3.1. Datasets
Two publicly available datasets were used as examples to demonstrate the ability of OmiEmbed: the Genomic Data Commons (GDC) pan-cancer multi-omics dataset [
17] and the DNA methylation dataset of human central nervous system tumours (GSE109381) [
5]. The overview information of the two datasets was summarised in
Table 1.
The GDC pan-cancer dataset is one of the most comprehensive and widely used multi-omics dataset. It comprises high-dimensional omics data and corresponding phenotype data from two cancer genome programmes: The Cancer Genome Atlas (TCGA) [
24] and Therapeutically Applicable Research to Generate Effective Treatment (TARGET). The TARGET programme mainly focuses on pediatric cancers. Three types of omics data from the GDC dataset were used in our experiments, including RNA-Seq gene expression profiling, DNA methylation profiling and miRNA expression profiling. The dimensionalities of the three types of omics data are 60,483, 485,577 and 1881 respectively. This dataset consists of 36 different types of tumour samples, along with corresponding normal control samples, among which 33 tumour types are from TCGA and 3 tumour types are from TARGET. The detailed tumour type information was tabulated in
Supplementary Table S1. A wide range of phenotype features are also available in the GDC dataset including demographics (e.g., age and gender), clinical sample information (e.g., primary site and disease stage of the sample) and the survival information (recorded time of death or censoring).
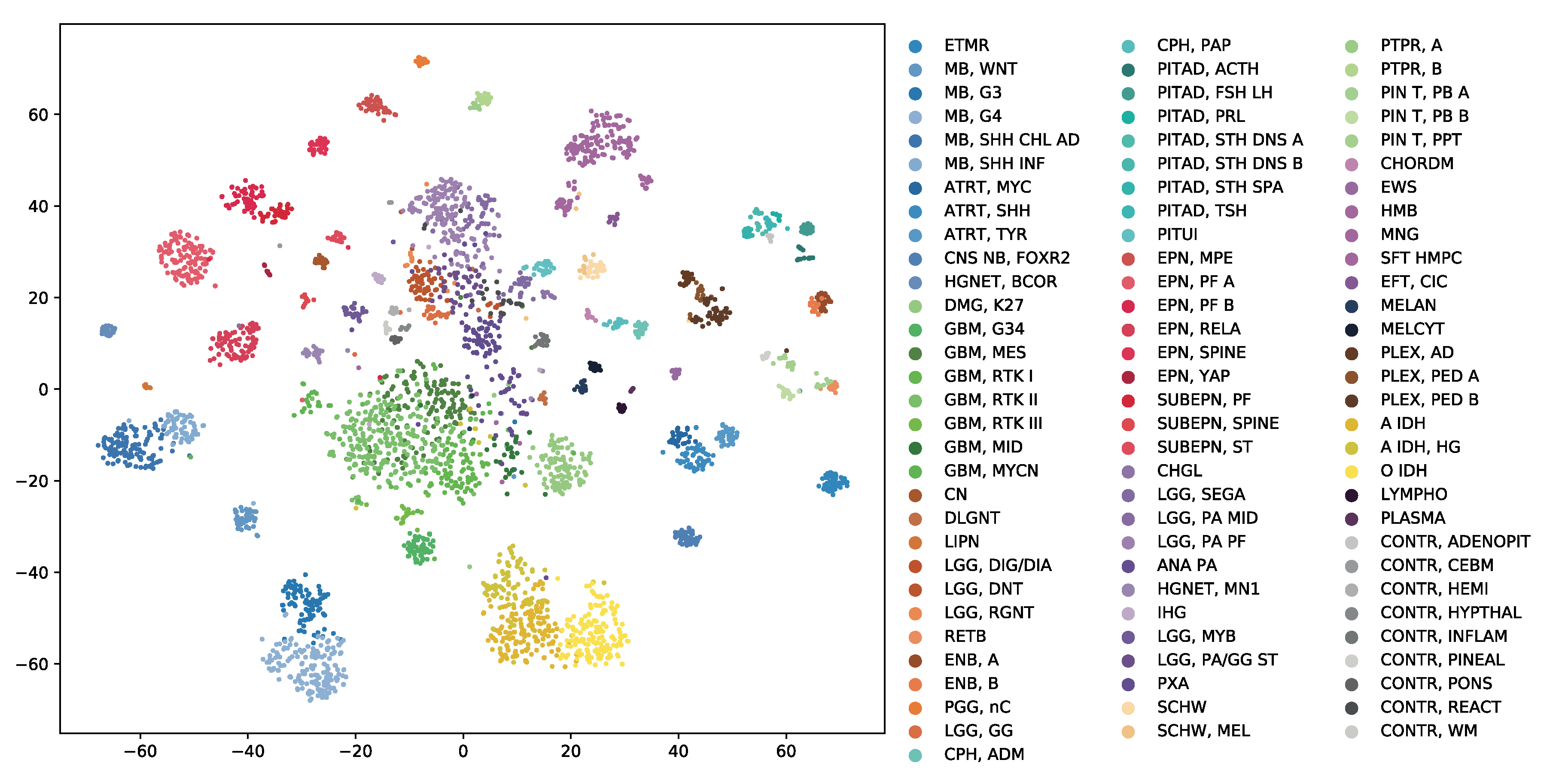
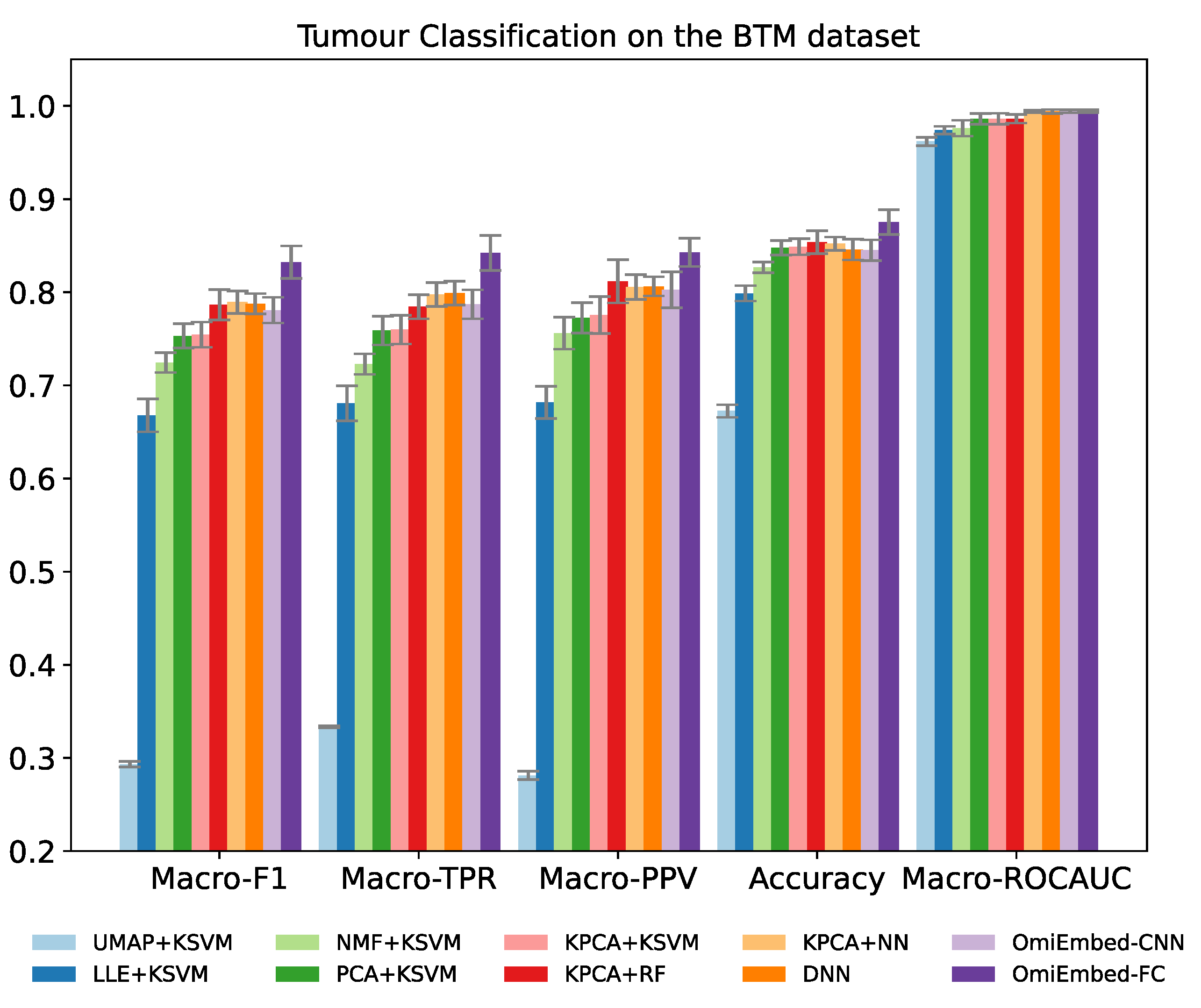
The GSE109381 brain tumour methylation (BTM) dataset from the Gene Expression Omnibus (GEO) is one of the largest DNA methylation datasets specifically targeting brain tumours. We integrated both the reference set and validation set of this dataset and the whole dataset consists of 3905 samples, with almost all WHO-defined central nervous system (CNS) tumour entities [
3] and eight non-neoplastic control CNS regions. The genome-wide DNA methylation profile for each sample was generated using Infinium HumanMethylation450 BeadChip (450 K) arrays, which is the same platform used for the GDC DNA methylation data. Each sample in this dataset has two types of diagnostic label, the histopathological class label defined by the latest 2016 WHO classification of CNS tumours [
3] and the methylation class label defined by the original paper of this dataset [
5]. The detailed tumour type information of the two label systems was listed in
Supplementary Tables S2 and S3. Other phenotypic information is also available in this dataset, including age, gender and the disease stage of each sample.
3.2. Omics Data Process
Raw data of the GSE109381 BTM dataset downloaded from GEO (
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE109381, accessed on 30 August 2019) were first processed by the Bioconductor R package minfi [
25] to obtain the beta value of each CpG probe. Beta value is the ratio of methylated signal intensities and the overall signal intensities, which indicates the methylation level of a specific CpG site. The DNA methylation profile generated by the 450 K array has 485,577 probes in total. Certain probes were removed during the feature filtering step according to the following criteria: probes targeting the Y chromosome (
n = 416), probes containing the dbSNP132Common single-nucleotide polymorphism (SNP) (
n = 7998), probes not mapping to the human reference genome (hg19) uniquely (one mismatch allowed) (
n = 3965), probes not included in the latest Infinium MethylationEPIC BeadChip (EPIC) array (
n = 32,260), the SNP assay probes (
n = 65), the non-CpG loci probes (
n = 3091) and probes with missing values (N/A) in more than 10% of samples (
n = 2). We followed some of the criteria mentioned in the original paper of this dataset [
5]. Overall, 46,746 probes were filtered out, which results in a final DNA methylation feature set of 438,831 CpG sites.
For the GDC pan-cancer dataset, the harmonised data of three omics types were downloaded from the UCSC Xena data portal (
https://xenabrowser.net/datapages/, accessed on 1 May 2019) with the original data dimensionality. Each RNA-Seq gene expression profile is comprised of 60,483 identifiers referring to corresponding genes. The expression level is quantified by the fragments per kilobase of transcript per million mapped reads (FPKM) value, which has been
-transformed. Feature filtering was applied to the gene expression data: targeting Y chromosome (
n = 594) and zero expression in all samples (
n = 1904). In total, 2440 genes were removed, leaving 58,043 molecular features for further analyses. As for miRNA expression profiles, the expression level of each miRNA stem-loop identifier was measured by the
-transformed reads per million mapped reads (RPM) value. All of the miRNA identifiers were kept in our experiments. For both the gene expression and miRNA expression profiles, the expression values were normalised to the range of 0 to 1 due to the input requirement of the OmiEmbed framework. The DNA methylation data in the GDC dataset were filtered based on the same criteria used for the BTM dataset. The remaining missing values in all datasets mentioned above were imputed by the mean of corresponding molecular features.
3.3. Overall Architecture
OmiEmbed is a unified end-to-end multi-view multi-task deep learning framework designed for high-dimensional multi-omics data, with application to dimensionality reduction, tumour type classification, multi-omics integration, demographic and clinical feature reconstruction, and survival prediction. The overall architecture of OmiEmbed is comprised of a deep embedding module and one or multiple downstream task modules, which is illustrated in
Figure 1.
The role of the deep embedding module in OmiEmbed is to embed high-dimensional multi-omics profiles into a low-dimensional latent space for the downstream task modules. The backbone network that we used in the deep embedding module is variational autoencoder (VAE) [
26]. VAE is a deep generative model which is also effective to capture the data manifold from high-dimensional data. We assume each sample
in the multi-omics dataset
can be represented by, and generated from, a latent vector
, where
. In the generation process, each latent vector is first sampled from a prior distribution
, and then the multi-omics data of each sample are generated from the conditional distribution
, where
is the set of learnable parameters of the decoder. In order to address the intractability of the true posterior
, the variational distribution
is introduced to approximate
, where
is the set of learnable parameters of the encoder. As a result, the VAE network is optimised by maximizing the variational lower bound, formularised as below:
where
is the Kullback–Leibler (KL) divergence between two probability distributions [
8].
We applied the framework of VAE to our deep embedding module to obtain the low-dimensional latent vector that can represent the original high-dimensional omics data in the downstream task modules. For each type of omics data, the input profiles were first encoded into corresponding vectors with specific encoders. Those vectors of different omics types were then concatenated together in the subsequent hidden layer and encoded into one multi-omics vector. Based on the idea of VAE, the multi-omics vector was connected to two bottleneck layers in order to obtain the mean vector
and the standard deviation vector
. These two vectors defined the Gaussian distribution
of the latent variable
given the input sample
, which is the variational distribution
. Since sampling
from the learnt distribution is not differentiable and suitable for backpropagation, the reparameterisation trick is applied as follows:
where
is a random variable sampled from the standard normal distribution
. The latent variable
was then fed to the decoders with a symmetrical network structure to obtain the reconstructed multi-omics data
.
We provided two types of detailed network structure for the encoders and decoders in the deep embedding module, the one-dimensional convolutional neural network (CNN) and the fully connected neural network (FC). The network structures of the two types of deep embedding modules were illustrated in
Supplementary Figures S1 and S2. Both network types shared the same architecture, and other state-of-the-art or customised embedding networks can be easily added to the OmiEmbed framework with minimal modification using our open-source repository in Github (
https://github.com/zhangxiaoyu11/OmiEmbed/, accessed on 19 May 2021). With the deep embedding module, we can obtain the low-dimensional representation of the input omics data. This new representation can directly replace the original omics data as the input of any downstream task. For instance, when the latent dimension is set to 2 or 3, the new representation can be used for visualisation purpose. Nevertheless, we can also attach one or multiple downstream task networks to the bottleneck layer of the deep embedding module to obtain an end-to-end multi-task model, which is able to guide the embedding step with objectives and share information among different tasks.
Three main types of end-to-end downstream tasks were provided in the OmiEmbed framework: classification task, regression task and survival prediction task. Each downstream task fell into one of these categories can be trained individually or collaboratively with other downstream tasks using the multi-task training strategy discussed in later sections. A multi-layer fully connected network was applied to classification-type downstream tasks, including diagnostic tasks such as tumour type classification, primary site prediction and disease stage (i.e., primary tumour, recurrent tumour, metastatic tumour or normal control) prediction and demographic tasks, e.g., the prediction of gender. The output dimension of the classification downstream network was set to the number of classes. For the regression task, a similar network was attached to the deep embedding module, but only one neuron was kept in the output layer to predict the target scalar value (e.g., age of the subject). The survival prediction downstream network is more complicated and addressed separately in a subsequent section. The downstream networks add further regularisation to the low dimensional latent representation and urge the deep embedding module to learn the omics embedding related to certain downstream tasks. With the downstream modules, a single well-trained multi-task OmiEmbed network is able to reconstruct a comprehensive phenotype profile, including diagnostic, prognostic and demographic information from omics data.
3.4. Training Strategy
The same as the overall structure, the joint loss function is also comprised of two main components: the loss of the deep embedding and the loss of the downstream tasks.
We denote each type of input omics profile as
, and the corresponding reconstructed profile as
, where
j is the omics type index and there are
M omics types in total. The deep embedding loss can then be defined as follows:
where
is the binary cross-entropy to measure the distance between input data and reconstructed data, and the second term is the KL divergence between the learnt distribution and a standard Gaussian distribution.
In the downstream modules, each downstream task has its specific loss function
and a corresponding weight
. For the classification type task, the loss function can be defined as:
where
y is the ground truth,
is the predicted label and
is the cross-entropy loss. Similar to the classification loss, the loss function of regression type task is
where
is the mean squared error between the real value and the predicted value. The loss function of the survival prediction task is discussed separately in the next section. The overall loss function of the downstream modules is the weighted sum of all downstream losses, i.e.,
where
K is the number of downstream tasks,
is the loss for a certain task and
is the corresponding weight.
can be manually set as hyperparameters or used as learnable parameters during the training process. In conclusion, the joint loss function of the end-to-end OmiEmbed network is
and depends on
, which balances the two terms in the overall loss function.
Based on the aforementioned loss functions, three training phases were designed in OmiEmbed. Phase 1 was the unsupervised phase that only focused on the deep embedding module. In this training phase, only the deep embedding loss was backpropagated and only the parameters in the deep embedding network were updated based on the gradients. No label was required in the first training phase and this phase can be used separately as a dimensionality reduction or visualisation method. In Phase 2, the pre-trained embedding network was fixed whilst the downstream networks were being trained. The joint downstream loss was backpropagated and only the downstream networks were updated during this phase. After the embedding network and the downstream networks were pre-trained separately, the overall loss function defined in Equation (
7) was calculated and backpropagated during Phase 3. In this final training phase the whole OmiEmbed network, including the deep embedding module and the downstream modules, was fine-tuned to obtain better performance.
3.5. Survival Prediction
Survival prediction is the most complicated downstream task implemented in OmiEmbed. The objective of this task is to obtain individual survival function and hazard function data for each subject, based on the high-dimensional omics data. The survival function can be denoted by
where
T is time elapsed between the sample collection time and the time of event occurring. The survival function signifies the probability that the failure event, i.e., death, has not occurred by time
t. The hazard function can be defined as:
which represents the instantaneous rate of occurrence for the failure event. A large hazard value manifests a great risk of death at specific time
t. However, the original form of hazard function is infrequently used, and the risk score of each sample
is more commonly applied by subdividing the time axis into
m time intervals, such that:
In order to train a survival prediction downstream network, besides the omics data , two elements of the survival labels are required: the event time T and the event indicator E. The indicator was set to 1 when the failure event was observed during the study and 0 when the event was not observed, which is termed censoring. In the case of censoring, the event time T is the time elapsed between the time when the sample was collected and the time of the last contact with the subject. Both T and E are available in the GDC dataset.
The multi-task logistic regression (MTLR) model [
27] was applied and adapted to the OmiEmbed framework for the survival prediction downstream task. In the first step, the time axis was divided into
m time intervals
. Each time interval was defined as
, where
= 0 and
. The number of time intervals
m is a hyperparameter. A larger
m results in more fine-grained output, but requires more computation resources. We applied the multi-layer fully connected network as the backbone of our survival prediction network and the dimension of the output layer is the number of time intervals. As a result, the output of our survival prediction network is an
m-dimensional vector
. Similarly, the survival label of each subject was also encoded into an
m-dimensional vector
, where
signifies the survival status of this subject at the time point
. The likelihood of observing
on the condition of sample
with the network parameters
can be defined as follows:
The objective of this survival network is to find a set of parameters
that maximises the log-likelihood, hence the loss function of the survival prediction downstream task is defined as,
which can be directly applied to the downstream module and included in the joint loss function of OmiEmbed.
3.6. Multi-Task Strategy
With the joint loss function (
6) of the multi-task downstream modules, we aimed to train multiple downstream networks in OmiEmbed simultaneously and efficiently instead of separate training, so as to obtain a unified model that can reconstruct a comprehensive phenotype profile for each subject. In order to balance the optimisation of different tasks, we adapted the multi-task optimisation method gradient normalisation (GradNorm) [
28] to our OmiEmbed framework.
In Equation (
6),
is the weight of each downstream loss, and the weight can also be regarded as a trainable parameter that varies at each training iteration. The idea of GradNorm is to penalise the network if gradients of any downstream task are too large or too small, which makes all the tasks train at similar rates [
28]. Firstly, the gradient norm of each downstream task is calculated by
where
is the parameters of the last encoding layer in the deep embedding module of OmiEmbed. The average gradient norm among all tasks can then be calculated by
where
K is the number of downstream tasks. The relative inverse training rate of each task can be defined as:
where
, which is the ratio of the current loss to the initial loss of the downstream task
k. Then, the loss of GradNorm can be defined as:
where
is the hyperparameter that represents strength pulling tasks back to a common training rate. A separate backpropagation process was conducted during each training iteration on
, which was only used for updating
.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}