
Advances in Immunotherapy for Adult Glioblastoma

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
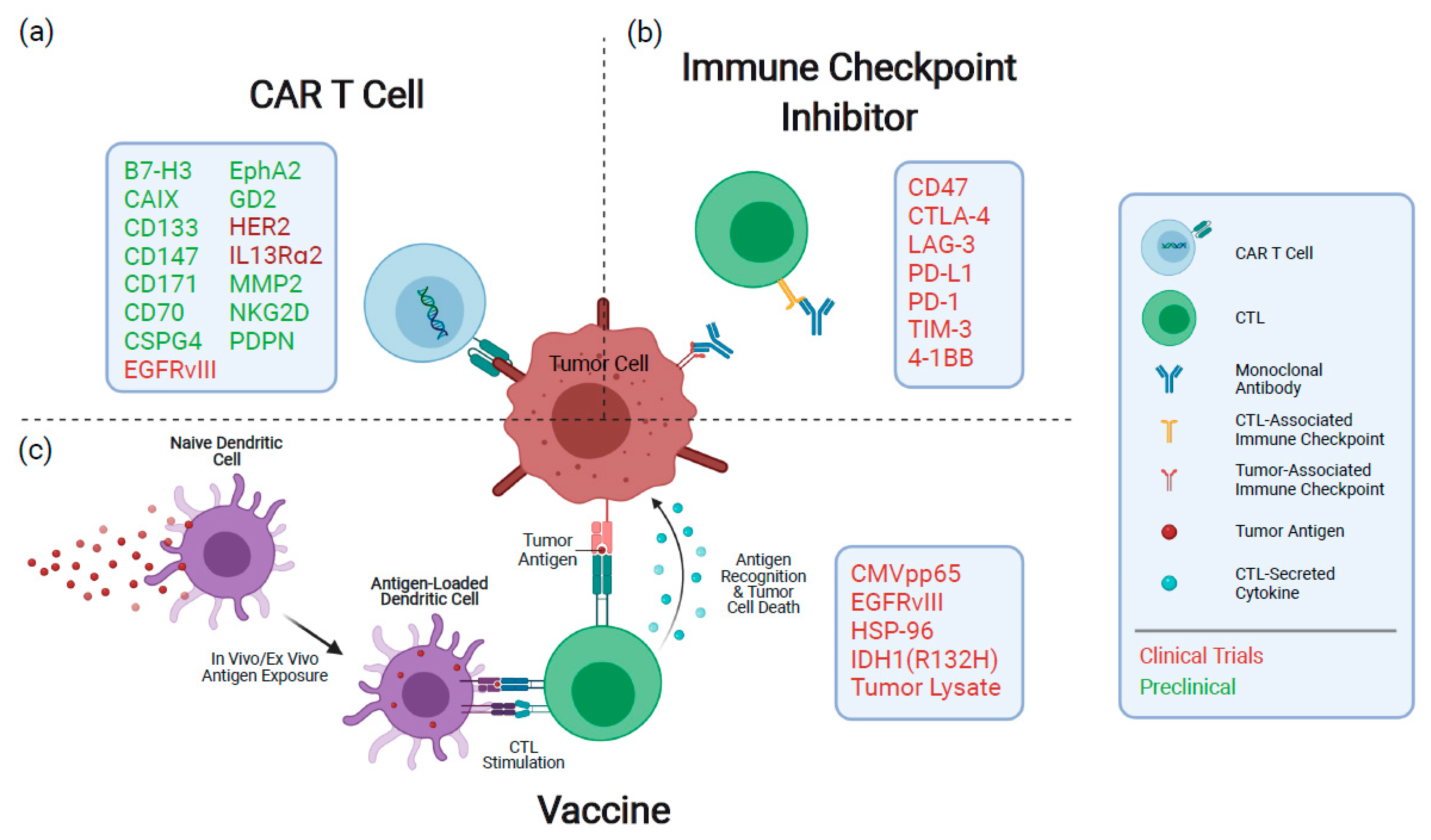
2. Vaccines
2.1. Single-Target Vaccines
2.2. Multi-Target Vaccines
3. Antibodies Modulating the Tumor Immune Microenvironment
3.1. Immune Checkpoint Inhibitors
3.2. Macrophage-Targeted Antibodies
4. Chimeric Antigen Receptor (CAR) T Cells
4.1. IL13Rα2-Specific CAR T Cells
4.2. EGFRvIII-Specific CAR T Cells
4.3. HER2-Specific CAR T Cells
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Xu, J.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009–2013. Neuro-oncology 2016, 18, v1–v75. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary brain tumours in adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Kim, J.; Lee, I.H.; Cho, H.J.; Park, C.K.; Jung, Y.S.; Kim, Y.; Nam, S.H.; Kim, B.S.; Johnson, M.D.; Kong, D.S.; et al. Spatiotemporal Evolution of the Primary Glioblastoma Genome. Cancer Cell 2015, 28, 318–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e821. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.J.; et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.; Reimand, J.; Lan, X.; Head, R.; Zhu, X.; Kushida, M.; Bayani, J.; Pressey, J.C.; Lionel, A.C.; Clarke, I.D.; et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc. Natl. Acad. Sci. USA 2015, 112, 851–856. [Google Scholar] [CrossRef] [Green Version]
- Rius-Rocabert, S.; Garcia-Romero, N.; Garcia, A.; Ayuso-Sacido, A.; Nistal-Villan, E. Oncolytic Virotherapy in Glioma Tumors. Int. J. Mol. Sci. 2020, 21, 7604. [Google Scholar] [CrossRef]
- Mitchell, D.A.; Xie, W.; Schmittling, R.; Learn, C.; Friedman, A.; McLendon, R.E.; Sampson, J.H. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro-oncology 2008, 10, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Bleeker, F.E.; Lamba, S.; Leenstra, S.; Troost, D.; Hulsebos, T.; Vandertop, W.P.; Frattini, M.; Molinari, F.; Knowles, M.; Cerrato, A.; et al. IDH1 mutations at residue p.R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum. Mutat. 2009, 30, 7–11. [Google Scholar] [CrossRef]
- Sampson, J.H.; Heimberger, A.B.; Archer, G.E.; Aldape, K.D.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E., 2nd; McLendon, R.E.; Mitchell, D.A.; et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2010, 28, 4722–4729. [Google Scholar] [CrossRef]
- Sampson, J.H.; Aldape, K.D.; Archer, G.E.; Coan, A.; Desjardins, A.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E.; McLendon, R.E.; et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro-oncology 2011, 13, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Schuster, J.; Lai, R.K.; Recht, L.D.; Reardon, D.A.; Paleologos, N.A.; Groves, M.D.; Mrugala, M.M.; Jensen, R.; Baehring, J.M.; Sloan, A.; et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: The ACT III study. Neuro-oncology 2015, 17, 854–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; O’Rourke, D.M.; Tran, D.D.; Fink, K.L.; Nabors, L.B.; Li, G.; Bota, D.A.; Lukas, R.V.; et al. Rindopepimut with Bevacizumab for Patients with Relapsed EGFRvIII-Expressing Glioblastoma (ReACT): Results of a Double-Blind Randomized Phase II Trial. Clin. Cancer Res. 2020, 26, 1586–1594. [Google Scholar] [CrossRef]
- Mitchell, D.A.; Batich, K.A.; Gunn, M.D.; Huang, M.N.; Sanchez-Perez, L.; Nair, S.K.; Congdon, K.L.; Reap, E.A.; Archer, G.E.; Desjardins, A.; et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015, 519, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Batich, K.A.; Reap, E.A.; Archer, G.E.; Sanchez-Perez, L.; Nair, S.K.; Schmittling, R.J.; Norberg, P.; Xie, W.; Herndon, J.E., 2nd; Healy, P.; et al. Long-term Survival in Glioblastoma with Cytomegalovirus pp65-Targeted Vaccination. Clin. Cancer Res. 2017, 23, 1898–1909. [Google Scholar] [CrossRef] [Green Version]
- Batich, K.A.; Mitchell, D.A.; Healy, P.; Herndon, J.E., 2nd; Sampson, J.H. Once, Twice, Three Times a Finding: Reproducibility of Dendritic Cell Vaccine Trials Targeting Cytomegalovirus in Glioblastoma. Clin. Cancer Res. 2020, 26, 5297–5303. [Google Scholar] [CrossRef]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crane, C.A.; Han, S.J.; Ahn, B.; Oehlke, J.; Kivett, V.; Fedoroff, A.; Butowski, N.; Chang, S.M.; Clarke, J.; Berger, M.S.; et al. Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin. Cancer Res. 2013, 19, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Bloch, O.; Crane, C.A.; Fuks, Y.; Kaur, R.; Aghi, M.K.; Berger, M.S.; Butowski, N.A.; Chang, S.M.; Clarke, J.L.; McDermott, M.W.; et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: A phase II, single-arm trial. Neuro-oncology 2014, 16, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Ji, N.; Zhang, Y.; Liu, Y.; Xie, J.; Wang, Y.; Hao, S.; Gao, Z. Heat shock protein peptide complex-96 vaccination for newly diagnosed glioblastoma: A phase I, single-arm trial. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Bloch, O.; Lim, M.; Sughrue, M.E.; Komotar, R.J.; Abrahams, J.M.; O’Rourke, D.M.; D’Ambrosio, A.; Bruce, J.N.; Parsa, A.T. Autologous Heat Shock Protein Peptide Vaccination for Newly Diagnosed Glioblastoma: Impact of Peripheral PD-L1 Expression on Response to Therapy. Clin. Cancer Res. 2017, 23, 3575–3584. [Google Scholar] [CrossRef] [Green Version]
- Heimberger, A.B.; Hlatky, R.; Suki, D.; Yang, D.; Weinberg, J.; Gilbert, M.; Sawaya, R.; Aldape, K. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 1462–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batra, S.K.; Castelino-Prabhu, S.; Wikstrand, C.J.; Zhu, X.; Humphrey, P.A.; Friedman, H.S.; Bigner, D.D. Epidermal growth factor ligand-independent, unregulated, cell-transforming potential of a naturally occurring human mutant EGFRvIII gene. Cell Growth Differ. 1995, 6, 1251–1259. [Google Scholar]
- Nagane, M.; Coufal, F.; Lin, H.; Bogler, O.; Cavenee, W.K.; Huang, H.J. A common mutant epidermal growth factor receptor confers enhanced tumorigenicity on human glioblastoma cells by increasing proliferation and reducing apoptosis. Cancer Res. 1996, 56, 5079–5086. [Google Scholar]
- Sampson, J.H.; Archer, G.E.; Mitchell, D.A.; Heimberger, A.B.; Herndon, J.E., 2nd; Lally-Goss, D.; McGehee-Norman, S.; Paolino, A.; Reardon, D.A.; Friedman, A.H.; et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol. Cancer Ther. 2009, 8, 2773–2779. [Google Scholar] [CrossRef] [Green Version]
- Heimberger, A.B.; Crotty, L.E.; Archer, G.E.; Hess, K.R.; Wikstrand, C.J.; Friedman, A.H.; Friedman, H.S.; Bigner, D.D.; Sampson, J.H. Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clin Cancer Res. 2003, 9, 4247–4254. [Google Scholar]
- Schafer, N.; Gielen, G.H.; Rauschenbach, L.; Kebir, S.; Till, A.; Reinartz, R.; Simon, M.; Niehusmann, P.; Kleinschnitz, C.; Herrlinger, U.; et al. Longitudinal heterogeneity in glioblastoma: Moving targets in recurrent versus primary tumors. J. Transl. Med. 2019, 17, 96. [Google Scholar] [CrossRef] [Green Version]
- Brock, C.S.; Newlands, E.S.; Wedge, S.R.; Bower, M.; Evans, H.; Colquhoun, I.; Roddie, M.; Glaser, M.; Brampton, M.H.; Rustin, G.J. Phase I trial of temozolomide using an extended continuous oral schedule. Cancer Res. 1998, 58, 4363–4367. [Google Scholar] [PubMed]
- Mansfield, A.S.; Nevala, W.K.; Lieser, E.A.; Leontovich, A.A.; Markovic, S.N. The immunomodulatory effects of bevacizumab on systemic immunity in patients with metastatic melanoma. Oncoimmunology 2013, 2, e24436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wills, M.R.; Carmichael, A.J.; Mynard, K.; Jin, X.; Weekes, M.P.; Plachter, B.; Sissons, J.G. The human cytotoxic T-lymphocyte (CTL) response to cytomegalovirus is dominated by structural protein pp65: Frequency, specificity, and T-cell receptor usage of pp65-specific CTL. J. Virol. 1996, 70, 7569–7579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arruda, L.B.; Sim, D.; Chikhlikar, P.R.; Maciel, M., Jr.; Akasaki, K.; August, J.T.; Marques, E.T. Dendritic cell-lysosomal-associated membrane protein (LAMP) and LAMP-1-HIV-1 gag chimeras have distinct cellular trafficking pathways and prime T and B cell responses to a diverse repertoire of epitopes. J. Immunol. 2006, 177, 2265–2275. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, D.A.; Cui, X.; Schmittling, R.J.; Sanchez-Perez, L.; Snyder, D.J.; Congdon, K.L.; Archer, G.E.; Desjardins, A.; Friedman, A.H.; Friedman, H.S.; et al. Monoclonal antibody blockade of IL-2 receptor alpha during lymphopenia selectively depletes regulatory T cells in mice and humans. Blood 2011, 118, 3003–3012. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, T.; Bunse, L.; Pusch, S.; Sahm, F.; Wiestler, B.; Quandt, J.; Menn, O.; Osswald, M.; Oezen, I.; Ott, M.; et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014, 512, 324–327. [Google Scholar] [CrossRef]
- Blass, E.; Ott, P.A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 2021, 18, 215–229. [Google Scholar] [CrossRef]
- Liau, L.M.; Black, K.L.; Prins, R.M.; Sykes, S.N.; DiPatre, P.L.; Cloughesy, T.F.; Becker, D.P.; Bronstein, J.M. Treatment of intracranial gliomas with bone marrow-derived dendritic cells pulsed with tumor antigens. J. Neurosurg. 1999, 90, 1115–1124. [Google Scholar] [CrossRef]
- Mitsuya, K.; Akiyama, Y.; Iizuka, A.; Miyata, H.; Deguchi, S.; Hayashi, N.; Maeda, C.; Kondou, R.; Kanematsu, A.; Watanabe, K.; et al. Alpha-type-1 Polarized Dendritic Cell-based Vaccination in Newly Diagnosed High-grade Glioma: A Phase II Clinical Trial. Anticancer. Res. 2020, 40, 6473–6484. [Google Scholar] [CrossRef]
- Akiyama, Y.; Oshita, C.; Kume, A.; Iizuka, A.; Miyata, H.; Komiyama, M.; Ashizawa, T.; Yagoto, M.; Abe, Y.; Mitsuya, K.; et al. alpha-type-1 polarized dendritic cell-based vaccination in recurrent high-grade glioma: A phase I clinical trial. BMC Cancer 2012, 12, 623. [Google Scholar] [CrossRef] [Green Version]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [Green Version]
- Davidson, T.B.; Lee, A.; Hsu, M.; Sedighim, S.; Orpilla, J.; Treger, J.; Mastall, M.; Roesch, S.; Rapp, C.; Galvez, M.; et al. Expression of PD-1 by T Cells in Malignant Glioma Patients Reflects Exhaustion and Activation. Clin. Cancer Res. 2019, 25, 1913–1922. [Google Scholar] [CrossRef]
- Contardi, E.; Palmisano, G.L.; Tazzari, P.L.; Martelli, A.M.; Fala, F.; Fabbi, M.; Kato, T.; Lucarelli, E.; Donati, D.; Polito, L.; et al. CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. Int. J. Cancer 2005, 117, 538–550. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bahr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Bristol Myers Squibb Announces Update on Phase 3 CheckMate -548 Trial Evaluating Patients with Newly Diagnosed MGMT-Methylated Glioblastoma Multiforme. Available online: https://news.bms.com/news/corporate-financial/2020/Bristol-Myers-Squibb-Announces-Update-on-Phase-3-CheckMate--548-Trial-Evaluating-Patients-with-Newly-Diagnosed-MGMT-Methylated-Glioblastoma-Multiforme/default.aspx (accessed on 2 June 2021).
- Sahebjam, S.; Forsyth, P.A.; Tran, N.D.; Arrington, J.A.; Macaulay, R.; Etame, A.B.; Walko, C.M.; Boyle, T.; Peguero, E.N.; Jaglal, M.; et al. Hypofractionated stereotactic re-irradiation with pembrolizumab and bevacizumab in patients with recurrent high-grade gliomas: Results from a phase I study. Neuro-oncology 2021, 23, 677–686. [Google Scholar] [CrossRef]
- Reardon, D.A. Phase II study of pembrolizumab or pembrolizumab plus bevacizumab for recurrent glioblastoma (rGBM) patients. J. Clin. Oncol. 2006, 36. [Google Scholar] [CrossRef]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; Lopez-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inoges, S.; de Andrea, C.; Lopez-Diaz de Cerio, A.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef]
- Reardon, D.A. Atim-12. Phase 2 study to evaluate the clinical efficacy and safety of medi4736 (durvalumab [dur]) in patients with bevacizumab (bev)-refractory recurrent glioblastoma (GBM). Neuro-oncology 2017, 19. [Google Scholar] [CrossRef]
- Lim, M.; Ye, X.; Piotrowski, A.F.; Desai, A.S.; Ahluwalia, M.S.; Walbert, T.; Fisher, J.D.; Desideri, S.; Belcaid, Z.; Jackson, C.; et al. Updated phase I trial of anti-LAG-3 or anti-CD137 alone and in combination with anti-PD-1 in patients with recurrent GBM. J. Clin. Oncol. 2019, 37, 2017. [Google Scholar] [CrossRef]
- Zeng, J.; See, A.P.; Phallen, J.; Jackson, C.M.; Belcaid, Z.; Ruzevick, J.; Durham, N.; Meyer, C.; Harris, T.J.; Albesiano, E.; et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neuro-oncology 2018, 20, 674–686. [Google Scholar] [CrossRef]
- Mathios, D.; Kim, J.E.; Mangraviti, A.; Phallen, J.; Park, C.K.; Jackson, C.M.; Garzon-Muvdi, T.; Kim, E.; Theodros, D.; Polanczyk, M.; et al. Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci. Transl. Med. 2016, 8, 370ra180. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Blake, S.J.; Yong, M.C.; Harjunpaa, H.; Ngiow, S.F.; Takeda, K.; Young, A.; O’Donnell, J.S.; Allen, S.; Smyth, M.J.; et al. Improved Efficacy of Neoadjuvant Compared to Adjuvant Immunotherapy to Eradicate Metastatic Disease. Cancer Discov. 2016, 6, 1382–1399. [Google Scholar] [CrossRef] [Green Version]
- Blank, C.U.; Rozeman, E.A.; Fanchi, L.F.; Sikorska, K.; van de Wiel, B.; Kvistborg, P.; Krijgsman, O.; van den Braber, M.; Philips, D.; Broeks, A.; et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat. Med. 2018, 24, 1655–1661. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Fecci, P.E.; Ochiai, H.; Mitchell, D.A.; Grossi, P.M.; Sweeney, A.E.; Archer, G.E.; Cummings, T.; Allison, J.P.; Bigner, D.D.; Sampson, J.H. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin. Cancer Res. 2007, 13, 2158–2167. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.A.; Gokhale, P.C.; Klein, S.R.; Ligon, K.L.; Rodig, S.J.; Ramkissoon, S.H.; Jones, K.L.; Conway, A.S.; Liao, X.; Zhou, J.; et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol. Res. 2016, 4, 124–135. [Google Scholar] [CrossRef] [Green Version]
- Harris-Bookman, S.; Mathios, D.; Martin, A.M.; Xia, Y.; Kim, E.; Xu, H.; Belcaid, Z.; Polanczyk, M.; Barberi, T.; Theodros, D.; et al. Expression of LAG-3 and efficacy of combination treatment with anti-LAG-3 and anti-PD-1 monoclonal antibodies in glioblastoma. Int. J. Cancer 2018, 143, 3201–3208. [Google Scholar] [CrossRef] [Green Version]
- Mair, M.J.; Kiesel, B.; Feldmann, K.; Widhalm, G.; Dieckmann, K.; Wohrer, A.; Mullauer, L.; Preusser, M.; Berghoff, A.S. LAG-3 expression in the inflammatory microenvironment of glioma. J. Neurooncol. 2021, 152, 533–539. [Google Scholar] [CrossRef]
- Kuhnol, C.; Herbarth, M.; Foll, J.; Staege, M.S.; Kramm, C. CD137 stimulation and p38 MAPK inhibition improve reactivity in an in vitro model of glioblastoma immunotherapy. Cancer Immunol. Immunother. 2013, 62, 1797–1809. [Google Scholar] [CrossRef]
- Newcomb, E.W.; Lukyanov, Y.; Kawashima, N.; Alonso-Basanta, M.; Wang, S.C.; Liu, M.; Jure-Kunkel, M.; Zagzag, D.; Demaria, S.; Formenti, S.C. Radiotherapy enhances antitumor effect of anti-CD137 therapy in a mouse Glioma model. Radiat. Res. 2010, 173, 426–432. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Sai, K.; Wang, X.L.; Ye, S.Q.; Liang, L.J.; Zhou, Y.; Chen, Z.J.; Hu, W.M.; Liu, J.M. Tim-3 Expression and MGMT Methylation Status Association With Survival in Glioblastoma. Front. Pharmacol. 2020, 11, 584652. [Google Scholar] [CrossRef]
- Kim, J.E.; Patel, M.A.; Mangraviti, A.; Kim, E.S.; Theodros, D.; Velarde, E.; Liu, A.; Sankey, E.W.; Tam, A.; Xu, H.; et al. Combination Therapy with Anti-PD-1, Anti-TIM-3, and Focal Radiation Results in Regression of Murine Gliomas. Clin. Cancer Res. 2017, 23, 124–136. [Google Scholar] [CrossRef] [Green Version]
- Goswami, S.; Walle, T.; Cornish, A.E.; Basu, S.; Anandhan, S.; Fernandez, I.; Vence, L.; Blando, J.; Zhao, H.; Yadav, S.S.; et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat. Med. 2020, 26, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Hutter, G.; Kahn, S.A.; Azad, T.D.; Gholamin, S.; Xu, C.Y.; Liu, J.; Achrol, A.S.; Richard, C.; Sommerkamp, P.; et al. Anti-CD47 Treatment Stimulates Phagocytosis of Glioblastoma by M1 and M2 Polarized Macrophages and Promotes M1 Polarized Macrophages In Vivo. PLoS ONE 2016, 11, e0153550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fan, J.; Wang, S.; Li, Y.; Wang, Y.; Li, S.; Luan, J.; Wang, Z.; Song, P.; Chen, Q.; et al. Targeting CD47 and Autophagy Elicited Enhanced Antitumor Effects in Non-Small Cell Lung Cancer. Cancer Immunol. Res. 2017, 5, 363–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edris, B.; Weiskopf, K.; Volkmer, A.K.; Volkmer, J.P.; Willingham, S.B.; Contreras-Trujillo, H.; Liu, J.; Majeti, R.; West, R.B.; Fletcher, J.A.; et al. Antibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc. Natl. Acad. Sci. USA 2012, 109, 6656–6661. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Lv, B.; Liu, Y.; Hua, T.; Han, J.; Sun, C.; Xu, L.; Zhang, Z.; Feng, Z.; Cai, Y.; et al. Blocking the CD47-SIRPalpha axis by delivery of anti-CD47 antibody induces antitumor effects in glioma and glioma stem cells. Oncoimmunology 2018, 7, e1391973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gholamin, S.; Youssef, O.A.; Rafat, M.; Esparza, R.; Kahn, S.; Shahin, M.; Giaccia, A.J.; Graves, E.E.; Weissman, I.; Mitra, S.; et al. Irradiation or temozolomide chemotherapy enhances anti-CD47 treatment of glioblastoma. Innate. Immun. 2020, 26, 130–137. [Google Scholar] [CrossRef] [PubMed]
- von Roemeling, C.A.; Wang, Y.; Qie, Y.; Yuan, H.; Zhao, H.; Liu, X.; Yang, Z.; Yang, M.; Deng, W.; Bruno, K.A.; et al. Therapeutic modulation of phagocytosis in glioblastoma can activate both innate and adaptive antitumour immunity. Nat. Commun. 2020, 11, 1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Chen, W.; Fan, J.; Wang, S.; Xian, Z.; Luan, J.; Li, Y.; Wang, Y.; Nan, Y.; Luo, M.; et al. Disrupting CD47-SIRPalpha axis alone or combined with autophagy depletion for the therapy of glioblastoma. Carcinogenesis 2018, 39, 689–699. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, S.; Nan, Y.; Fan, J.; Chen, W.; Luan, J.; Wang, Y.; Liang, Y.; Li, S.; Tian, W.; et al. Inhibition of autophagy potentiated the anti-tumor effects of VEGF and CD47 bispecific therapy in glioblastoma. Appl. Microbiol. Biotechnol. 2018, 102, 6503–6513. [Google Scholar] [CrossRef]
- Sockolosky, J.T.; Dougan, M.; Ingram, J.R.; Ho, C.C.; Kauke, M.J.; Almo, S.C.; Ploegh, H.L.; Garcia, K.C. Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proc. Natl. Acad. Sci. USA 2016, 113, E2646–E2654. [Google Scholar] [CrossRef] [Green Version]
- Sadelain, M.; Brentjens, R.; Riviere, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Finney, H.M.; Lawson, A.D.; Bebbington, C.R.; Weir, A.N. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J. Immunol. 1998, 161, 2791–2797. [Google Scholar] [PubMed]
- Finney, H.M.; Akbar, A.N.; Lawson, A.D. Activation of resting human primary T cells with chimeric receptors: Costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J. Immunol. 2004, 172, 104–113. [Google Scholar] [CrossRef]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef] [Green Version]
- Hombach, A.; Wieczarkowiecz, A.; Marquardt, T.; Heuser, C.; Usai, L.; Pohl, C.; Seliger, B.; Abken, H. Tumor-specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J. Immunol. 2001, 167, 6123–6131. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Bagley, S.J.; Desai, A.S.; Linette, G.P.; June, C.H.; O’Rourke, D.M. CAR T-cell therapy for glioblastoma: Recent clinical advances and future challenges. Neuro-oncology 2018, 20, 1429–1438. [Google Scholar] [CrossRef] [Green Version]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-oncology 2018, 20, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Watkins, S.; Robel, S.; Kimbrough, I.F.; Robert, S.M.; Ellis-Davies, G.; Sontheimer, H. Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nat. Commun. 2014, 5, 4196. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N.; Salsman, V.S.; Kew, Y.; Shaffer, D.; Powell, S.; Zhang, Y.J.; Grossman, R.G.; Heslop, H.E.; Gottschalk, S. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 474–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keu, K.V.; Witney, T.H.; Yaghoubi, S.; Rosenberg, J.; Kurien, A.; Magnusson, R.; Williams, J.; Habte, F.; Wagner, J.R.; Forman, S.; et al. Reporter gene imaging of targeted T cell immunotherapy in recurrent glioma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Starr, R.; Chang, W.C.; Aguilar, B.; Alizadeh, D.; Wright, S.L.; Yang, X.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Tang, X.; Zhao, S.; Zhang, Y.; Wang, Y.; Zhang, Z.; Yang, M.; Zhu, Y.; Zhang, G.; Guo, G.; Tong, A.; et al. B7-H3 as a Novel CAR-T Therapeutic Target for Glioblastoma. Mol. Ther. Oncolytics 2019, 14, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Wang, Y.; Huang, J.; Zhang, Z.; Liu, F.; Xu, J.; Guo, G.; Wang, W.; Tong, A.; Zhou, L. Administration of B7-H3 targeted chimeric antigen receptor-T cells induce regression of glioblastoma. Signal Transduct. Target. Ther. 2021, 6, 125. [Google Scholar] [CrossRef]
- Nehama, D.; Di Ianni, N.; Musio, S.; Du, H.; Patane, M.; Pollo, B.; Finocchiaro, G.; Park, J.J.H.; Dunn, D.E.; Edwards, D.S.; et al. B7-H3-redirected chimeric antigen receptor T cells target glioblastoma and neurospheres. EBioMedicine 2019, 47, 33–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Li, X.; Zhang, J.; Mao, L. Novel cellular immunotherapy using NKG2D CAR-T for the treatment of cervical cancer. Biomed. Pharmacother. Biomed. Pharmacother. 2020, 131, 110562. [Google Scholar] [CrossRef]
- Yang, D.; Sun, B.; Dai, H.; Li, W.; Shi, L.; Zhang, P.; Li, S.; Zhao, X. T cells expressing NKG2D chimeric antigen receptors efficiently eliminate glioblastoma and cancer stem cells. J. Immunother. Cancer 2019, 7, 171. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.C.; Lu, L.; Zheng, Z.; et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-transduced T Cells Targeting EGFRvIII in Patients With Glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef]
- Brown, C.E.; Warden, C.D.; Starr, R.; Deng, X.; Badie, B.; Yuan, Y.C.; Forman, S.J.; Barish, M.E. Glioma IL13Ralpha2 is associated with mesenchymal signature gene expression and poor patient prognosis. PLoS ONE 2013, 8, e77769. [Google Scholar] [CrossRef]
- Debinski, W.; Gibo, D.M.; Hulet, S.W.; Connor, J.R.; Gillespie, G.Y. Receptor for interleukin 13 is a marker and therapeutic target for human high-grade gliomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1999, 5, 985–990. [Google Scholar]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; de Carvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2018, 33, 152. [Google Scholar] [CrossRef] [Green Version]
- Kong, S.; Sengupta, S.; Tyler, B.; Bais, A.J.; Ma, Q.; Doucette, S.; Zhou, J.; Sahin, A.; Carter, B.S.; Brem, H.; et al. Suppression of human glioma xenografts with second-generation IL13R-specific chimeric antigen receptor-modified T cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 5949–5960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.E.; Aguilar, B.; Starr, R.; Yang, X.; Chang, W.C.; Weng, L.; Chang, B.; Sarkissian, A.; Brito, A.; Sanchez, J.F.; et al. Optimization of IL13Ralpha2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 31–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krenciute, G.; Krebs, S.; Torres, D.; Wu, M.F.; Liu, H.; Dotti, G.; Li, X.N.; Lesniak, M.S.; Balyasnikova, I.V.; Gottschalk, S. Characterization and Functional Analysis of scFv-based Chimeric Antigen Receptors to Redirect T Cells to IL13Ralpha2-positive Glioma. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Krenciute, G.; Prinzing, B.L.; Yi, Z.; Wu, M.F.; Liu, H.; Dotti, G.; Balyasnikova, I.V.; Gottschalk, S. Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Ralpha2-CAR T Cells but Results in Antigen Loss Variants. Cancer Immunol. Res. 2017, 5, 571–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pituch, K.C.; Miska, J.; Krenciute, G.; Panek, W.K.; Li, G.; Rodriguez-Cruz, T.; Wu, M.; Han, Y.; Lesniak, M.S.; Gottschalk, S.; et al. Adoptive Transfer of IL13Ralpha2-Specific Chimeric Antigen Receptor T Cells Creates a Pro-inflammatory Environment in Glioblastoma. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 986–995. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Aguilar, B.; Starr, R.; Alizadeh, D.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; Forman, S.J.; Brown, C.E. Glioblastoma-targeted CD4+ CAR T cells mediate superior antitumor activity. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padfield, E.; Ellis, H.P.; Kurian, K.M. Current Therapeutic Advances Targeting EGFR and EGFRvIII in Glioblastoma. Front. Oncol. 2015, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.A.; Scholler, J.; Ohkuri, T.; Kosaka, A.; Patel, P.R.; McGettigan, S.E.; Nace, A.K.; Dentchev, T.; Thekkat, P.; Loew, A.; et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med. 2015, 7, 275ra222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, M.; Ohkuri, T.; Kosaka, A.; Tanahashi, K.; June, C.H.; Natsume, A.; Okada, H. Expression of miR-17-92 enhances anti-tumor activity of T-cells transduced with the anti-EGFRvIII chimeric antigen receptor in mice bearing human GBM xenografts. J. Immunother. Cancer 2013, 1, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, B.D.; Suryadevara, C.M.; Gedeon, P.C.; Herndon, J.E., 2nd; Sanchez-Perez, L.; Bigner, D.D.; Sampson, J.H. Intracerebral delivery of a third generation EGFRvIII-specific chimeric antigen receptor is efficacious against human glioma. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2014, 21, 189–190. [Google Scholar] [CrossRef] [Green Version]
- Sampson, J.H.; Choi, B.D.; Sanchez-Perez, L.; Suryadevara, C.M.; Snyder, D.J.; Flores, C.T.; Schmittling, R.J.; Nair, S.K.; Reap, E.A.; Norberg, P.K.; et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 972–984. [Google Scholar] [CrossRef] [Green Version]
- Miao, H.; Choi, B.D.; Suryadevara, C.M.; Sanchez-Perez, L.; Yang, S.; De Leon, G.; Sayour, E.J.; McLendon, R.; Herndon, J.E., 2nd; Healy, P.; et al. EGFRvIII-specific chimeric antigen receptor T cells migrate to and kill tumor deposits infiltrating the brain parenchyma in an invasive xenograft model of glioblastoma. PLoS ONE 2014, 9, e94281. [Google Scholar] [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Pyo, K.H.; Soo, R.A.; Cho, B.C. The promise of bispecific antibodies: Clinical applications and challenges. Cancer Treat. Rev. 2021, 99, 102240. [Google Scholar] [CrossRef] [PubMed]
- Choe, J.H.; Watchmaker, P.B.; Simic, M.S.; Gilbert, R.D.; Li, A.W.; Krasnow, N.A.; Downey, K.M.; Yu, W.; Carrera, D.A.; Celli, A.; et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Haynik, D.M.; Roma, A.A.; Prayson, R.A. HER-2/neu expression in glioblastoma multiforme. Appl. Immunohistochem. Mol. Morphol. 2007, 15, 56–58. [Google Scholar] [CrossRef]
- Koka, V.; Potti, A.; Forseen, S.E.; Pervez, H.; Fraiman, G.N.; Koch, M.; Levitt, R. Role of Her-2/neu overexpression and clinical determinants of early mortality in glioblastoma multiforme. Am. J. Clin. Oncol. 2003, 26, 332–335. [Google Scholar] [CrossRef]
- Jackson, C.M.; Choi, J.; Lim, M. Mechanisms of immunotherapy resistance: Lessons from glioblastoma. Nat. Immunol. 2019, 20, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, J.; Guo, G.; Yu, J. Immunotherapy for recurrent glioblastoma: Practical insights and challenging prospects. Cell Death Dis. 2021, 12, 299. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.W.; Quail, D.F. Immunotherapy for Glioblastoma: Current Progress and Challenge. Front. Immunol. 2021, 12, 676301. [Google Scholar] [CrossRef] [PubMed]
- Hodges, T.R.; Ott, M.; Xiu, J.; Gatalica, Z.; Swensen, J.; Zhou, S.; Huse, J.T.; de Groot, J.; Li, S.; Overwijk, W.W.; et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: Implications for immune checkpoint immunotherapy. Neuro-oncology 2017, 19, 1047–1057. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cockle, J.V.; Rajani, K.; Zaidi, S.; Kottke, T.; Thompson, J.; Diaz, R.M.; Shim, K.; Peterson, T.; Parney, I.F.; Short, S.; et al. Combination viroimmunotherapy with checkpoint inhibition to treat glioma, based on location-specific tumor profiling. Neuro-oncology 2016, 18, 518–527. [Google Scholar] [CrossRef]
- Jiang, H.; Rivera-Molina, Y.; Gomez-Manzano, C.; Clise-Dwyer, K.; Bover, L.; Vence, L.M.; Yuan, Y.; Lang, F.F.; Toniatti, C.; Hossain, M.B.; et al. Oncolytic Adenovirus and Tumor-Targeting Immune Modulatory Therapy Improve Autologous Cancer Vaccination. Cancer Res. 2017, 77, 3894–3907. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.Y.; Hutzen, B.; Wedekind, M.F.; Cripe, T.P. Oncolytic virus and PD-1/PD-L1 blockade combination therapy. Oncolytic Virother. 2018, 7, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Antonios, J.P.; Soto, H.; Everson, R.G.; Orpilla, J.; Moughon, D.; Shin, N.; Sedighim, S.; Yong, W.H.; Li, G.; Cloughesy, T.F.; et al. PD-1 blockade enhances the vaccination-induced immune response in glioma. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [Green Version]
- Murty, S.; Haile, S.T.; Beinat, C.; Aalipour, A.; Alam, I.S.; Murty, T.; Shaffer, T.M.; Patel, C.B.; Graves, E.E.; Mackall, C.L.; et al. Intravital imaging reveals synergistic effect of CAR T-cells and radiation therapy in a preclinical immunocompetent glioblastoma model. Oncoimmunology 2020, 9, 1757360. [Google Scholar] [CrossRef]
- Yang, M.; Tang, X.; Zhang, Z.; Gu, L.; Wei, H.; Zhao, S.; Zhong, K.; Mu, M.; Huang, C.; Jiang, C.; et al. Tandem CAR-T cells targeting CD70 and B7-H3 exhibit potent preclinical activity against multiple solid tumors. Theranostics 2020, 10, 7622–7634. [Google Scholar] [CrossRef]
- Jin, L.; Ge, H.; Long, Y.; Yang, C.; Chang, Y.E.; Mu, L.; Sayour, E.J.; De Leon, G.; Wang, Q.J.; Yang, J.C.; et al. CD70, a novel target of CAR T-cell therapy for gliomas. Neuro-oncology 2018, 20, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Vora, P.; Venugopal, C.; Salim, S.K.; Tatari, N.; Bakhshinyan, D.; Singh, M.; Seyfrid, M.; Upreti, D.; Rentas, S.; Wong, N.; et al. The Rational Development of CD133-Targeting Immunotherapies for Glioblastoma. Cell Stem Cell 2020, 26, 832–844.e836. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Zhang, Q.; Song, Q.; Wang, H.; Dmitriev, P.; Sun, M.Y.; Cao, X.; Wang, Y.; Guo, L.; Indig, I.H.; et al. Targeting hypoxia downstream signaling protein, CAIX, for CAR T-cell therapy against glioblastoma. Neuro-oncology 2019, 21, 1436–1446. [Google Scholar] [CrossRef]
- Yi, Z.; Prinzing, B.L.; Cao, F.; Gottschalk, S.; Krenciute, G. Optimizing EphA2-CAR T Cells for the Adoptive Immunotherapy of Glioma. Mol. Ther. Methods Clin. Dev. 2018, 9, 70–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, K.K.; Naik, S.; Kakarla, S.; Brawley, V.S.; Shaffer, D.R.; Yi, Z.; Rainusso, N.; Wu, M.F.; Liu, H.; Kew, Y.; et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 629–637. [Google Scholar] [CrossRef] [Green Version]
- Shiina, S.; Ohno, M.; Ohka, F.; Kuramitsu, S.; Yamamichi, A.; Kato, A.; Motomura, K.; Tanahashi, K.; Yamamoto, T.; Watanabe, R.; et al. CAR T Cells Targeting Podoplanin Reduce Orthotopic Glioblastomas in Mouse Brains. Cancer Immunol. Res. 2016, 4, 259–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beard, R.E.; Zheng, Z.; Lagisetty, K.H.; Burns, W.R.; Tran, E.; Hewitt, S.M.; Abate-Daga, D.; Rosati, S.F.; Fine, H.A.; Ferrone, S.; et al. Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J. Immunother. Cancer 2014, 2, 25. [Google Scholar] [CrossRef] [Green Version]
- Geldres, C.; Savoldo, B.; Hoyos, V.; Caruana, I.; Zhang, M.; Yvon, E.; Del Vecchio, M.; Creighton, C.J.; Ittmann, M.; Ferrone, S.; et al. T lymphocytes redirected against the chondroitin sulfate proteoglycan-4 control the growth of multiple solid tumors both in vitro and in vivo. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 962–971. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.; Stastny, M.; Brown, C.; Chang, W.C.; Ostberg, J.R.; Forman, S.J.; Jensen, M.C. Diverse solid tumors expressing a restricted epitope of L1-CAM can be targeted by chimeric antigen receptor redirected T lymphocytes. J. Immunother. 2014, 37, 93–104. [Google Scholar] [CrossRef]
- Pasquale, E.B. Eph-ephrin bidirectional signaling in physiology and disease. Cell 2008, 133, 38–52. [Google Scholar] [CrossRef] [Green Version]
- Qazi, M.A.; Vora, P.; Venugopal, C.; Adams, J.; Singh, M.; Hu, A.; Gorelik, M.; Subapanditha, M.K.; Savage, N.; Yang, J.; et al. Cotargeting Ephrin Receptor Tyrosine Kinases A2 and A3 in Cancer Stem Cells Reduces Growth of Recurrent Glioblastoma. Cancer Res. 2018, 78, 5023–5037. [Google Scholar] [CrossRef] [Green Version]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.; et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.F.; Orange, J.S.; Sumazin, P.; Man, T.K.; et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro-oncology 2018, 20, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Shaim, H.; Shanley, M.; Basar, R.; Daher, M.; Gumin, J.; Zamler, D.B.; Uprety, N.; Wang, F.; Huang, Y.; Gabrusiewicz, K.; et al. Targeting the alphav integrin-TGF-beta axis improves natural killer cell function against glioblastoma stem cells. J. Clin. Investig. 2021. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| NCT Number | Treatment | Summary of Results | Indication | References |
|---|---|---|---|---|
| NCT00643097 | EGFRvIII peptide vaccine + DI-TMZ | EGFRvIII-expressing cells eradicated and vaccine immunogenic, with DI-TMZ cohort having enhanced humoral response. Median overall survival of 23.6 months. | Primary GBM | Sampson et al. [13] Sampson et al. [14] |
| NCT00458601 | EGFRvIII peptide vaccine + TMZ | Median overall survival of 21.8 months and 36-month survival of 26%. Anti-EGFRvIII antibodies increased ≥4-fold in 85% of patients with duration of treatment. | Primary GBM | Schuster et al. [15] |
| NCT01480479 | EGFRvIII peptide vaccine + TMZ | Strong humoral responses; however, no survival advantage and loss of EGFRvIII expression upon recurrence. | Primary GBM | Weller et al. [16] |
| NCT01498328 | EGFRvIII peptide vaccine + bevacizumab | 24-month survival of 20% compared to 3% for controls. | Recurrent GBM | Reardon et al. [17] |
| NCT00639639 | CMV pp65 DC vaccine + Td Toxoid + TMZ | Td toxoid pre-conditioning enhanced DC migration to the lymph nodes and improved survival. 3/6 Td toxoid patients were alive and progression-free at time of survival analysis (>36.6 months), while controls had median overall survival of 18.5 months. | Primary GBM | Mitchell et al. [18] |
| NCT00639639 | CMV pp65 DC vaccine + DI-TMZ | Antigen-specific immune responses and median overall survival of 41.1 months in DI-TMZ cohort. A total of 36% survival 5 years from diagnosis, with four patients remaining progression-free at 59–64 months from diagnosis. | Primary GBM | Batich et al. [19] |
| NCT02366728 | CMV pp65 DC vaccine + 111In-labeled DC vaccine + Td Toxoid + basiliximab | Ongoing, have reported increased DC migration to lymph nodes following Td toxoid pre-conditioning. | Primary GBM | Batich et al. [20] |
| NCT02454634 | IDH1 peptide vaccine | A total of 93% vaccine-specific response rate, 84% survival >3 years. | High-grade glioma | Platten et al. [21] |
| NCT00045968 | DCVax-L vaccine | Median overall survival of 23.1 months, with large group (n = 100) reaching 40.5 months. | Primary GBM | Liau et al. [22] |
| NCT00293423 | HSPPC-96 peptide vaccine | Specific immune response in 11 of the 12 patients, responders had median overall survival of 11.8 months. | Recurrent GBM | Crane et al. [23] Bloch et al. [24] |
| NCT02122822 | HSPPC-96 peptide vaccine + TMZ + radiotherapy | Median overall survival of 31.4 months. Patients with high tumor-specific immune responses had median overall survival of >40.5 months compared to 14.6 months for low responders. | Primary GBM | Ji et al. [25] |
| NCT00905060 | HSPPC-96 vaccine + TMZ | Median overall survival of 23.8 months. Patients with low PD-L1 expression in myeloid cells had median overall survival of 44.7 months compared to 18 months for those with high expression. | Primary GBM | Bloch et al. [26] |
| NCT Number | Treatment | Summary of Results | Indication | References |
|---|---|---|---|---|
| NCT02017717 | Nivolumab (anti-PD-1) or bevacizumab | Median overall survival was around 10 months for both groups; 12-month survival rates were identical between treatments at 42%. | Recurrent GBM | Reardon et al. [46] |
| NCT02617589 | Nivolumab + radiotherapy or TMZ + radiotherapy | No survival advantage over TMZ, median overall survival of 13.4 months for nivolumab cohort and 14.88 months for TMZ. | Primary GBM | No Reference |
| NCT02667587 | Nivolumab + TMZ + radiotherapy | Nivolumab provided no survival advantage over placebo, trial still ongoing. | Primary GBM | Squibb et al. [47] |
| NCT02313272 | Hypofractionated stereotactic irradiation + pembrolizumab (anti-PD-1) + bevacizumab | >50% patients had significant response; median overall survival of 13.5 months. | Recurrent high-grade glioma | Sahebjam et al. [48] |
| NCT02337491 | Pembrolizumab or pembrolizumab + bevacizumab | Median overall survival of 8.8 months for pembrolizumab with bevacizumab, 10.3 months for pembrolizumab alone. | Recurrent GBM | Reardon et al. [49] |
| NCT02550249 | Neoadjuvant nivolumab | Neoadjuvant nivolumab enhanced chemokine expression, TCR clonal diversity among TILs and immune cell infiltration of the tumor; however, median overall survival was only 7.3 months. | GBM | Schalper et al. [50] |
| NCT02336165 | Durvalumab (anti-PD-L1) alone, with bevacizumab or with radiotherapy | Preliminary results of recurrent, bevacizumab-refractory cohort had 36% survival at 5.5 months. Trial still ongoing. | GBM | Reardon et al. [51] |
| NCT02658981 | Anti-LAG-3 or anti-4-1BB alone or with anti-PD-1 | Median overall survival of 8 months for anti-LAG-3, 7 months for anti-LAG-3, anti-PD-1 combination and 14 months for anti-4-1BB. Trial still ongoing. | Recurrent GBM | Lim et al. [52] |
| NCT Number | Treatment | Summary of Results | Indication | References |
|---|---|---|---|---|
| NCT00730613 | IL13(E13Y)-CD3ζ CAR T cells (first generation) | Transient inflammation at tumor site and a significant decrease in IL13Rα2 expression post-treatment were observed. Two grade 3 adverse events were observed. A median survival of 11 months after tumor relapse was noted. | Recurrent GBM | Brown et al. [90] |
| NCT02208362 | IL13(E13Y)-41BBζ CAR T cells (second generation) | A single patient with multifocal relapsed GBM was treated, resulting in 77–100% decrease in tumor burden and 7.5 months of progression-free survival. Increased presence of inflammatory cytokines at tumor site with no adverse events related to CAR T cell therapy. | Recurrent GBM | Brown et al. [91] |
| NCT01109095 | HER2-CD28ζ CAR T cells (second generation) | No dose-limiting toxicity was observed and CAR T cells persisted for 12 months post-infusion. No significant increase in survival was noted, with a median overall survival of 11.1 months. | GBM | Ahmed et al. [98] |
| NCT02209376 | EGFRvIII-41BBζ CAR T cells (second generation) | No dose-limiting toxicity was observed and EGFRvIII expression was reduced post-treatment. No significant increase in survival was noted, with a median overall survival of 8 months post-treatment. | Recurrent GBM | O’Rourke et al. [87] |
| NCT01454596 | EGFRvIII-CD28-41BBζ CAR T cells (third generation) | At highest dose, 2 patients suffered dose-limiting toxicity. A median overall survival of 6.9 months was noted, with one patient alive at 59 months. | Recurrent GBM | Goff et al. [99] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chokshi, C.R.; Brakel, B.A.; Tatari, N.; Savage, N.; Salim, S.K.; Venugopal, C.; Singh, S.K. Advances in Immunotherapy for Adult Glioblastoma. Cancers 2021, 13, 3400. https://doi.org/10.3390/cancers13143400
Chokshi CR, Brakel BA, Tatari N, Savage N, Salim SK, Venugopal C, Singh SK. Advances in Immunotherapy for Adult Glioblastoma. Cancers. 2021; 13(14):3400. https://doi.org/10.3390/cancers13143400
Chicago/Turabian StyleChokshi, Chirayu R., Benjamin A. Brakel, Nazanin Tatari, Neil Savage, Sabra K. Salim, Chitra Venugopal, and Sheila K. Singh. 2021. "Advances in Immunotherapy for Adult Glioblastoma" Cancers 13, no. 14: 3400. https://doi.org/10.3390/cancers13143400
APA StyleChokshi, C. R., Brakel, B. A., Tatari, N., Savage, N., Salim, S. K., Venugopal, C., & Singh, S. K. (2021). Advances in Immunotherapy for Adult Glioblastoma. Cancers, 13(14), 3400. https://doi.org/10.3390/cancers13143400