma |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jordan, F.; Huber, S.; Sommer, S.; Schenkirsch, G.; Frühwald, M.C.; Trepel, M.; Claus, R.; Kuhlen, M. A Retrospective 5-Year Single Center Study Highlighting the Risk of Cancer Predisposition in Adolescents and Young Adults. Cancers 2021, 13, 3033. https://doi.org/10.3390/cancers13123033
Jordan F, Huber S, Sommer S, Schenkirsch G, Frühwald MC, Trepel M, Claus R, Kuhlen M. A Retrospective 5-Year Single Center Study Highlighting the Risk of Cancer Predisposition in Adolescents and Young Adults. Cancers. 2021; 13(12):3033. https://doi.org/10.3390/cancers13123033
Chicago/Turabian StyleJordan, Frank, Simon Huber, Sebastian Sommer, Gerhard Schenkirsch, Michael C. Frühwald, Martin Trepel, Rainer Claus, and Michaela Kuhlen. 2021. "A Retrospective 5-Year Single Center Study Highlighting the Risk of Cancer Predisposition in Adolescents and Young Adults" Cancers 13, no. 12: 3033. https://doi.org/10.3390/cancers13123033
APA StyleJordan, F., Huber, S., Sommer, S., Schenkirsch, G., Frühwald, M. C., Trepel, M., Claus, R., & Kuhlen, M. (2021). A Retrospective 5-Year Single Center Study Highlighting the Risk of Cancer Predisposition in Adolescents and Young Adults. Cancers, 13(12), 3033. https://doi.org/10.3390/cancers13123033