
Review of PP2A Tumor Biology and Antitumor Effects of PP2A Inhibitor LB100 in the Nervous System


Abstract
:Simple Summary
Abstract
1. Introduction
2. Role of PP2A in Cellular Signaling Pathways
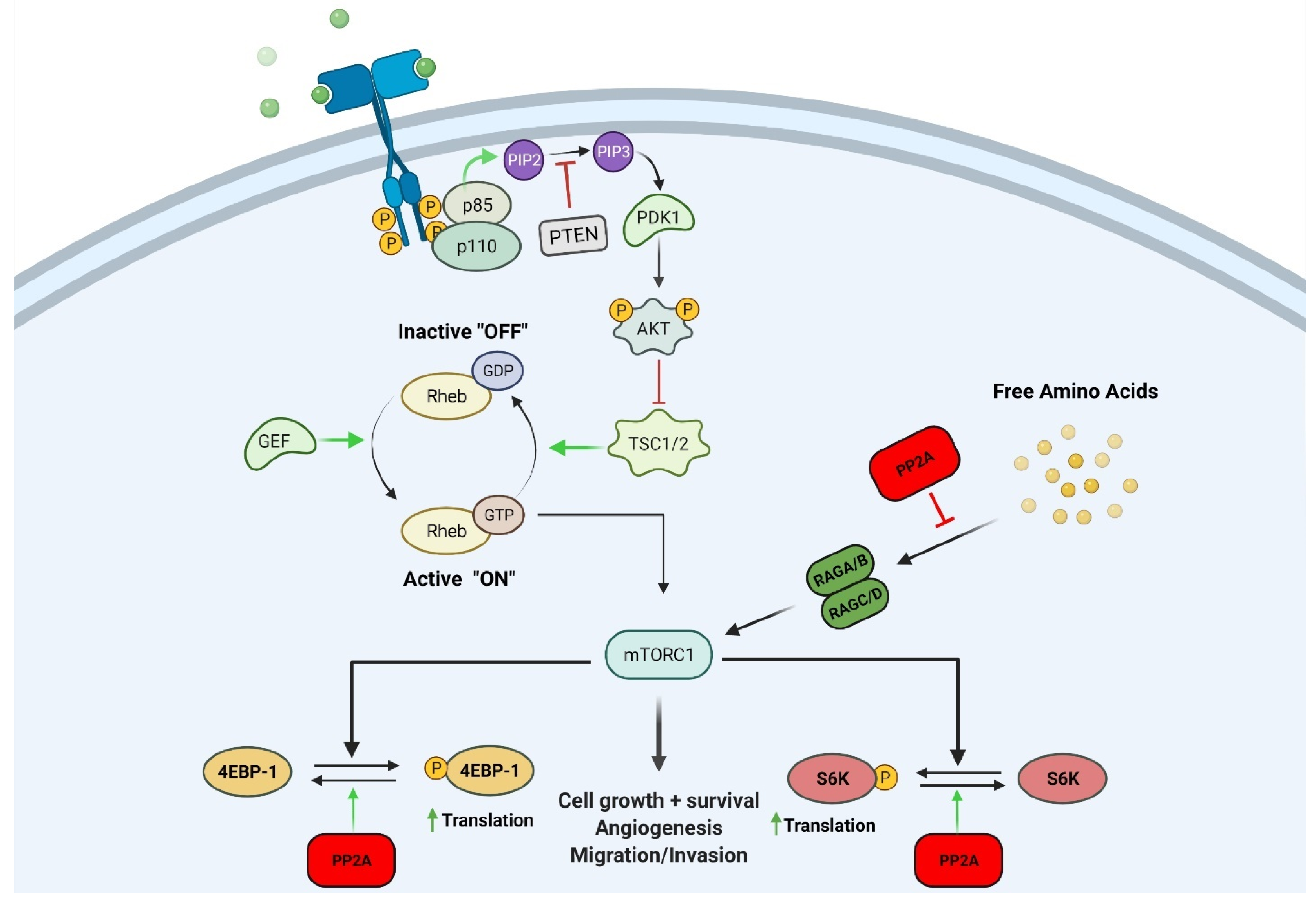
2.1. Mechanistic Target of Rapamycin (mTOR)
2.2. Wnt Signaling Pathway
2.3. Mitogen-Activated Protein Kinase (MAPK) Signaling Pathway
3. Role of PP2A in Neurodevelopment and Neurophysiology
4. LB100 as Therapy for Solid Nervous System Tumors
4.1. Glioblastoma
4.2. Pheochromocytoma
4.3. Medulloblastoma
4.4. Diffuse Intrinsic Pontine Glioma
4.5. Neuroblastoma
5. Biological Insights and Future Directions
6. Conclusions
Author Contributions
Funding
Declaration of Interest
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perrotti, D.; Neviani, P. Protein phosphatase 2A: A target for anticancer therapy. Lancet Oncol. 2013, 14, e229–e238. [Google Scholar] [CrossRef] [Green Version]
- Leslie, S.; Nairn, A.C. cAMP regulation of protein phosphatases PP1 and PP2A in brain. Biochim. Biophys. Acta Bioenerg. 2019, 1866, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Wlodarchak, N.; Xing, Y. PP2A as a master regulator of the cell cycle. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 162–184. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Lee, K.; Park, E.S.; Oh, S.; Yan, R.; Zhang, J.; Beach, T.G.; Adler, C.H.; Voronkov, M.; Braithwaite, S.P.; et al. Dysregulation of protein phosphatase 2A in parkinson disease and dementia with lewy bodies. Ann. Clin. Transl. Neurol. 2016, 3, 769–780. [Google Scholar] [CrossRef]
- Gong, C.-X.; Singh, T.J.; Grundke-Iqbal, I.; Iqbal, K. Phosphoprotein Phosphatase Activities in Alzheimer Disease Brain. J. Neurochem. 1993, 61, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Sontag, E.; Luangpirom, A.; Hladik, C.; Mudrak, I.; Ogris, E.; Speciale, S.; White, C.L., III. Altered Expression Levels of the Protein Phosphatase 2A ABαC Enzyme Are Associated with Alzheimer Disease Pathology. J. Neuropathol. Exp. Neurol. 2004, 63, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Javadpour, P.; Dargahi, L.; Ahmadiani, A.; Ghasemi, R. To be or not to be: PP2A as a dual player in CNS functions, its role in neurodegeneration, and its interaction with brain insulin signaling. Cell. Mol. Life Sci. 2019, 76, 2277–2297. [Google Scholar] [CrossRef] [PubMed]
- Narla, G.; Sangodkar, J.; Ryder, C.B. The impact of phosphatases on proliferative and survival signaling in cancer. Cell. Mol. Life Sci. 2018, 75, 2695–2718. [Google Scholar] [CrossRef]
- Meeusen, B.; Janssens, V. Tumor suppressive protein phosphatases in human cancer: Emerging targets for therapeutic intervention and tumor stratification. Int. J. Biochem. Cell Biol. 2018, 96, 98–134. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.S.; Ho, W.; Zhang, C.; Yang, C.; Elder, J.B.; Zhuang, Z. LB100, a small molecule inhibitor of PP2A with potent chemo- and radio-sensitizing potential. Cancer Biol. Ther. 2015, 16, 821–833. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Showkat, M.; Beigh, M.A.; Andrabi, K.I. mTOR Signaling in Protein Translation Regulation: Implications in Cancer Genesis and Therapeutic Interventions. Mol. Biol. Int. 2014, 2014, 686984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murugan, A.K. mTOR: Role in cancer, metastasis and drug resistance. Semin. Cancer Biol. 2019, 59, 92–111. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, J.L.; Garcia, E.; Pieper, R.O. S6K1 Plays a Key Role in Glial Transformation. Cancer Res. 2008, 68, 6516–6523. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Guo, J.; Li, H.; Wang, J. Meta-analysis of the prognostic value of p-4EBP1 in human malignancies. Oncotarget 2017, 9, 2761–2769. [Google Scholar] [CrossRef] [Green Version]
- Peterson, R.T.; Desai, B.N.; Hardwick, J.S.; Schreiber, S.L. Protein phosphatase 2A interacts with the 70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycinassociated protein. Proc. Natl. Acad. Sci. USA 1999, 96, 4438–4442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, A.; Tanimura-Ito, K.; Oshiro, N.; Eguchi, S.; Miyamoto, T.; Momonami, A.; Kamada, S.; Yonezawa, K.; Kikkawa, U. A positive role of mammalian Tip41-like protein, TIPRL, in the amino-acid dependent mTORC1-signaling pathway through interaction with PP2A. FEBS Lett. 2013, 587, 2924–2929. [Google Scholar] [CrossRef]
- Pachow, D.; Wick, W.; Gutmann, D.H.; Mawrin, C. The mTOR signaling pathway as a treatment target for intracranial neoplasms. Neuro-Oncology 2014, 17, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Plamper, M.; Born, M.; Gohlke, B.; Schreiner, F.; Schulte, S.; Splittstößer, V.; Woelfle, J.; Michaela, P.; Mark, B.; Bettina, G.; et al. Cerebral MRI and Clinical Findings in Children with PTEN Hamartoma Tumor Syndrome: Can Cerebral MRI Scan Help to Establish an Earlier Diagnosis of PHTS in Children? Cells 2020, 9, 1668. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; Wang, M.Y.; Vivanco, I.; Haas-Kogan, D.A.; Zhu, S.; Dia, E.Q.; Lu, K.V.; Yoshimoto, K.; Huang, J.H.; Chute, D.J.; et al. Molecular Determinants of the Response of Glioblastomas to EGFR Kinase Inhibitors. N. Engl. J. Med. 2005, 353, 2012–2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masica, D.L.; Karchin, R. Correlation of Somatic Mutation and Expression Identifies Genes Important in Human Glioblastoma Progression and Survival. Cancer Res. 2011, 71, 4550–4561. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H. Wnt/β-Catenin Signaling in Development and Disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julius, M.; Schelbert, B.; Hsu, W.; Fitzpatrick, E.; Jho, E.-H.; Fagotto, F.; Costantini, F.; Kitajewski, J. Domains of Axin and Disheveled Required for Interaction and Function in Wnt Signaling. Biochem. Biophys. Res. Commun. 2000, 276, 1162–1169. [Google Scholar] [CrossRef]
- Behrens, J.; Von Kries, J.P.; Kühl, M.; Bruhn, L.; Wedlich, D. Functional interaction of β-catenin with the transcription factor LEF-1. Nat. Cell Biol. 1996, 382, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, N.; Kurzrock, R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 2018, 62, 50–60. [Google Scholar] [CrossRef]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nat. Cell Biol. 2005, 434, 843–850. [Google Scholar] [CrossRef]
- Vermeulen, L.; Melo, F.D.S.E.; Van Der Heijden, M.; Cameron, K.; De Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef]
- Latres, E.; Chiaur, D.S.; Pagano, M. The human F box protein β-Trcp associates with the Cul1/Skp1 complex and regulates the stability of β-catenin. Oncogene 1999, 18, 849–854. [Google Scholar] [CrossRef] [Green Version]
- Ha, N.-C.; Tonozuka, T.; Stamos, J.L.; Choi, H.-J.; Weis, W.I. Mechanism of Phosphorylation-Dependent Binding of APC to β-Catenin and Its Role in β-Catenin Degradation. Mol. Cell 2004, 15, 511–521. [Google Scholar] [CrossRef]
- Jho, E.-H.; Lomvardas, S.; Costantini, F. A GSK3β Phosphorylation Site in Axin Modulates Interaction with β-Catenin and Tcf-Mediated Gene Expression. Biochem. Biophys. Res. Commun. 1999, 266, 28–35. [Google Scholar] [CrossRef]
- Jiang, Y.; Luo, W.; Howe, P.H. Dab2 stabilizes Axin and attenuates Wnt/β-catenin signaling by preventing protein phosphatase 1 (PP1)–Axin interactions. Oncogene 2009, 28, 2999–3007. [Google Scholar] [CrossRef] [Green Version]
- Seshacharyulu, P.; Pandey, P.; Datta, K.; Batra, S.K. Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett. 2013, 335, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Yang, J.; Liu, Y.; Chen, X.; Yu, T.; Jia, J.; Liu, C. PR55α, a Regulatory Subunit of PP2A, Specifically Regulates PP2A-mediated β-Catenin Dephosphorylation. J. Biol. Chem. 2009, 284, 22649–22656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hein, A.L.; Seshacharyulu, P.; Rachagani, S.; Sheinin, Y.M.; Ouellette, M.M.; Ponnusamy, M.P.; Mumby, M.C.; Batra, S.K.; Yan, Y. PR55α Subunit of Protein Phosphatase 2A Supports the Tumorigenic and Metastatic Potential of Pancreatic Cancer Cells by Sustaining Hyperactive Oncogenic Signaling. Cancer Res. 2016, 76, 2243–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, N.; Malbon, C.C. Phosphoprotein phosphatase-2A docks to Dishevelled and counterregulates Wnt3a/β-catenin signaling. J. Mol. Signal. 2007, 2, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, A.; Menezes, M.E.; Pannell, L.K.; Mulekar, M.S.; Honkanen, R.E.; Shevde, L.A.; Samant, R.S. DNAJB6 chaperones PP2A mediated dephosphorylation of GSK3β to downregulate β-catenin transcription target, osteopontin. Oncogene 2012, 31, 4472–4483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago, L.; Daniels, G.; Wang, D.; Deng, F.-M.; Lee, P. Wnt signaling pathway protein LEF1 in cancer, as a biomarker for prognosis and a target for treatment. Am. J. Cancer Res. 2017, 7, 1389–1406. [Google Scholar]
- Arnés, M.; Tintó, S.C. Aberrant Wnt signaling: A special focus in CNS diseases. J. Neurogenet. 2017, 31, 216–222. [Google Scholar] [CrossRef]
- Auger, N.; Thillet, J.; Wanherdrick, K.; Idbaih, A.; Legrier, M.-E.; Dutrillaux, B.; Sanson, M.; Poupon, M.-F. Genetic alterations associated with acquired temozolomide resistance in SNB-19, a human glioma cell line. Mol. Cancer Ther. 2006, 5, 2182–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Jeon, H.-Y.; Joo, K.M.; Kim, J.-K.; Jin, J.; Kim, S.H.; Kang, B.G.; Beck, S.; Lee, S.J.; Kim, J.K.; et al. Frizzled 4 Regulates Stemness and Invasiveness of Migrating Glioma Cells Established by Serial Intracranial Transplantation. Cancer Res. 2011, 71, 3066–3075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Liu, W.-Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.-F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal. Transduct. 2015, 35, 600–604. [Google Scholar] [CrossRef]
- Guo, Y.; Pan, W.; Liu, S.; Shen, Z.; Xu, Y.; Hu, L. ERK/MAPK signalling pathway and tumorigenesis (Review). Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.X.; Xiong, H.F.; Wang, S.; Wang, J.; Nie, X.; Guo, Q.; Li, X.; Qi, Y.; Liu, J.J.; Lin, B. Overexpression of TEM8 promotes ovarian cancer progression via Rac1/Cdc42/JNK and MEK/ERK/STAT3 signaling pathways. Am. J. Transl. Res. 2020, 12, 3557–3576. [Google Scholar]
- Ugi, S.; Imamura, T.; Ricketts, W.; Olefsky, J.M. Protein Phosphatase 2A Forms a Molecular Complex with Shc and Regulates Shc Tyrosine Phosphorylation and Downstream Mitogenic Signaling. Mol. Cell. Biol. 2002, 22, 2375–2387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lao, D.-H.; Yusoff, P.; Chandramouli, S.; Philp, R.J.; Fong, C.W.; Jackson, R.A.; Saw, T.Y.; Yu, C.Y.; Guy, G.R. Direct Binding of PP2A to Sprouty2 and Phosphorylation Changes Are a Prerequisite for ERK Inhibition Downstream of Fibroblast Growth Factor Receptor Stimulation. J. Biol. Chem. 2007, 282, 9117–9126. [Google Scholar] [CrossRef] [Green Version]
- Zwaenepoel, K.; Goris, J.; Erneux, C.; Parker, P.J.; Janssens, V. Protein phosphatase 2A PR130/B”α:1 subunit binds to the SH2 domain-containing inositol polyphosphate 5-phosphatase 2 and prevents epidermal growth factor (EGF)-induced EGF receptor degradation sustaining EGF-mediated signaling. FASEB J. 2009, 24, 538–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaumot, M.; Hancock, J.F. Protein phosphatases 1 and 2A promote Raf-1 activation by regulating 14-3-3 interactions. Oncogene 2001, 20, 3949–3958. [Google Scholar] [CrossRef]
- DeClue, J.E.; Papageorge, A.G.; Fletcher, J.A.; Diehl, S.R.; Ratner, N.; Vass, W.C.; Lowy, D.R. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell 1992, 69, 265–273. [Google Scholar] [CrossRef]
- Lewis, R.A.; Gerson, L.P.; Axelson, K.A.; Riccardi, V.M.; Whitford, R.P. von Recklinghausen Neurofibromatosis. Ophthalmology 1984, 91, 929–935. [Google Scholar] [CrossRef]
- Daniel, P.M.; Filiz, G.; Tymms, M.J.; Ramsay, R.G.; Kaye, A.H.; Stylli, S.S.; Mantamadiotis, T. Intratumor MAPK and PI3K signaling pathway heterogeneity in glioblastoma tissue correlates with CREB signaling and distinct target gene signatures. Exp. Mol. Pathol. 2018, 105, 23–31. [Google Scholar] [CrossRef]
- Skinner, M.; Ward, S.M.; Nilsson, C.L.; Emrick, T. Augmenting Glioblastoma Chemotherapy with Polymers. ACS Chem. Neurosci. 2017, 9, 8–10. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Sun, L.-H.; Huang, Y.-F.; Guo, L.-J.; Luo, L.-S. Protein phosphatase 2ACα gene knock-out results in cortical atrophy through activating hippo cascade in neuronal progenitor cells. Int. J. Biochem. Cell Biol. 2018, 95, 53–62. [Google Scholar] [CrossRef]
- Yamashita, T.; Inui, S.; Maeda, K.; Hua, D.R.; Takagi, K.; Fukunaga, K.; Sakaguchi, N. Regulation of CaMKII by α4/PP2Ac contributes to learning and memory. Brain Res. 2006, 1082, 1–10. [Google Scholar] [CrossRef]
- Rossetti, T.; Banerjee, S.; Kim, C.; Leubner, M.; Lamar, C.; Gupta, P.; Lee, B.; Neve, R.; Lisman, J. Memory Erasure Experiments Indicate a Critical Role of CaMKII in Memory Storage. Neuron 2017, 96, 207–216.e2. [Google Scholar] [CrossRef] [Green Version]
- Monroe, J.D.; Heathcote, R.D. Protein phosphatases regulate the growth of developing neurites. Int. J. Dev. Neurosci. 2013, 31, 250–257. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Torii, T.; Yamamori, N.; Ogata, T.; Tanoue, A.; Yamauchi, J. Akt and PP2A Reciprocally Regulate the Guanine Nucleotide Exchange Factor Dock6 to Control Axon Growth of Sensory Neurons. Sci. Signal. 2013, 6, ra15. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.-Q.; Zheng, H.-Y.; Peng, C.-X.; Liu, D.; Li, H.-L.; Wang, Q.; Wang, J.-Z. Protein Phosphatase 2A Facilitates Axonogenesis by Dephosphorylating CRMP2. J. Neurosci. 2010, 30, 3839–3848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nematullah; Hoda, M.N.; Khan, F. Protein Phosphatase 2A: A Double-Faced Phosphatase of Cellular System and Its Role in Neurodegenerative Disorders. Mol. Neurobiol. 2017, 55, 1750–1761. [Google Scholar] [CrossRef] [PubMed]
- Sim, A.T.; Lloyd, H.G.; Jarvie, P.E.; Morrison, M.; Rostas, J.A.; Dunkley, P.R. Synaptosomal amino acid release: Effect of inhibiting protein phosphatases with okadaic acid. Neurosci. Lett. 1993, 160, 181–184. [Google Scholar] [CrossRef]
- Beaulieu, J.-M.; Sotnikova, T.D.; Marion, S.; Lefkowitz, R.J.; Gainetdinov, R.; Caron, M.G. An Akt/β-Arrestin 2/PP2A Signaling Complex Mediates Dopaminergic Neurotransmission and Behavior. Cell 2005, 122, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Avila, J.; Lucas, J.J.; Pérez, M.; Hernandez, F. Role of Tau Protein in Both Physiological and Pathological Conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [CrossRef]
- Kimura, T.; Sharma, G.; Ishiguro, K.; Hisanaga, S.-I. Phospho-Tau Bar Code: Analysis of Phosphoisotypes of Tau and Its Application to Tauopathy. Front. Neurosci. 2018, 12, 44. [Google Scholar] [CrossRef]
- Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.-X. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur. J. Neurosci. 2005, 22, 1942–1950. [Google Scholar] [CrossRef] [PubMed]
- Sontag, E.; Nunbhakdi-Craig, V.; Lee, G.; Bloom, G.S.; Mumby, M.C. Regulation of the Phosphorylation State and Microtubule-Binding Activity of Tau by Protein Phosphatase 2A. Neuron 1996, 17, 1201–1207. [Google Scholar] [CrossRef] [Green Version]
- Schild, A.; Ittner, L.M.; Götz, J. Altered phosphorylation of cytoskeletal proteins in mutant protein phosphatase 2A transgenic mice. Biochem. Biophys. Res. Commun. 2006, 343, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, A.; Taleski, G.; Sontag, E. The protein serine/threonine phosphatases PP2A, PP1 and calcineurin: A triple threat in the regulation of the neuronal cytoskeleton. Mol. Cell. Neurosci. 2017, 84, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-J.; Lee, K.-W.; Oh, S.; Yan, R.; Zhang, J.; Beach, T.G.; Adler, C.H.; Voronkov, M.; Braithwaite, S.P.; Stock, J.B.; et al. Protein Phosphatase 2A and Its Methylation Modulating Enzymes LCMT-1 and PME-1 Are Dysregulated in Tauopathies of Progressive Supranuclear Palsy and Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2018, 77, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Arif, M.; Kazim, S.F.; Grundke-Iqbal, I.; Garruto, R.M.; Iqbal, K. Tau pathology involves protein phosphatase 2A in Parkinsonism-dementia of Guam. Proc. Natl. Acad. Sci. USA 2014, 111, 1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sontag, J.-M.; Sontag, E. Protein phosphatase 2A dysfunction in Alzheimer’s disease. Front. Mol. Neurosci. 2014, 7, 16. [Google Scholar] [CrossRef]
- Chen, W.; Arroyo, J.D.; Timmons, J.C.; Possemato, R.; Hahn, W.C. Cancer-Associated PP2A Aα Subunits Induce Functional Haploinsufficiency and Tumorigenicity. Cancer Res. 2005, 65, 8183–8192. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.C.; Liu, W.; Kim, S.-T.; Sun, J.; Lu, L.; Sun, J.; Zheng, S.L.; Isaacs, W.B.; Xu, J. Evaluation of PPP2R2A as a prostate cancer susceptibility gene: A comprehensive germline and somatic study. Cancer Genet. 2011, 204, 375–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haesen, D.; Asbagh, L.A.; Derua, R.; Hubert, A.; Schrauwen, S.; Hoorne, Y.; Amant, F.; Waelkens, E.; Sablina, A.; Janssens, V. Recurrent PPP2R1A Mutations in Uterine Cancer Act through a Dominant-Negative Mechanism to Promote Malignant Cell Growth. Cancer Res. 2016, 76, 5719–5731. [Google Scholar] [CrossRef] [Green Version]
- Nobumori, Y.; Shouse, G.P.; Wu, Y.; Lee, K.J.; Shen, B.; Liu, X. B56γ Tumor-Associated Mutations Provide New Mechanisms for B56γ-PP2A Tumor Suppressor Activity. Mol. Cancer Res. 2013, 11, 995–1003. [Google Scholar] [CrossRef] [Green Version]
- Saddoughi, S.A.; Gencer, S.; Peterson, Y.K.; Ward, K.E.; Mukhopadhyay, A.; Oaks, J.; Bielawski, J.; Szulc, Z.M.; Thomas, R.J.; Selvam, S.P.; et al. Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol. Med. 2012, 5, 105–121. [Google Scholar] [CrossRef]
- Shouse, G.; De Necochea-Campion, R.; Mirshahidi, S.; Liu, X.; Chen, C.-S. Novel B55α-PP2A mutations in AML promote AKT T308 phosphorylation and sensitivity to AKT inhibitor-induced growth arrest. Oncotarget 2016, 7, 61081–61092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grech, G.; Baldacchino, S.; Saliba, C.; Grixti, M.P.; Gauci, R.; Petroni, V.; Fenech, A.G.; Scerri, C. Deregulation of the protein phosphatase 2A, PP2A in cancer: Complexity and therapeutic options. Tumor Biol. 2016, 37, 11691–11700. [Google Scholar] [CrossRef] [PubMed]
- Janssens, V.; Zwaenepoel, K.; Rossé, C.; Petit, M.M.R.; Goris, J.; Parker, P.J. PP2A binds the LIM-domains of Lipoma Preferred Partner via its PR130/B” subunit to regulate cell adhesion and migration. J. Cell Sci. 2016, 129, 1605–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Fang, G.; Guo, F.; Zhang, H.; Chen, X.; An, L.; Chen, M.; Zhou, L.; Wang, W.; Ye, T.; et al. Selective Inhibition of STRN3-Containing PP2A Phosphatase Restores Hippo Tumor-Suppressor Activity in Gastric Cancer. Cancer Cell 2020, 38, 115–128.e9. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.-Y.; Guan, K.-L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF -TRCP. Genes Dev. 2010, 24, 72–85. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Liu, Y.; Zheng, Y.; Dong, J.; Pan, D. The TEAD/TEF Family Protein Scalloped Mediates Transcriptional Output of the Hippo Growth-Regulatory Pathway. Dev. Cell 2008, 14, 388–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalev, P.; Simicek, M.; Vazquez, I.; Munck, S.; Chen, L.; Soin, T.; Danda, N.; Chen, W.; Sablina, A. Loss of PPP2R2A Inhibits Homologous Recombination DNA Repair and Predicts Tumor Sensitivity to PARP Inhibition. Cancer Res. 2012, 72, 6414–6424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, D.; Parsels, L.A.; Karnak, D.; Davis, M.A.; Parsels, J.D.; Marsh, A.C.; Zhao, L.; Maybaum, J.; Lawrence, T.S.; Sun, Y.; et al. Inhibition of Protein Phosphatase 2A Radiosensitizes Pancreatic Cancers by Modulating CDC25C/CDK1 and Homologous Recombination Repair. Clin. Cancer Res. 2013, 19, 4422–4432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazhar, S.; Taylor, S.E.; Sangodkar, J.; Narla, G. Targeting PP2A in cancer: Combination therapies. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Qiu, P.; Wang, S.; Liu, M.; Ma, H.; Zeng, X.; Zhang, M.; Xu, L.; Cui, Y.; Xu, H.; Tang, Y.; et al. Norcantharidin Inhibits cell growth by suppressing the expression and phosphorylation of both EGFR and c-Met in human colon cancer cells. BMC Cancer 2017, 17, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Ho, W.S.; Feldman, M.J.; Maric, D.; Amable, L.; Hall, M.D.; Feldman, G.M.; Ray-Chaudhury, A.; Lizak, M.J.; Vera, J.-C.; Robison, R.A.; et al. PP2A inhibition with LB100 enhances cisplatin cytotoxicity and overcomes cisplatin resistance in medulloblastoma cells. Oncotarget 2016, 7, 12447–12463. [Google Scholar] [CrossRef] [Green Version]
- Swingle, M.; Ni, L.; Honkanen, R.E. Small-molecule inhibitors of ser/thr protein phosphatases: Specificity, use and common forms of abuse. Breast Cancer 2007, 365, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Honkanen, R.; Golden, T. Regulators of Serine / Threonine Protein Phosphatases at the Dawn of a Clinical Era? Curr. Med. Chem. 2002, 9, 2055–2075. [Google Scholar] [CrossRef]
- Han, K.; Gan, Z.; Lin, S.; Hu, H.; Shen, Z.; Min, D. Elevated expression of serine/threonine phosphatase type 5 correlates with malignant proliferation in human osteosarcoma. Acta Biochim. Pol. 2017, 64, 11–16. [Google Scholar] [CrossRef]
- Zhi, X.; Zhang, H.; He, C.; Wei, Y.; Bian, L.; Li, G. Serine/Threonine Protein Phosphatase-5 Accelerates Cell Growth and Migration in Human Glioma. Cell. Mol. Neurobiol. 2015, 35, 669–677. [Google Scholar] [CrossRef]
- Golden, T.; Aragon, I.V.; Zhou, G.; Cooper, S.R.; Dean, N.M.; Honkanen, R.E. Constitutive over expression of serine/threonine protein phosphatase 5 (PP5) augments estrogen-dependent tumor growth in mice. Cancer Lett. 2004, 215, 95–100. [Google Scholar] [CrossRef]
- D’Arcy, B.M.; Swingle, M.R.; Papke, C.M.; Abney, K.A.; Bouska, E.S.; Prakash, A.; Honkanen, R.E. The Antitumor Drug LB-100 Is a Catalytic Inhibitor of Protein Phosphatase 2A (PPP2CA) and 5 (PPP5C) Coordinating with the Active-Site Catalytic Metals in PPP5C. Mol. Cancer Ther. 2019, 18, 556–566. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Kovach, J.S.; Johnson, F.; Chiang, J.; Hodes, R.; Lonser, R.; Zhuang, Z. Inhibition of serine/threonine phosphatase PP2A enhances cancer chemotherapy by blocking DNA damage induced defense mechanisms. Proc. Natl. Acad. Sci. USA 2009, 106, 11697–11702. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Zhuang, Z.; Song, D.K.; Mehta, G.; Ikejiri, B.; Mushlin, H.; Park, D.M.; Lonser, R.R. The effect of a PP2A inhibitor on the nuclear receptor corepressor pathway in glioma. J. Neurosurg. 2010, 113, 225–233. [Google Scholar] [CrossRef]
- Park, D.; Li, J.; Okamoto, H.; Akeju, O.; Kim, S.; Lubensky, I. N-CoR pathway targeting induces glioblastoma derived cancer stem cell differentiation. Cell Cycle 2007, 6, 467–470. [Google Scholar] [CrossRef]
- Gordon, I.K.; Lu, J.; Graves, C.A.; Huntoon, K.; Frerich, J.M.; Hanson, R.H.; Wang, X.; Hong, C.S.; Ho, W.; Feldman, M.J.; et al. Protein Phosphatase 2A Inhibition with LB100 Enhances Radiation-Induced Mitotic Catastrophe and Tumor Growth Delay in Glioblastoma. Mol. Cancer Ther. 2015, 14, 1540–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, J.; Wang, H.; Medina, R.; Zhang, Q.; Xu, C.; Indig, I.H.; Zhou, J.; Song, Q.; Dmitriev, P.; Sun, M.Y.; et al. Inhibition of PP2A with LB-100 Enhances Efficacy of CAR-T Cell Therapy Against Glioblastoma. Cancers 2020, 12, 139. [Google Scholar] [CrossRef] [Green Version]
- Maggio, D.; Ho, W.S.; Breese, R.; Walbridge, S.; Wang, H.; Cui, J.; Heiss, J.D.; Gilbert, M.R.; Kovach, J.S.; Lu, R.O.; et al. Inhibition of protein phosphatase-2A with LB-100 enhances antitumor immunity against glioblastoma. J. Neuro-Oncol. 2020, 148, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Otani, Y.; Sur, H.; Rachaiah, G.; Namagiri, S.; Chowdhury, A.; Lewis, C.T.; Shimizu, T.; Gangaplara, A.; Wang, X.; Vézina, A.; et al. Inhibiting protein phosphatase 2A increases the antitumor effect of protein arginine methyltransferase 5 inhibition in models of glioblastoma. Neuro-Oncology 2021. [Google Scholar] [CrossRef] [PubMed]
- Martiniova, L.; Lu, J.; Chiang, J.; Bernardo, M.; Lonser, R.; Zhuang, Z.; Pacak, K. Pharmacologic Modulation of Serine/Threonine Phosphorylation Highly Sensitizes PHEO in a MPC Cell and Mouse Model to Conventional Chemotherapy. PLoS ONE 2011, 6, e14678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corssmit, E.P.; Snel, M.; Kapiteijn, E. Malignant pheochromocytoma and paraganglioma: Management options. Curr. Opin. Oncol. 2020, 32, 20–26. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Menyhárt, O.; Győrffy, B. Molecular stratifications, biomarker candidates and new therapeutic options in current medulloblastoma treatment approaches. Cancer Metastasis Rev. 2020, 39, 211–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juraschka, K.; Taylor, M.D. Medulloblastoma in the age of molecular subgroups: A review. J. Neurosurg. Pediatr. 2019, 24, 353–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, R.J.; Zhou, T.; Holmes, E.; Vezina, G.; Gajjar, A. Survival and secondary tumors in children with medulloblastoma receiving radiotherapy and adjuvant chemotherapy: Results of Children’s Oncology Group trial A9961. Neuro-Oncology 2013, 15, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Łastowska, M.; Trubicka, J.; Niemira, M.; Paczkowska-Abdulsalam, M.; Karkucińska-Więckowska, A.; Kaleta, M.; Drogosiewicz, M.; Perek-Polnik, M.; Kretowski, A.; Cukrowska, B.; et al. Medulloblastoma with transitional features between Group 3 and Group 4 is associated with good prognosis. J. Neuro-Oncol. 2018, 138, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Vanan, M.I.; Eisenstat, D.D. DIPG in Children—What Can We Learn from the Past? Front. Oncol. 2015, 5, 237. [Google Scholar] [CrossRef] [Green Version]
- Schramm, K.; Iskar, M.; Statz, B.; Jäger, N.; Haag, D.; Słabicki, M.; Pfister, S.M.; Zapatka, M.; Gronych, J.; Jones, D.T.W.; et al. DECIPHER pooled shRNA library screen identifies PP2A and FGFR signaling as potential therapeutic targets for diffuse intrinsic pontine gliomas. Neuro-Oncology 2019, 21, 867–877. [Google Scholar] [CrossRef]
- Ishola, T.A.; Chung, D.H. Neuroblastoma. Surg. Oncol. 2007, 16, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Chung, V.; Mansfield, A.S.; Braiteh, F.; Richards, D.; Durivage, H.; Ungerleider, R.S.; Johnson, F.; Kovach, J.S. Safety, Tolerability, and Preliminary Activity of LB-100, an Inhibitor of Protein Phosphatase 2A, in Patients with Relapsed Solid Tumors: An Open-Label, Dose Escalation, First-in-Human, Phase I Trial. Clin. Cancer Res. 2016, 23, 3277–3284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Dube, C.; Gibert, J.M., Jr.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The p53 Pathway in Glioblastoma. Cancers 2018, 10, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Subunit Family | Protein Isoform | Other Associated Name(s) | Nervous System Tissue Expression |
|---|---|---|---|
| A | Aα | PR65α | Anterior cingulate cortex |
| Aβ | PR65β | Corpus callosum | |
| B”’/Striatin | B”’ | Striatin | Corpus callosum |
| B/PR55 | Bα | B55α/PR55α | Corpus callosum |
| Bβ1 | B55β1/PR55β1 | Corpus callosum | |
| Bβ2 | B55β2/PR55β2 | Corpus callosum | |
| Bγ | B55γ/PR55γ | Caudate nucleus | |
| Bδ | B55δ/PR55δ | Dorsal/ventral thalamus | |
| B’/PR61 | Bα | B56α/PR61α | Corpus callosum |
| Bβ | B56β/PR61β | Right cerebellar hemisphere | |
| B’γ1 | B56γ1/PR61γ1 | Caudate nucleus | |
| B’γ2 | B56γ2/PR61γ2 | Caudate nucleus | |
| B’γ3 | B56γ3/PR61γ3 | Caudate nucleus | |
| B’δ | B56δ1/PR61δ | Dorsal/ventral thalamus | |
| B’ε | B56ε/PR61ε | Forebrain | |
| B”/Pr72 | B”α | PR130 | Forebrain |
| B”α | PR72 | Forebrain | |
| B”β | PR70 | Hypothalamus | |
| B”γ | G5PR | C1 segment of cervical spinal cord | |
| C | Cα | PP2Acα | Frontal cortex |
| Cβ | PP2Acβ | Dorsal/ventral thalamus |
| Investigators (Year) | Tumor Type | Treatment Method | Outcome |
|---|---|---|---|
| Lu et al. [95] (2010) | Glioblastoma | LB100 only | LB100 inhibited PP2A and caused dose-dependent antiproliferative activity in two GBM cell lines. LB100 treatment resulted in a significant reduction in tumor volume compared to controls (p < 0.001) in vivo. In vivo experiments also resulted in decreased nuclear N-CoR expression. |
| Gordon et al. [97] (2015) | Glioblastoma | LB100 and radiation therapy | LB100 resulted in radiation dose enhancement and increased mitotic catastrophe. Combination therapy significantly enhanced tumor growth delay while decreasing p53 in vivo. Combination therapy also increased the overall survival of mouse xenografts. |
| Cui et al. [98] (2020) | Glioblastoma | LB100 and CAR-T cells | Anti-CAIX CAR-T cell and LB100 combination therapy resulted in significant cytotoxicity against GBM tumor cells and increased cytokine production compared to control T-cell treatment in vitro. Combination therapy significantly increased tumor regression compared to monotherapy in vivo (p < 0.05) and significantly prolonged survival (p < 0.001). |
| Maggio et al. [99] (2020) | Glioblastoma | LB100 and PD-1 inhibition | Combination therapy significantly improved survival compared to monotherapy (p < 0.005) and controls (p < 0.001). Complete tumor regression was seen in 25% of combination-treated mice but no other subgroups. |
| Otani et al. [100] (2021) | Glioblastoma | LB100 and PRMT5 knockdown | LB100 administration significantly reduced viability in PRMT5-depleted GBMNS compared to PRMT5-intact GBMNS. PRMT5 knockdown and LB100 combination therapy increased the expression of phospho-MLKL. Combination therapy significantly decreased tumor size and prolonged survival in in vivo mouse xenografts. |
| Lu et al. [94] (2009) | Neuroblastoma and GBM | LB102 and TMZ | LB102 treatment in U87MG GBM cells resulted in morphological features of mitotic catastrophe. LB102 caused complete regression of GBM xenografts with no recurrence in 50% of animals and inhibited the growth of NB xenografts. |
| Martiniova et al. [101] (2011) | Pheochromocytoma | LB100 and TMZ | Combination therapy resulted in significantly greater tumor cell inhibition in vitro compared to monotherapy. PHEO mouse xenografts treated with combination therapy had significantly prolonged survival compared to monotherapy (p < 0.0001). Combination therapy significantly delayed the appearance of hepatic tumors compared to monotherapy alone (p < 0.0001). |
| Ho et al. [87] (2016) | Medulloblastoma | LB100 and cisplatin | LB100 alone had a potent antitumor effect of multiple medulloblastoma cell lines. Combination therapy enhanced cisplatin cytotoxicity and significantly decreased medulloblastoma cell viability as compared to controls (p < 0.005). Combination therapy significantly reduced tumor burden on POD64 compared to cisplatin treatment alone (p < 0.05) in vivo. |
| Schramm et al. [110] (2019) | DIPG | LB100 only | Investigators used a large-scale gene knockdown approach using shRNA and DNA sequencing to identify susceptibilities of DIPG tumor cells. Screening resulted in FGFR and PP2A deemed as candidate targets. LB100 therapy induced apoptosis in two DIPG cell lines in a dose-dependent manner and increased pAkt expression in vitro. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bryant, J.-P.; Levy, A.; Heiss, J.; Banasavadi-Siddegowda, Y.K. Review of PP2A Tumor Biology and Antitumor Effects of PP2A Inhibitor LB100 in the Nervous System. Cancers 2021, 13, 3087. https://doi.org/10.3390/cancers13123087
Bryant J-P, Levy A, Heiss J, Banasavadi-Siddegowda YK. Review of PP2A Tumor Biology and Antitumor Effects of PP2A Inhibitor LB100 in the Nervous System. Cancers. 2021; 13(12):3087. https://doi.org/10.3390/cancers13123087
Chicago/Turabian StyleBryant, Jean-Paul, Adam Levy, John Heiss, and Yeshavanth Kumar Banasavadi-Siddegowda. 2021. "Review of PP2A Tumor Biology and Antitumor Effects of PP2A Inhibitor LB100 in the Nervous System" Cancers 13, no. 12: 3087. https://doi.org/10.3390/cancers13123087
APA StyleBryant, J. -P., Levy, A., Heiss, J., & Banasavadi-Siddegowda, Y. K. (2021). Review of PP2A Tumor Biology and Antitumor Effects of PP2A Inhibitor LB100 in the Nervous System. Cancers, 13(12), 3087. https://doi.org/10.3390/cancers13123087