
Myeloid-Specific Acly Deletion Alters Macrophage Phenotype In Vitro and In Vivo without Affecting Tumor Growth

 , , ,
, , ,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice and Bone Marrow-Derived Macrophage Culture
2.2. Tumor Models
2.3. Sample Preparation and Antibody Staining
2.4. Flow Cytometry and Analysis
2.5. Transcriptomics
3. Results
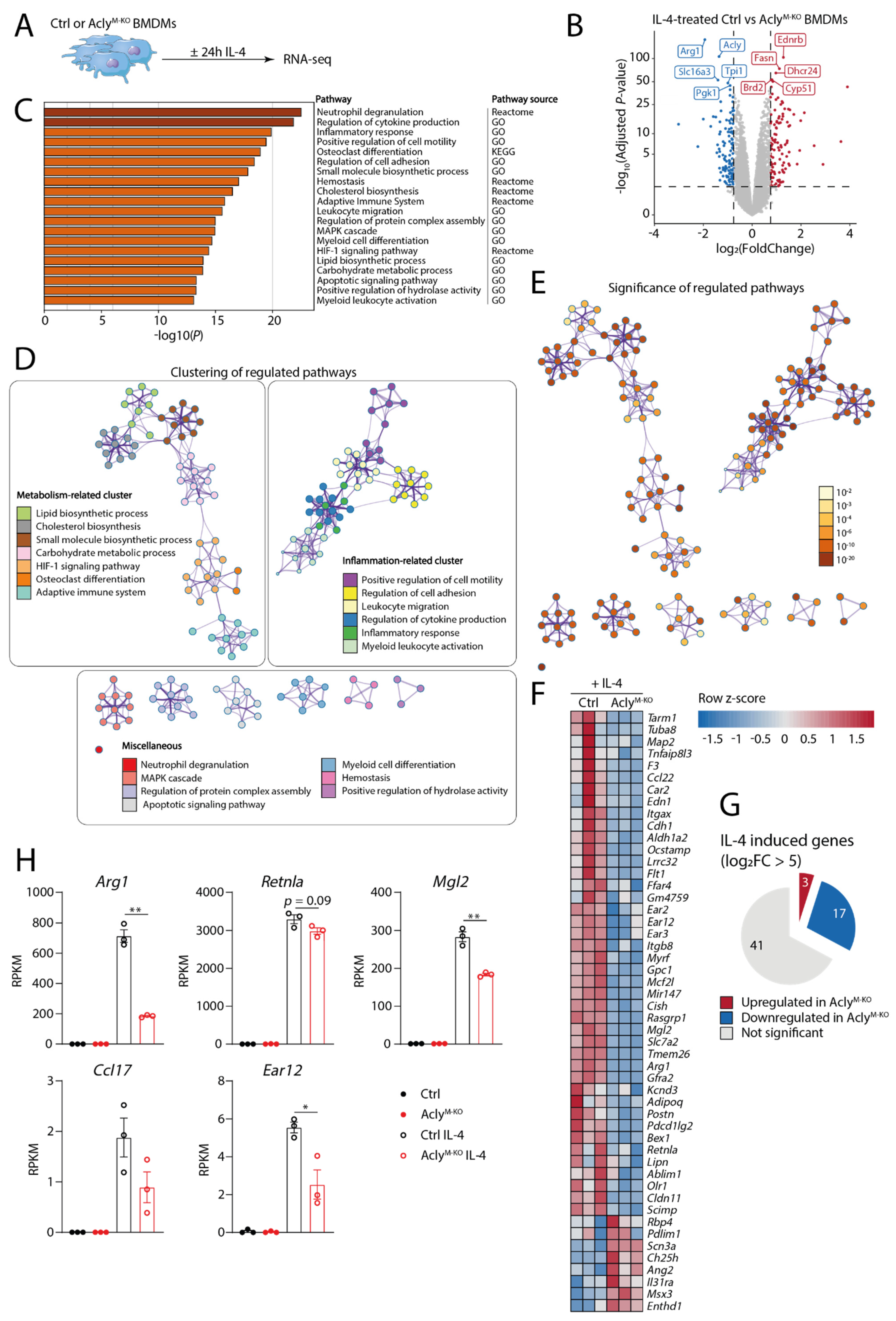
3.1. Transcriptomics Analysis of Myeloid-Specific Acly-Deficient Mice Validates the Involvement of Acly in IL-4-Induced Macrophage Activation
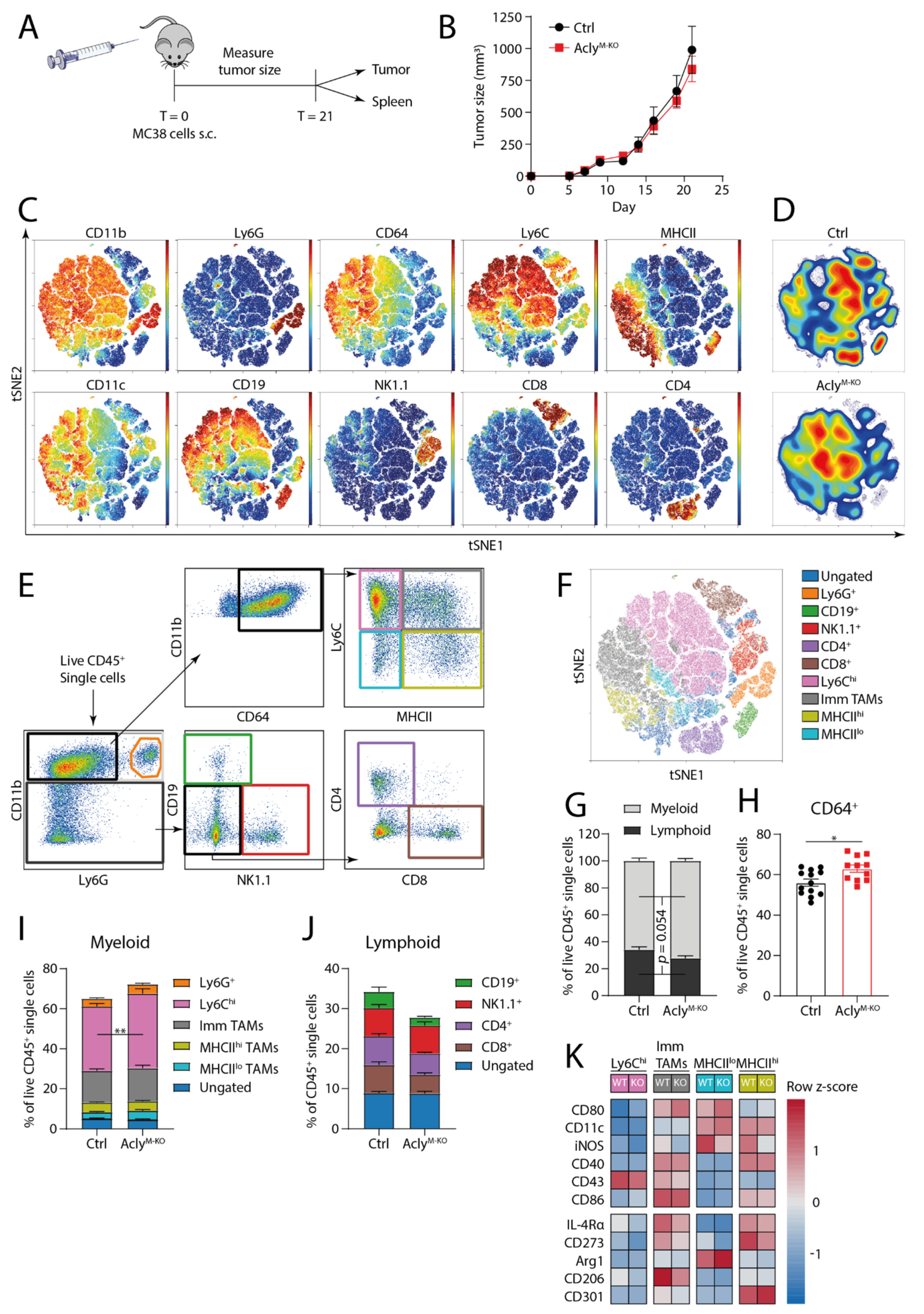
3.2. AclyM-KO Macrophages Do Not Improve In Vivo Anti-Tumor Responses
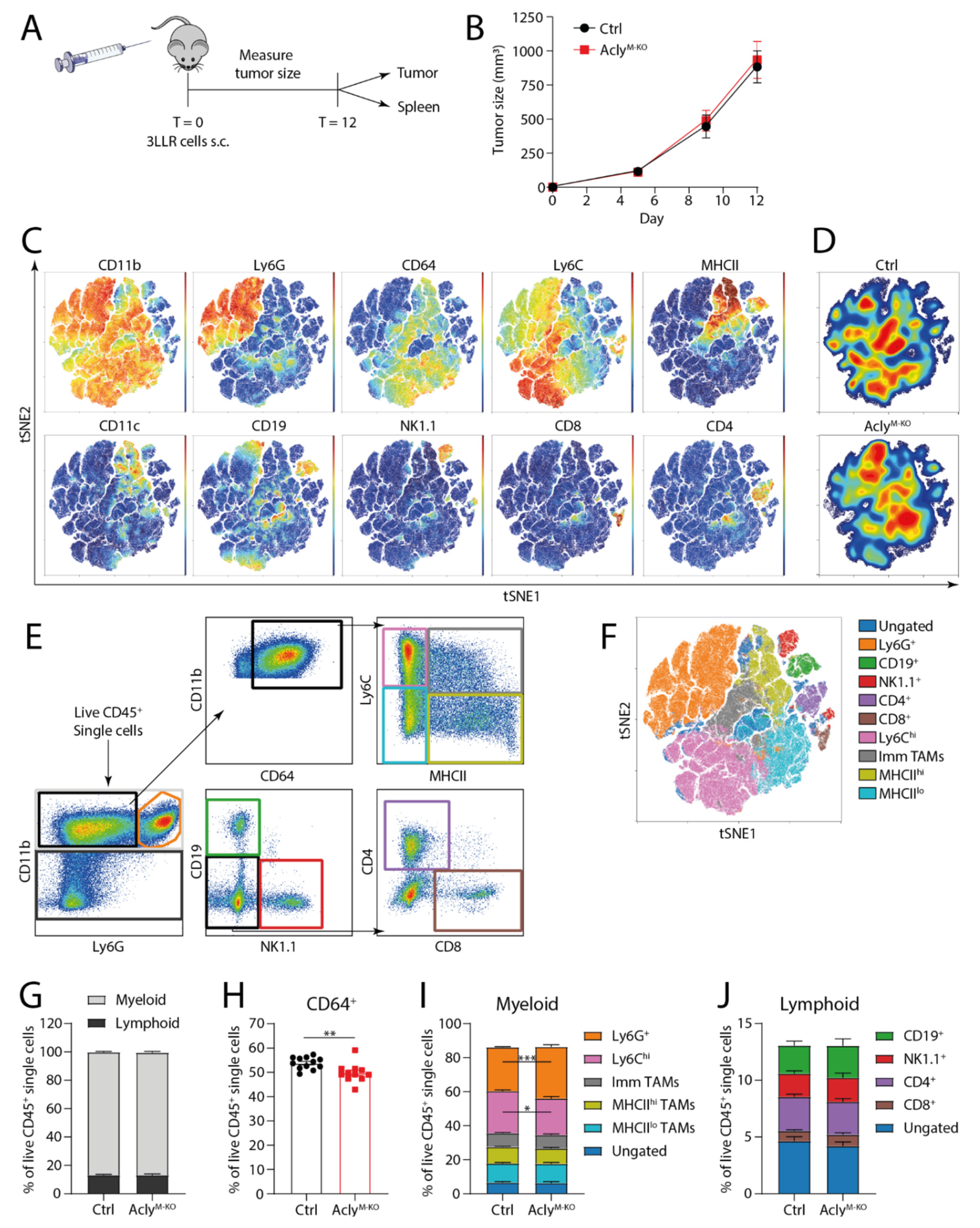
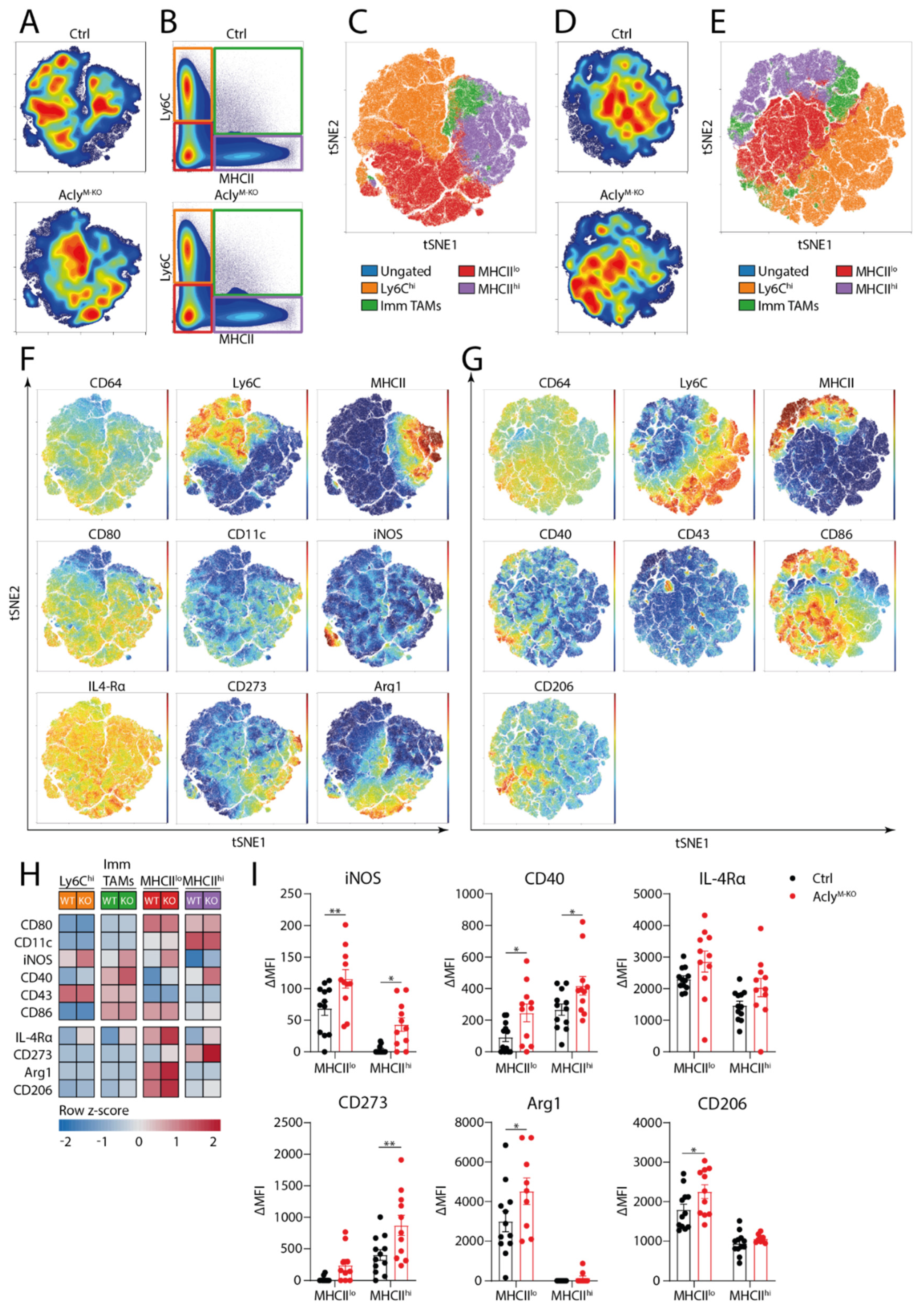
3.3. Acly Modulates In Vivo TAM Phenotype in 3LLR Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wen, J.; Min, X.; Shen, M.; Hua, Q.; Han, Y.; Zhao, L.; Liu, L.; Huang, G.; Liu, J.; Zhao, X. ACLY facilitates colon cancer cell metastasis by CTNNB1. J. Exp. Clin. Cancer Res. 2019, 38, 401. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Li, K.; Gong, D.; Zhang, J.; Li, Q.; Zhao, G.; Lin, P. ACLY: A biomarker of recurrence in breast cancer. Pathol Res. Pract. 2020, 216, 153076. [Google Scholar] [CrossRef]
- Bauer, D.E.; Hatzivassiliou, G.; Zhao, F.; Andreadis, C.; Thompson, C.B. ATP citrate lyase is an important component of cell growth and transformation. Oncogene 2005, 24, 6314–6322. [Google Scholar] [CrossRef] [Green Version]
- Hatzivassiliou, G.; Zhao, F.; Bauer, D.E.; Andreadis, C.; Shaw, A.N.; Dhanak, D.; Hingorani, S.R.; Tuveson, D.A.; Thompson, C.B. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005, 8, 311–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrer, A.; Trefely, S.; Zhao, S.; Campbell, S.L.; Norgard, R.J.; Schultz, K.C.; Sidoli, S.; Parris, J.L.D.; Affronti, H.C.; Sivanand, S.; et al. Acetyl-coa metabolism supports multistep pancreatic tumorigenesis. Cancer Discov. 2019, 9, 416–435. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.; Carriveau, W.J.; Li, J.; Campbell, S.L.; Kopinski, P.K.; Lim, H.W.; Daurio, N.; Trefely, S.; Won, K.J.; Wallace, D.C.; et al. Targeting ACLY sensitizes castration-resistant prostate cancer cells to AR antagonism by impinging on an ACLY-AMPK-AR feedback mechanism. Oncotarget 2016, 7, 43713–43730. [Google Scholar] [CrossRef] [Green Version]
- Khwairakpam, A.D.; Shyamananda, M.S.; Sailo, B.L.; Rathnakaram, S.R.; Padmavathi, G.; Kotoky, J.; Kunnumakkara, A.B. ATP citrate lyase (ACLY): A promising target for cancer prevention and treatment. Curr. Drug Targets 2015, 16, 156–163. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and cancer: Triggers, mechanisms, and consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- De Goede, K.E.; Harber, K.J.; Van den Bossche, J. Let’s enter the wonderful world of immunometabolites. Trends Endocrinol. Metab. 2019, 30, 329–331. [Google Scholar] [CrossRef]
- Van den Bossche, J.; Baardman, J.; Otto, N.A.; van der Velden, S.; Neele, A.E.; van den Berg, S.M.; Luque-Martin, R.; Chen, H.J.; Boshuizen, M.C.; Ahmed, M.; et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 2016, 17, 684–696. [Google Scholar] [CrossRef] [Green Version]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Ryan, D.G.; Murphy, M.P.; Frezza, C.; Prag, H.A.; Chouchani, E.T.; O’Neill, L.A.; Mills, E.L. Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat. Metab. 2019, 1, 16–33. [Google Scholar] [CrossRef]
- Van den Bossche, J.; O’Neill, L.A.; Menon, D. Macrophage immunometabolism: Where are we (going)? Trends Immunol. 2017, 38, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Aksoylar, H.I.; Yu, J.; Snyder, N.W.; Worth, A.J.; Iyer, S.S.; Wang, J.; Ben-Sahra, I.; Byles, V.; Polynne-Stapornkul, T.; et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Lauterbach, M.A.; Hanke, J.E.; Serefidou, M.; Mangan, M.S.J.; Kolbe, C.C.; Hess, T.; Rothe, M.; Kaiser, R.; Hoss, F.; Gehlen, J.; et al. Toll-like receptor signaling rewires macrophage metabolism and promotes histone acetylation via atp-citrate lyase. Immunity 2019, 51, 997–1011. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, J.; van der Windt, G.J.W. Fatty acid oxidation in macrophages and t cells: Time for reassessment? Cell Metab. 2018, 28, 538–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Wagner, R.A.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1 beta attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Divakaruni, A.S.; Hsieh, W.Y.; Minarrieta, L.; Duong, T.N.; Kim, K.K.O.; Desousa, B.R.; Andreyev, A.Y.; Bowman, C.E.; Caradonna, K.; Dranka, B.P.; et al. Etomoxir inhibits macrophage polarization by disrupting coa homeostasis. Cell Metab. 2018, 28, 490–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, M.; Liu, J.; Rovira, I.I.; Gonzalez-Hurtado, E.; Lee, J.; Wolfgang, M.J.; Finkel, T. Fatty acid oxidation in macrophage polarization. Nat. Immunol. 2016, 17, 216–217. [Google Scholar] [CrossRef] [PubMed]
- Namgaladze, D.; Zukunft, S.; Schnutgen, F.; Kurrle, N.; Fleming, I.; Fuhrmann, D.; Brune, B. Polarization of human macrophages by Interleukin-4 does not require ATP-Citrate lyase. Front. Immunol. 2018, 9, 2858. [Google Scholar] [CrossRef]
- Baardman, J.; Verberk, S.G.S.; van der Velden, S.; Gijbels, M.J.J.; van Roomen, C.; Sluimer, J.C.; Broos, J.Y.; Griffith, G.R.; Prange, K.H.M.; van Weeghel, M.; et al. Macrophage ATP citrate lyase deficiency stabilizes atherosclerotic plaques. Nat. Commun. 2020, 11, 6296. [Google Scholar] [CrossRef]
- Geeraerts, X.; Bolli, E.; Fendt, S.M.; Van Ginderachter, J.A. Macrophage metabolism as therapeutic target for cancer, atherosclerosis, and obesity. Front. Immunol. 2017, 8, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonelli, S.; Geeraerts, X.; Bolli, E.; Keirsse, J.; Kiss, M.; Pombo Antunes, A.R.; Van Damme, H.; De Vlaminck, K.; Movahedi, K.; Laoui, D.; et al. Beyond the M-CSF receptor—Novel therapeutic targets in tumor-associated macrophages. FEBS J. 2018, 285, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Turkowski, K.; Mora, J.; Brune, B.; Seeger, W.; Weigert, A.; Savai, R. Redirecting tumor-associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget 2017, 8, 48436–48452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagemann, T.; Lawrence, T.; McNeish, I.; Charles, K.A.; Kulbe, H.; Thompson, R.G.; Robinson, S.C.; Balkwill, F.R. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J. Exp. Med. 2008, 205, 1261–1268. [Google Scholar] [CrossRef]
- Kiss, M.; Vande Walle, L.; Saavedra, P.H.V.; Lebegge, E.; Van Damme, H.; Murgaski, A.; Qian, J.; Ehling, M.; Pretto, S.; Bolli, E.; et al. IL1beta promotes immune suppression in the tumor microenvironment independent of the inflammasome and gasdermin D. Cancer Immunol. Res. 2020. [Google Scholar] [CrossRef]
- De Goede, K.E.; Driessen, A.J.M.; Van den Bossche, J. Metabolic Cancer-Macrophage Crosstalk in the Tumor Microenvironment. Biology 2020, 9, 380. [Google Scholar] [CrossRef]
- Honkanen, T.J.; Tikkanen, A.; Karihtala, P.; Makinen, M.; Vayrynen, J.P.; Koivunen, J.P. Prognostic and predictive role of tumour-associated macrophages in HER2 positive breast cancer. Sci. Rep. 2019, 9, 10961. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Torres, A.; Henry, R.A.; Trefely, S.; Wallace, M.; Lee, J.V.; Carrer, A.; Sengupta, A.; Campbell, S.L.; Kuo, Y.M.; et al. ATP-Citrate lyase controls a glucose-to-acetate metabolic switch. Cell Rep. 2016, 17, 1037–1052. [Google Scholar] [CrossRef] [Green Version]
- Remels, L.M.; De Baetselier, P.C. Characterization of 3LL-tumor variants generated by in vitro macrophage-mediated selection. Int. J. Cancer 1987, 39, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Movahedi, K.; Laoui, D.; Gysemans, C.; Baeten, M.; Stange, G.; Van den Bossche, J.; Mack, M.; Pipeleers, D.; In’t Veld, P.; De Baetselier, P.; et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010, 70, 5728–5739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schetters, S.T.T.; Rodriguez, E.; Kruijssen, L.J.W.; Crommentuijn, M.H.W.; Boon, L.; Van den Bossche, J.; Den Haan, J.M.M.; Van Kooyk, Y. Monocyte-derived APCs are central to the response of PD1 checkpoint blockade and provide a therapeutic target for combination therapy. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Baardman, J.; Verberk, S.G.S.; Prange, K.H.M.; van Weeghel, M.; van der Velden, S.; Ryan, D.G.; Wust, R.C.I.; Neele, A.E.; Speijer, D.; Denis, S.W.; et al. A defective pentose phosphate pathway reduces inflammatory macrophage responses during hypercholesterolemia. Cell Rep. 2018, 25, 2044–2052. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, J.; Lamers, W.H.; Koehler, E.S.; Geuns, J.M.; Alhonen, L.; Uimari, A.; Pirnes-Karhu, S.; Van Overmeire, E.; Morias, Y.; Brys, L.; et al. Pivotal advance: Arginase-1-independent polyamine production stimulates the expression of IL-4-induced alternatively activated macrophage markers while inhibiting LPS-induced expression of inflammatory genes. J. Leukoc. Biol. 2012, 91, 685–699. [Google Scholar] [CrossRef]
- Van den Bossche, J.; Laoui, D.; Morias, Y.; Movahedi, K.; Raes, G.; De Baetselier, P.; Van Ginderachter, J.A. Claudin-1, claudin-2 and claudin-11 genes differentially associate with distinct types of anti-inflammatory macrophages in vitro and with parasite- and tumour-elicited macrophages in vivo. Scand. J. Immunol. 2012, 75, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Langston, P.K.; Nambu, A.; Jung, J.; Shibata, M.; Aksoylar, H.I.; Lei, J.; Xu, P.; Doan, M.T.; Jiang, H.; MacArthur, M.R.; et al. Glycerol phosphate shuttle enzyme GPD2 regulates macrophage inflammatory responses. Nat. Immunol. 2019, 20, 1186–1195. [Google Scholar] [CrossRef]
- Rhee, J.; Solomon, L.A.; DeKoter, R.P. A role for ATP Citrate Lyase in cell cycle regulation during myeloid differentiation. Blood Cells Mol. Dis. 2019, 76, 82–90. [Google Scholar] [CrossRef]
- Gubin, M.M.; Esaulova, E.; Ward, J.P.; Malkova, O.N.; Runci, D.; Wong, P.; Noguchi, T.; Arthur, C.D.; Meng, W.; Alspach, E.; et al. High-dimensional analysis delineates myeloid and lymphoid compartment remodeling during successful immune-checkpoint cancer therapy. Cell 2018, 175, 1014–1030.e1019. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [Green Version]
- Benner, B.; Scarberry, L.; Suarez-Kelly, L.P.; Duggan, M.C.; Campbell, A.R.; Smith, E.; Lapurga, G.; Jiang, K.; Butchar, J.P.; Tridandapani, S.; et al. Generation of monocyte-derived tumor-associated macrophages using tumor-conditioned media provides a novel method to study tumor-associated macrophages in vitro. J. Immunother. Cancer 2019, 7, 140. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.D.; Tse, M.J.; Read, E.L.; Liu, W.F. Regulation of macrophage polarization and plasticity by complex activation signals. Integr. Biol. 2016, 8, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Brancato, S.K.; Thomay, A.A.; Reichner, J.S.; Albina, J.E. The phenotype of murine wound macrophages. J. Leukoc. Biol. 2010, 87, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 2018, 174, 1293–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Rojas, A.R.; Kelsey, I.; Pappalardo, J.L.; Chen, M.; Miller-Jensen, K. Co-stimulation with opposing macrophage polarization cues leads to orthogonal secretion programs in individual cells. Nat. Commun. 2021, 12, 301. [Google Scholar] [CrossRef]
- Ham, S.; Lima, L.G.; Lek, E.; Moller, A. The impact of the cancer microenvironment on macrophage phenotypes. Front. Immunol. 2020, 11, 1308. [Google Scholar] [CrossRef]
- Guilliams, M.; Scott, C.L. Does niche competition determine the origin of tissue-resident macrophages? Nat. Rev. Immunol. 2017, 17, 451–460. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Goede, K.E.; Verberk, S.G.S.; Baardman, J.; Harber, K.J.; van Kooyk, Y.; de Winther, M.P.J.; Schetters, S.T.T.; Van den Bossche, J. Myeloid-Specific Acly Deletion Alters Macrophage Phenotype In Vitro and In Vivo without Affecting Tumor Growth. Cancers 2021, 13, 3054. https://doi.org/10.3390/cancers13123054
de Goede KE, Verberk SGS, Baardman J, Harber KJ, van Kooyk Y, de Winther MPJ, Schetters STT, Van den Bossche J. Myeloid-Specific Acly Deletion Alters Macrophage Phenotype In Vitro and In Vivo without Affecting Tumor Growth. Cancers. 2021; 13(12):3054. https://doi.org/10.3390/cancers13123054
Chicago/Turabian Stylede Goede, Kyra E., Sanne G. S. Verberk, Jeroen Baardman, Karl J. Harber, Yvette van Kooyk, Menno P. J. de Winther, Sjoerd T. T. Schetters, and Jan Van den Bossche. 2021. "Myeloid-Specific Acly Deletion Alters Macrophage Phenotype In Vitro and In Vivo without Affecting Tumor Growth" Cancers 13, no. 12: 3054. https://doi.org/10.3390/cancers13123054
APA Stylede Goede, K. E., Verberk, S. G. S., Baardman, J., Harber, K. J., van Kooyk, Y., de Winther, M. P. J., Schetters, S. T. T., & Van den Bossche, J. (2021). Myeloid-Specific Acly Deletion Alters Macrophage Phenotype In Vitro and In Vivo without Affecting Tumor Growth. Cancers, 13(12), 3054. https://doi.org/10.3390/cancers13123054