
Targeting PI3K/AKT/mTOR Signaling Pathway in Breast Cancer



{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Genetic Alterations of the PI3K Pathway in Breast Cancer and Clinical Implications
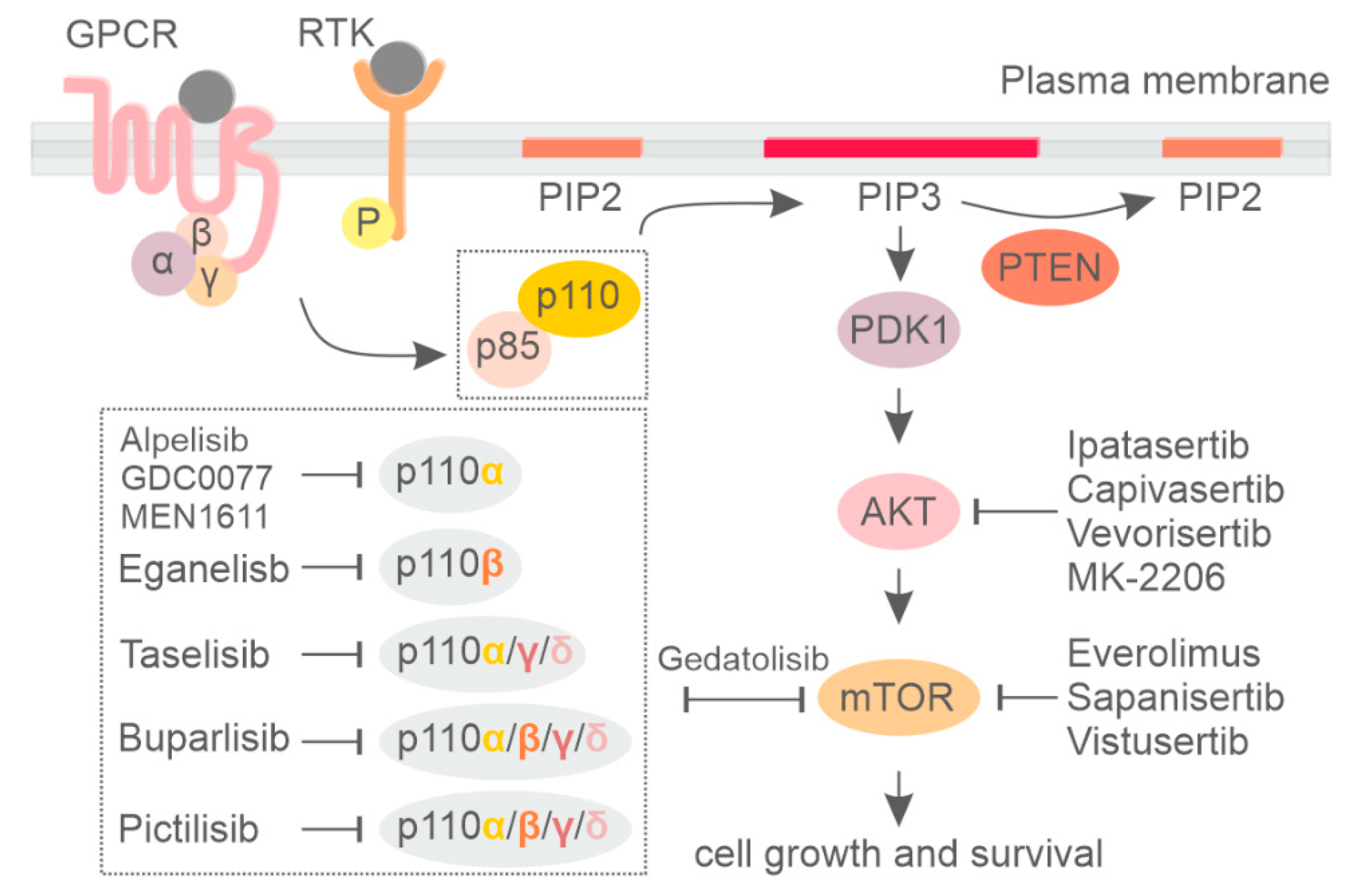
3. Clinical Usage of Pan-PI3K Inhibitors
- Buparlisib
- Pictilisib
4. PI3K Isoform-Specific Inhibitors
- Alpelisib
- Taselisib
5. PI3K Pathway Inhibition in HER2+ and Triple-Negative Breast Cancer Subtypes
- HER2-Positive Breast Cancer
- Triple-Negative Breast Cancer (TNBC)
6. Currently Available Inhibitors Acting on AKT and mTOR in Breast Cancer
- AKT Inhibitors
- mTOR Inhibitors
- Dual PI3K/mTOR Inhibitors
7. Rationale for Targeting Class II PI3K in Breast Cancer
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bilanges, B.; Posor, Y.; Vanhaesebroeck, B. PI3K isoforms in cell signalling and vesicle trafficking. Nat. Rev. Mol. Cell Biol. 2019, 20, 515–534. [Google Scholar] [CrossRef] [PubMed]
- Gulluni, F.; De Santis, M.C.; Margaria, J.P.; Martini, M.; Hirsch, E. Class II PI3K Functions in Cell Biology and Disease. Trends Cell Biol. 2019, 29, 339–359. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.; Gulluni, F.; Martini, M. Phosphoinositides in cell proliferation and metabolism. Adv. Biol. Regul. 2020, 75, 100693. [Google Scholar] [CrossRef]
- Maehama, T.; Dixon, J.E. The Tumor Suppressor, PTEN/MMAC1, Dephosphorylates the Lipid Second Messenger, Phosphatidylinositol 3,4,5-Trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geering, B.; Cutillas, P.R.; Nock, G.; Gharbi, S.I.; Vanhaesebroeck, B. Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc. Natl. Acad. Sci. USA 2007, 104, 7809–7814. [Google Scholar] [CrossRef] [Green Version]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Kok, K.; Geering, B.; Vanhaesebroeck, B. Regulation of phosphoinositide 3-kinase expression in health and disease. Trends Biochem. Sci. 2009, 34, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Kok, K.; Nock, G.E.; Verrall, E.A.G.; Mitchell, M.P.; Hommes, D.W.; Peppelenbosch, M.; Vanhaesebroeck, B. Regulation of p110δ PI 3-Kinase Gene Expression. PLoS ONE 2009, 4, e5145. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Velculescu, V. Oncogenic Mutations of PIK3CA in Human Cancers. Cell Cycle 2004, 3, 1221–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindhurst, M.J.; Parker, V.E.R.; Payne, F.; Sapp, J.; Rudge, S.; Harris, J.; Witkowski, A.M.; Zhang, Q.; Groeneveld, M.P.; Scott, C.E.; et al. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nat. Genet. 2012, 44, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Angulo, I.; Vadas, O.; Garçon, F.; Banham-Hall, E.; Plagnol, V.; Leahy, T.R.; Baxendale, H.; Coulter, T.; Curtis, J.; Wu, C.; et al. Phosphoinositide 3-Kinase Gene Mutation Predisposes to Respiratory Infection and Airway Damage. Science 2013, 342, 866–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, C.; Kuehn, H.S.; Zhao, F.; Niemela, J.; Deenick, E.K.; Palendira, U.; Avery, D.T.; Moens, L.; Cannons, J.L.; Biancalana, M.; et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat. Immunol. 2014, 15, 88–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, C.; Zhang, Y.; Venida, A.; Wang, Y.; Hughes, J.; McElwee, J.; Butrick, M.; Matthews, H.; Price, S.; Biancalana, M.; et al. Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J. Exp. Med. 2014, 211, 2537–2547. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Vanhaesebroeck, B.; Perry, M.W.D.; Brown, J.R.; André, F.; Okkenhaug, K. PI3K inhibitors are finally coming of age. Nat. Rev. Drug Discov. 2021, 1–29. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Burke, J.; Perisic, O.; Masson, G.; Vadas, O.; Williams, R. Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110α (PIK3CA). Proc. Natl. Acad. Sci. USA 2012, 109, 15259–15264. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Tenorio, G.; Alkhori, L.; Olsson, B.; Waltersson, M.A.; Nordenskjöld, B.; Rutqvist, L.E.; Skoog, L.; Stål, O. PIK3CA Mutations and PTEN Loss Correlate with Similar Prognostic Factors and Are Not Mutually Exclusive in Breast Cancer. Clin. Cancer Res. 2007, 13, 3577–3584. [Google Scholar] [CrossRef] [Green Version]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.-L.; Davies, M.; Carey, M.; Yinghui, G.; Guan, Y.; Sahin, A.; et al. An Integrative Genomic and Proteomic Analysis of PIK3CA, PTEN, and AKT Mutations in Breast Cancer. Cancer Res. 2008, 68, 6084–6091. [Google Scholar] [CrossRef] [Green Version]
- Campbell, I.G.; Russell, S.E.; Choong, D.Y.H.; Montgomery, K.G.; Ciavarella, M.L.; Hooi, C.S.F.; Cristiano, B.E.; Pearson, R.; Phillips, W. Mutation of the PIK3CA Gene in Ovarian and Breast Cancer. Cancer Res. 2004, 64, 7678–7681. [Google Scholar] [CrossRef] [Green Version]
- Ellis, M.J.; Lin, L.; Crowder, R.; Tao, Y.; Hoog, J.; Snider, J.; Davies, S.; DeSchryver, K.; Evans, D.B.; Steinseifer-Szabo, J.; et al. Phosphatidyl-inositol-3-kinase alpha catalytic subunit mutation and response to neoadjuvant endocrine therapy for estrogen receptor positive breast cancer. Breast Cancer Res. Treat. 2009, 119, 379–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Angulo, A.M.; Ferrer-Lozano, J.; Stemke-Hale, K.; Sahin, A.; Liu, S.; Barrera, J.A.; Burgues, O.; Lluch, A.; Chen, H.; Hortobagyi, G.N.; et al. PI3K Pathway Mutations and PTEN Levels in Primary and Metastatic Breast Cancer. Mol. Cancer Ther. 2011, 10, 1093–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennessy, B.T.; Gonzalez-Angulo, A.-M.; Stemke-Hale, K.; Gilcrease, M.Z.; Krishnamurthy, S.; Lee, J.-S.; Fridlyand, J.; A Sahin, A.; Agarwal, R.; Joy, C.; et al. Characterization of a Naturally Occurring Breast Cancer Subset Enriched in Epithelial-to-Mesenchymal Transition and Stem Cell Characteristics. Cancer Res. 2009, 69, 4116–4124. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Sáez, O.; Chic, N.; Pascual, T.; Adamo, B.; Vidal, M.; González-Farré, B.; Sanfeliu, E.; Schettini, F.; Conte, B.; Brasó-Maristany, F.; et al. Frequency and spectrum of PIK3CA somatic mutations in breast cancer. Breast Cancer Res. 2020, 22, 45. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Gatza, M.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [Green Version]
- Keraite, I.; Alvarez-Garcia, V.; Garcia-Murillas, I.; Beaney, M.; Turner, N.C.; Bartos, C.; Oikonomidou, O.; Kersaudy-Kerhoas, M.; Leslie, N.R. PIK3CA mutation enrichment and quantitation from blood and tissue. Sci. Rep. 2020, 10, 17082. [Google Scholar] [CrossRef]
- Bertucci, F.; Ng, C.K.Y.; Patsouris, A.; Droin, N.; Piscuoglio, S.; Carbuccia, N.; Soria, J.C.; Dien, A.T.; Adnani, Y.; Kamal, M.; et al. Genomic characterization of metastatic breast cancers. Nat. Cell Biol. 2019, 569, 560–564. [Google Scholar] [CrossRef]
- Crowder, R.J.; Phommaly, C.; Tao, Y.; Hoog, J.; Luo, J.; Perou, C.; Parker, J.S.; Miller, M.A.; Huntsman, D.G.; Lin, L.; et al. PIK3CA and PIK3CB Inhibition Produce Synthetic Lethality when Combined with Estrogen Deprivation in Estrogen Receptor–Positive Breast Cancer. Cancer Res. 2009, 69, 3955–3962. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, B.S.; Janakiraman, V.; Kljavin, N.M.; Chaudhuri, S.; Stern, H.M.; Wang, W.; Kan, Z.; Dbouk, H.; Peters, B.; Waring, P.; et al. Somatic Mutations in p85α Promote Tumorigenesis through Class IA PI3K Activation. Cancer Cell 2009, 16, 463–474. [Google Scholar] [CrossRef] [Green Version]
- Dbouk, H.; Khalil, B.D.; Wu, H.; Shymanets, A.; Nürnberg, B.; Backer, J.M. Characterization of a Tumor-Associated Activating Mutation of the p110β PI 3-Kinase. PLoS ONE 2013, 8, e63833. [Google Scholar] [CrossRef] [Green Version]
- Ciraolo, E.; Iezzi, M.; Marone, R.; Marengo, S.; Curcio, C.; Costa, C.; Azzolino, O.; Gonella, C.; Rubinetto, C.; Wu, H.; et al. Phosphoinositide 3-Kinase p110 Activity: Key Role in Metabolism and Mammary Gland Cancer but Not Development. Sci. Signal. 2008, 1, ra3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dbouk, H.A.; Vadas, O.; Shymanets, A.; Burke, J.E.; Salamon, R.S.; Khalil, B.D.; Barrett, M.O.; Waldo, G.L.; Surve, C.; Hsueh, C.; et al. G Protein-Coupled Receptor-Mediated Activation of p110 by G Is Required for Cellular Transformation and Invasiveness. Sci. Signal. 2012, 5, ra89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kan, Z.; Jaiswal, B.S.; Stinson, J.; Janakiraman, V.; Bhatt, D.; Stern, H.M.; Yue, P.; Haverty, P.M.; Bourgon, R.; Zheng, J.; et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nat. Cell Biol. 2010, 466, 869–873. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 1–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saal, L.; Johansson, P.; Holm, K.; Gruvberger-Saal, S.K.; She, Q.-B.; Maurer, M.; Koujak, S.; Ferrando, A.A.; Malmström, P.; Memeo, L.; et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7564–7569. [Google Scholar] [CrossRef] [Green Version]
- Shoman, N.; Klassen, S.; McFadden, A.; Bickis, M.G.; Torlakovic, E.; Chibbar, R. Reduced PTEN expression predicts relapse in patients with breast carcinoma treated by tamoxifen. Mod. Pathol. 2004, 18, 250–259. [Google Scholar] [CrossRef] [Green Version]
- Carbognin, L.; Miglietta, F.; Paris, I.; Dieci, M.V. Prognostic and Predictive Implications of PTEN in Breast Cancer: Unfulfilled Promises but Intriguing Perspectives. Cancers 2019, 11, 1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, L.M.; Batist, G.; Meric-Bernstam, F.; Kabos, P.; Spanggaard, I.; Lluch, A.; Jhaveri, K.; Varga, A.; Wong, A.; Schram, A.M.; et al. Selective AKT kinase inhibitor capivasertib in combination with fulvestrant in PTEN-mutant ER-positive metastatic breast cancer. Npj Breast Cancer 2021, 7, 1–7. [Google Scholar] [CrossRef]
- Saal, L.; Holm, K.; Maurer, M.; Memeo, L.; Su, T.; Wang, X.; Yu, J.S.; Malmström, P.-O.; Mansukhani, M.; Enoksson, J.; et al. PIK3CA Mutations Correlate with Hormone Receptors, Node Metastasis, and ERBB2, and Are Mutually Exclusive with PTEN Loss in Human Breast Carcinoma. Cancer Res. 2005, 65, 2554–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedele, C.G.; Ooms, L.M.; Ho, M.; Vieusseux, J.; O’Toole, S.A.; Millar, E.; Knowles, E.L.; Sriratana, A.; Gurung, R.; Baglietto, L.; et al. Inositol polyphosphate 4-phosphatase II regulates PI3K/Akt signaling and is lost in human basal-like breast cancers. Proc. Natl. Acad. Sci. USA 2010, 107, 22231–22236. [Google Scholar] [CrossRef] [Green Version]
- Gewinner, C.; Wang, Z.C.; Richardson, A.; Teruya-Feldstein, J.; Etemadmoghadam, D.; Bowtell, D.; Barretina, J.; Lin, W.M.; Rameh, L.; Salmena, L.; et al. Evidence that Inositol Polyphosphate 4-Phosphatase Type II Is a Tumor Suppressor that Inhibits PI3K Signaling. Cancer Cell 2009, 16, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Paddock, M.N.; Wang, H.; Murphy, C.J.; Geck, R.C.; Navarro, A.J.; Wulf, G.M.; Elemento, O.; Haucke, V.; Cantley, L.C.; et al. The INPP4B Tumor Suppressor Modulates EGFR Trafficking and Promotes Triple-Negative Breast Cancer. Cancer Discov. 2020, 10, 1226–1239. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, S.J.; Ooms, L.M.; Oorschot, V.M.J.; Schittenhelm, R.B.; Nguyen, E.V.; Hamila, S.A.; Rynkiewicz, N.; Gurung, R.; Eramo, M.J.; Sriratana, A.; et al. INPP4B promotes PI3Kα-dependent late endosome formation and Wnt/β-catenin signaling in breast cancer. Nat. Commun. 2021, 12, 3140. [Google Scholar] [CrossRef]
- Carpten, J.D.; Faber, A.L.; Horn, C.; Donoho, G.P.; Briggs, S.L.; Robbins, C.M.; Hostetter, G.; Boguslawski, S.; Moses, T.Y.; Savage, S.; et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 2007, 448, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Bellacosa, A.; De Feo, D.; Godwin, A.K.; Bell, D.W.; Cheng, J.Q.; Altomare, D.A.; Wan, M.; Dubeau, L.; Scambia, G.; Masciullo, V.; et al. Molecular alterations of theAKT2 oncogene in ovarian and breast carcinomas. Int. J. Cancer 1995, 64, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.; Su, T.; Saal, L.; Koujak, S.; Hopkins, B.D.; Barkley, C.R.; Wu, J.; Nandula, S.; Dutta, B.; Xie, Y.; et al. 3-Phosphoinositide–Dependent Kinase 1 Potentiates Upstream Lesions on the Phosphatidylinositol 3-Kinase Pathway in Breast Carcinoma. Cancer Res. 2009, 69, 6299–6306. [Google Scholar] [CrossRef] [Green Version]
- Monni, O.; Barlund, M.; Mousses, S.; Kononen, J.; Sauter, G.; Heiskanen, M.; Paavola, P.; Avela, K.; Chen, Y.; Bittner, M.L.; et al. Comprehensive copy number and gene expression profiling of the 17q23 amplicon in human breast cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 5711–5716. [Google Scholar] [CrossRef] [Green Version]
- Rochlitz, C.F.; Scott, G.K.; Dodson, J.M.; Liu, E.; Dollbaum, C.; Smith, H.S.; Benz, C.C.; Rochlitz, C.F.; Scott, G.K.; Dodson, J.M.; et al. Incidence of activating ras oncogene mutations associated with primary and metastatic human breast cancer. Cancer Res. 1989, 49, 357–360. [Google Scholar]
- Loi, S.; Michiels, S.; Lambrechts, D.; Fumagalli, D.; Claes, B.; Kellokumpu-Lehtinen, P.-L.; Bono, P.; Kataja, V.; Piccart, M.J.; Joensuu, H.; et al. Somatic Mutation Profiling and Associations With Prognosis and Trastuzumab Benefit in Early Breast Cancer. J. Natl. Cancer Inst. 2013, 105, 960–967. [Google Scholar] [CrossRef]
- Sabine, V.S.; Crozier, C.; Brookes, C.L.; Drake, C.; Piper, T.; Van De Velde, C.J.; Hasenburg, A.; Kieback, D.G.; Markopoulos, C.; Dirix, L.; et al. Mutational Analysis of PI3K/AKT Signaling Pathway in Tamoxifen Exemestane Adjuvant Multinational Pathology Study. J. Clin. Oncol. 2014, 32, 2951–2958. [Google Scholar] [CrossRef]
- Azim, H.A.; Brohée, S.; Peccatori, F.; Desmedt, C.; Loi, S.; Lambrechts, D.; Dell’Orto, P.; Majjaj, S.; Jose, V.; Rotmensz, N.; et al. Biology of breast cancer during pregnancy using genomic profiling. Endocr.Relat. Cancer 2014, 21, 545–554. [Google Scholar] [CrossRef]
- Boyault, S.; Drouet, Y.; Navarro, C.; Bachelot, T.; Lasset, C.; Treilleux, I.; Tabone, E.; Puisieux, A.; Wang, Q. Mutational characterization of individual breast tumors: TP53 and PI3K pathway genes are frequently and distinctively mutated in different subtypes. Breast Cancer Res. Treat. 2011, 132, 29–39. [Google Scholar] [CrossRef]
- Barbareschi, M.; Buttitta, F.; Felicioni, L.; Cotrupi, S.; Barassi, F.; Del Grammastro, M.; Ferro, A.; Palma, P.D.; Galligioni, E.; Marchetti, A. Different Prognostic Roles of Mutations in the Helical and Kinase Domains of the PIK3CA Gene in Breast Carcinomas. Clin. Cancer Res. 2007, 13, 6064–6069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaxoinis, G.; Kotoula, V.; Alexopoulou, Z.; Kalogeras, K.T.; Zagouri, F.; Timotheadou, E.; Gogas, H.; Pentheroudakis, G.; Christodoulou, C.; Koutras, A.; et al. Significance of PIK3CA Mutations in Patients with Early Breast Cancer Treated with Adjuvant Chemotherapy: A Hellenic Cooperative Oncology Group (HeCOG) Study. PLoS ONE 2015, 10, e0140293. [Google Scholar] [CrossRef] [Green Version]
- Jensen, J.D.; Knoop, A.; Laenkholm, A.V.; Grauslund, M.; Jensen, M.B.; Santoni-Rugiu, E.; Andersson, M.; Ewertz, M. PIK3CA mutations, PTEN, and pHER2 expression and impact on outcome in HER2-positive early-stage breast cancer patients treated with adjuvant chemotherapy and trastuzumab. Ann. Oncol. 2012, 23, 2034–2042. [Google Scholar] [CrossRef]
- Gallardo, A.; Lerma, E.; Escuin, D.; Tibau, A.; Muñoz, J.; Ojeda, B.; Barnadas, A.; Adrover, E.; Sanchez-Tejada, L.; Giner, D.; et al. Increased signalling of EGFR and IGF1R, and deregulation of PTEN/PI3K/Akt pathway are related with trastuzumab resistance in HER2 breast carcinomas. Br. J. Cancer 2012, 106, 1367–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinsky, K.; Jacks, L.M.; Heguy, A.; Patil, S.; Drobnjak, M.; Bhanot, U.; Hedvat, C.; Traina, T.A.; Solit, D.; Gerald, W.; et al. PIK3CA Mutation Associates with Improved Outcome in Breast Cancer. Clin. Cancer Res. 2009, 15, 5049–5059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruyama, N.; Miyoshi, Y.; Taguchi, T.; Tamaki, Y.; Monden, M.; Noguchi, S. Clinicopathologic Analysis of Breast Cancers with PIK3CA Mutations in Japanese Women. Clin. Cancer Res. 2007, 13, 408–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, E.L.; O’Toole, S.A.; McNeil, C.M.; Millar, E.; Qiu, M.R.; Crea, P.; Daly, R.; Musgrove, E.A.; Sutherland, R.L. PI3K pathway activation in breast cancer is associated with the basal-like phenotype and cancer-specific mortality. Int. J. Cancer 2009, 126, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Hudis, C.A.; Barlow, W.E.; Costantino, J.P.; Gray, R.J.; Pritchard, K.I.; Chapman, J.-A.W.; Sparano, J.A.; Hunsberger, S.; Enos, R.A.; Gelber, R.D.; et al. Proposal for Standardized Definitions for Efficacy End Points in Adjuvant Breast Cancer Trials: The STEEP System. J. Clin. Oncol. 2007, 25, 2127–2132. [Google Scholar] [CrossRef]
- Zardavas, D.; Marvelde, L.T.; Milne, R.L.; Fumagalli, D.; Fountzilas, G.; Kotoula, V.; Razis, E.; Papaxoinis, G.; Joensuu, H.; Moynahan, M.E.; et al. Tumor PIK3CA Genotype and Prognosis in Early-Stage Breast Cancer: A Pooled Analysis of Individual Patient Data. J. Clin. Oncol. 2018, 36, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Shao, Z.-M.; Liu, Y.-R.; Jiang, Y.-Z.; Yu, K.-D.; Zuo, W.-J. PIK3CA mutations define favorable prognostic biomarkers in operable breast cancer: A systematic review and meta-analysis. OncoTargets Ther. 2014, 7, 543–552. [Google Scholar] [CrossRef] [Green Version]
- Pang, B.; Cheng, S.; Sun, S.-P.; An, C.; Liu, Z.-Y.; Feng, X.; Liu, G.-J. Prognostic role of PIK3CA mutations and their association with hormone receptor expression in breast cancer: A meta-analysis. Sci. Rep. 2014, 4, srep06255. [Google Scholar] [CrossRef] [Green Version]
- Mosele, F.; Stefanovska, B.; Lusque, A.; Dien, A.T.; Garberis, I.; Droin, N.; Le Tourneau, C.; Sablin, M.-P.; Lacroix, L.; Enrico, D.; et al. Outcome and molecular landscape of patients with PIK3CA-mutated metastatic breast cancer. Ann. Oncol. 2020, 31, 377–386. [Google Scholar] [CrossRef] [Green Version]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Hortobagyi, G.N.; Stemmer, S.; Burris, H.; Yap, Y.; Sonke, G.; Paluch-Shimon, S.; Campone, M.; Petrakova, K.; Blackwell, K.; Winer, E.; et al. Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann. Oncol. 2018, 29, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Im, S.A.S.; Iwata, H.; Cortés, J.; De Laurentiis, M.; Jiang, Z.; Arteaga, C.C.; Jonat, W.; Clemons, M.J.; Ito, Y.Y.; et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 904–916. [Google Scholar] [CrossRef]
- Di Leo, A.; Johnston, S.; Lee, K.S.; Ciruelos, E.; E Lønning, P.; Janni, W.; O’Regan, R.; Mouret-Reynier, M.-A.; Kalev, D.; Egle, D.; et al. Buparlisib plus fulvestrant in postmenopausal women with hormone-receptor-positive, HER2-negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2018, 19, 87–100. [Google Scholar] [CrossRef]
- E Krop, E.I.; Mayer, I.I.; Ganju, V.V.; Dickler, M.M.; Johnston, S.; Morales, S.S.; Yardley, D.D.; Melichar, B.B.; Forero-Torres, A.A.; Lee, S.C.S.; et al. Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016, 17, 811–821. [Google Scholar] [CrossRef] [Green Version]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.-A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar] [CrossRef] [Green Version]
- Moynahan, M.E.; Chen, D.; He, W.; Sung, P.; Samoila, A.; You, D.; Bhatt, T.; Patel, P.; Ringeisen, F.; Hortobagyi, G.N.; et al. Correlation between PIK3CA mutations in cell-free DNA and everolimus efficacy in HR+, HER2− advanced breast cancer: Results from BOLERO-2. Br. J. Cancer 2017, 116, 726–730. [Google Scholar] [CrossRef] [Green Version]
- Baselga, J.; Cortes, J.; Im, S.-A.; Clark, E.; Ross, G.; Kiermaier, A.; Swain, S. Biomarker Analyses in CLEOPATRA: A Phase III, Placebo-Controlled Study of Pertuzumab in Human Epidermal Growth Factor Receptor 2–Positive, First-Line Metastatic Breast Cancer. J. Clin. Oncol. 2014, 32, 3753–3761. [Google Scholar] [CrossRef]
- Miller, T.W.; Hennessy, B.T.; González-Angulo, A.M.; Fox, E.M.; Mills, G.B.; Chen, H.; Higham, C.; García-Echeverría, C.; Shyr, Y.; Arteaga, C.L. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor–positive human breast cancer. J. Clin. Investig. 2010, 120, 2406–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simoncini, T.; Hafezi-Moghadam, A.; Brazil, D.; Ley, K.; Chin, W.W.; Liao, J.K. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nat. Cell Biol. 2000, 407, 538–541. [Google Scholar] [CrossRef]
- Maira, S.-M.; Pecchi, S.; Huang, A.; Burger, M.; Knapp, M.; Sterker, D.; Schnell, C.; Guthy, D.; Nagel, T.; Wiesmann, M.; et al. Identification and Characterization of NVP-BKM120, an Orally Available Pan-Class I PI3-Kinase Inhibitor. Mol. Cancer Ther. 2012, 11, 317–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín, M.; Chan, A.; Dirix, L.; O’Shaughnessy, J.; Hegg, R.; Manikhas, A.; Shtivelband, M.; Krivorotko, P.; López, N.B.; Campone, M.; et al. A randomized adaptive phase II/III study of buparlisib, a pan-class I PI3K inhibitor, combined with paclitaxel for the treatment of HER2– advanced breast cancer (BELLE-4). Ann. Oncol. 2017, 28, 313–320. [Google Scholar] [CrossRef]
- Bradford, L.S.; Rauh-Hain, A.; Clark, R.M.; Groeneweg, J.W.; Zhang, L.; Borger, D.; Zukerberg, L.R.; Growdon, W.B.; Foster, R.; Rueda, B.R. Assessing the efficacy of targeting the phosphatidylinositol 3-kinase/AKT/mTOR signaling pathway in endometrial cancer. Gynecol. Oncol. 2014, 133, 346–352. [Google Scholar] [CrossRef]
- Folkes, A.J.; Ahmadi, K.; Alderton, W.; Alix, S.; Baker, S.J.; Box, G.; Chuckowree, I.S.; Clarke, P.; Depledge, P.; Eccles, S.A.; et al. The Identification of 2-(1H-Indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a Potent, Selective, Orally Bioavailable Inhibitor of Class I PI3 Kinase for the Treatment of Cancer. J. Med. Chem. 2008, 51, 5522–5532. [Google Scholar] [CrossRef] [PubMed]
- Vuylsteke, P.; Huizing, M.; Petrakova, K.; Roylance, R.; Laing, R.; Chan, S.; Abell, F.; Gendreau, S.; Rooney, I.; Apt, D.; et al. Pictilisib PI3Kinase inhibitor (a phosphatidylinositol 3-kinase [PI3K] inhibitor) plus paclitaxel for the treatment of hormone receptor-positive, HER2-negative, locally recurrent, or metastatic breast cancer: Interim analysis of the multicentre, placebo-controlled, phase II randomised PEGGY study. Ann. Oncol. 2016, 27, 2059–2066. [Google Scholar] [CrossRef]
- Dienstmann, R.; Rodon, J.; Serra, V.; Tabernero, J. Picking the Point of Inhibition: A Comparative Review of PI3K/AKT/mTOR Pathway Inhibitors. Mol. Cancer Ther. 2014, 13, 1021–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsch, C.; Huang, A.; Chatenay-Rivauday, C.; Schnell, C.; Reddy, A.; Liu, M.; Kauffmann, A.; Guthy, D.; Erdmann, D.; De Pover, A.; et al. Characterization of the Novel and Specific PI3Kα Inhibitor NVP-BYL719 and Development of the Patient Stratification Strategy for Clinical Trials. Mol. Cancer Ther. 2014, 13, 1117–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsch, C.; Pfister, E.; Ebel, N.; Guthy, D.; Schnell, C.; Hofmann, F. Abstract 3934: Determination of the PI3Kα selective inhibitor alpelisib mechanism of action and efficacy in ER+/PIK3CA mutant breast cancer preclinical models. Exp. Mol. Ther. 2018, 78, 3934. [Google Scholar] [CrossRef]
- Juric, D.; Janku, F.; Rodón, J.; Burris, H.A.; Mayer, I.A.; Schuler, M.; Seggewiss-Bernhardt, R.; Gil-Martin, M.; Middleton, M.R.; Baselga, J.; et al. Alpelisib Plus Fulvestrant in PIK3CA-Altered and PIK3CA-Wild-Type Estrogen Receptor–Positive Advanced Breast Cancer. JAMA Oncol. 2019, 5, e184475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moehler, M.; Shitara, K.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Bragagnoli, A.C.; Liu, T.; et al. LBA6_PR Nivolumab (nivo) plus chemotherapy (chemo) versus chemo as first-line (1L) treatment for advanced gastric cancer/gastroesophageal junction cancer (GC/GEJC)/esophageal adenocarcinoma (EAC): First results of the CheckMate 649 study. Ann. Oncol. 2020, 31, S1191. [Google Scholar] [CrossRef]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.-A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]
- Im, S.-A.; Lu, Y.-S.; Bardia, A.; Harbeck, N.; Colleoni, M.; Franke, F.; Chow, L.; Sohn, J.; Lee, K.-S.; Campos-Gomez, S.; et al. Overall Survival with Ribociclib plus Endocrine Therapy in Breast Cancer. N. Engl. J. Med. 2019, 381, 307–316. [Google Scholar] [CrossRef]
- Rugo, H.S.; Lerebours, F.; Ciruelos, E.; Drullinsky, P.; Borrego, M.R.; Neven, P.; Park, Y.H.; Prat, A.; Bachelot, T.; Juric, D.; et al. Alpelisib (ALP) + fulvestrant (FUL) in patients (pts) with PIK3CA-mutated (mut) hormone receptor-positive (HR+), human epidermal growth factor receptor 2-negative (HER2–) advanced breast cancer (ABC) previously treated with cyclin-dependent kinase 4/6 inhibitor (CDKi) + aromatase inhibitor (AI): BYLieve study results. J. Clin. Oncol. 2020, 38, 1006. [Google Scholar] [CrossRef]
- Olivero, A.G.; Heffron, T.P.; Baumgardner, M.; Belvin, M.; Ross, L.B.; Blaquiere, N.; Bradley, E.; Castanedo, G.; Derynck, M.; Do, S.; et al. Abstract DDT02-01: Discovery of GDC-0032: A beta-sparing PI3K inhibitor active against PIK3CA mutant tumors. Cancer Chem. 2013, 73. [Google Scholar] [CrossRef]
- Juric, D.; Krop, I.; Ramanathan, R.K.; Wilson, T.R.; Ware, J.A.; Bohorquez, S.S.; Savage, H.M.; Sampath, D.; Salphati, L.; Lin, R.S.; et al. Phase I Dose-Escalation Study of Taselisib, an Oral PI3K Inhibitor, in Patients with Advanced Solid Tumors. Cancer Discov. 2017, 7, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Baselga, J.; Dent, S.F.; Cortés, J.; Im, Y.-H.; Diéras, V.; Harbeck, N.; Krop, I.E.; Verma, S.; Wilson, T.R.; Jin, H.; et al. Phase III study of taselisib (GDC-0032) + fulvestrant (FULV) v FULV in patients (pts) with estrogen receptor (ER)-positive, PIK3CA-mutant (MUT), locally advanced or metastatic breast cancer (MBC): Primary analysis from SANDPIPER. J. Clin. Oncol. 2018, 36, LBA1006. [Google Scholar] [CrossRef]
- Ndubaku, C.O.; Heffron, T.P.; Staben, S.T.; Baumgardner, M.; Blaquiere, N.; Bradley, E.; Bull, R.; Do, S.; Dotson, J.; Dudley, D.; et al. Discovery of 2-{3-[2-(1-Isopropyl-3-methyl-1H-1,2–4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl}-2-methylpropanamide (GDC-0032): A β-Sparing Phosphoinositide 3-Kinase Inhibitor with High Unbound Exposure and Robust in Vivo Antitumor Activity. J. Med. Chem. 2013, 56, 4597–4610. [Google Scholar] [CrossRef] [PubMed]
- Hanker, A.; Pfefferle, A.D.; Balko, J.M.; Kuba, M.G.; Young, C.D.; Sánchez, V.; Sutton, C.R.; Cheng, H.; Perou, C.; Zhao, J.J.; et al. Mutant PIK3CA accelerates HER2-driven transgenic mammary tumors and induces resistance to combinations of anti-HER2 therapies. Proc. Natl. Acad. Sci. USA 2013, 110, 14372–14377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loibl, S.; Majewski, I.; Guarneri, V.; Nekljudova, V.; Holmes, E.; Bria, E.; Denkert, C.; Schem, C.; Sotiriou, C.; Loi, S.; et al. Corrections to “PIK3CA mutations are associated with reduced pathological complete response rates in primary HER2-positive breast cancer: Pooled analysis of 967 patients from five prospective trials investigating lapatinib and trastuzumab”. Ann. Oncol. 2019, 30, 1180. [Google Scholar] [CrossRef] [PubMed]
- Guerin, M.; Rezai, K.; Isambert, N.; Campone, M.; Autret, A.; Pakradouni, J.; Provansal, M.; Camerlo, J.; Sabatier, R.; Bertucci, F.; et al. PIKHER2: A phase IB study evaluating buparlisib in combination with lapatinib in trastuzumab-resistant HER2-positive advanced breast cancer. Eur. J. Cancer 2017, 86, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Pistilli, B.; Pluard, T.; Urruticoechea, A.; Farci, D.; Kong, A.; Bachelot, T.; Chan, S.; Han, H.S.; Jerusalem, G.; Urban, P.; et al. Phase II study of buparlisib (BKM120) and trastuzumab in patients with HER2+ locally advanced or metastatic breast cancer resistant to trastuzumab-based therapy. Breast Cancer Res. Treat. 2018, 168, 357–364. [Google Scholar] [CrossRef]
- Loibl, S.; de la Pena, L.; Nekljudova, V.; Zardavas, D.; Michiels, S.; Denkert, C.; Rezai, M.; Bermejo, B.; Untch, M.; Lee, S.C.; et al. Neoadjuvant buparlisib plus trastuzumab and paclitaxel for women with HER2+ primary breast cancer: A randomised, double-blind, placebo-controlled phase II trial (NeoPHOEBE). Eur. J. Cancer 2017, 85, 133–145. [Google Scholar] [CrossRef]
- Jain, S.; Shah, A.N.; Santa-Maria, C.A.; Siziopikou, K.; Rademaker, A.; Helenowski, I.; Cristofanilli, M.; Gradishar, W.J. Phase I study of alpelisib (BYL-719) and trastuzumab emtansine (T-DM1) in HER2-positive metastatic breast cancer (MBC) after trastuzumab and taxane therapy. Breast Cancer Res. Treat. 2018, 171, 371–381. [Google Scholar] [CrossRef]
- Barok, M.; Tanner, M.; Köninki, K.; Isola, J. Trastuzumab-DM1 causes tumour growth inhibition by mitotic catastrophe in trastuzumab-resistant breast cancer cells in vivo. Breast Cancer Res. 2011, 13, R46. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Jang, H.; Nussinov, R. PI3K inhibitors: Review and new strategies. Chem. Sci. 2020, 11, 5855–5865. [Google Scholar] [CrossRef]
- Hong, R.; Edgar, K.; Song, K.; Steven, S.; Young, A.; Hamilton, P.; Arrazate, A.; De La Cruz, C.; Chan, C.; Pang, J.; et al. Abstract PD4-14: GDC-0077 is a selective PI3Kalpha inhibitor that demonstrates robust efficacy in PIK3CA mutant breast cancer models as a single agent and in combination with standard of care therapies. Poster Discuss. Abstr. 2018, 78. [Google Scholar] [CrossRef]
- Turner, N.; Dent, R.; O’Shaughnessy, J.; Kim, S.-B.; Isakoff, S.; Barrios, C.; Saji, S.; Bondarenko, I.; Nowecki, Z.; Lian, Q.; et al. 283MO Ipatasertib (IPAT) + paclitaxel (PAC) for PIK3CA/AKT1/PTEN-altered hormone receptor-positive (HR+) HER2-negative advanced breast cancer (aBC): Primary results from Cohort B of the IPATunity130 randomised phase III trial. Ann. Oncol. 2020, 31, S354–S355. [Google Scholar] [CrossRef]
- Merlino, G.; Fiascarelli, A.; Bigioni, M.; Bressan, A.; Irrissuto, C.; Pellacani, A.; Scaltriti, M.; Binaschi, M. Abstract 2160: MEN1611, a novel α-selective PI3K inhibitor in solid tumors. Tumor Biol. 2018, 78, 2160. [Google Scholar] [CrossRef]
- Janku, F.; Huang, H.; Treskova, I.; Pivovarcikova, K.; Call, S.; Meric-Bernstam, F.; Pesta, M.; Polivka, J. Ultra-sensitive detection of circulating tumor DNA identifies patients in high risk of recurrence in early stages melanoma. Ann. Oncol. 2019, 30, v767. [Google Scholar] [CrossRef]
- Hansen, A.R.; Shapiro, G.; Do, K.T.; Kumar, R.; Martin-Liberal, J.; Higano, C.S.; Wisinski, K.B.; Dean, E.J.; Heath, E.I.; Rathkopf, D.E.; et al. A first in human phase I study of AZD8186, a potent and selective inhibitor of PI3K in patients with advanced solid tumours as monotherapy and in combination with the dual mTORC1/2 inhibitor vistusertib (AZD2014) or abiraterone acetate. J. Clin. Oncol. 2017, 35, 2570. [Google Scholar] [CrossRef]
- Owusu-Brackett, N.; Zhao, M.; Akcakanat, A.; Evans, K.W.; Yuca, E.; Dumbrava, E.I.; Janku, F.; Meric-Bernstam, F. Targeting PI3Kβ alone and in combination with chemotherapy or immunotherapy in tumors with PTEN loss. Oncotarget 2020, 11, 969–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Richmond, A. The Role of PI3K Inhibition in the Treatment of Breast Cancer, Alone or Combined With Immune Checkpoint Inhibitors. Front. Mol. Biosci. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Szekely, B.; Bossuyt, V.; Li, X.; Wali, V.; Patwardhan, G.; Frederick, C.; Silber, A.; Park, T.; Harigopal, M.; Pelekanou, V.; et al. Immunological differences between primary and metastatic breast cancer. Ann. Oncol. 2018, 29, 2232–2239. [Google Scholar] [CrossRef]
- Hamilton, E.; Lee, A.; Swart, R.; Newton, G.; O’Connell, B.; Roberts, J.; Zhang, H.; Soliman, H. Abstract PS11-32: Mario-3 phase II study safety run-in evaluating a novel triplet combination of eganelisib (formerly IPI-549), atezolizumab (atezo), and nab-paclitaxel (nab-pac) as first-line (1L) therapy for locally advanced or metastatic triple-negative breast cancer (TNBC). Poster Sess. Abstr. 2021, 81, PS11-32. [Google Scholar]
- Nitulescu, G.M.; Van De Venter, M.; Nitulescu, G.; Ungurianu, A.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Grădinaru, D.; Tsatsakis, A.; Tsoukalas, D.; et al. The Akt pathway in oncology therapy and beyond (Review). Int. J. Oncol. 2018, 53, 2319–2331. [Google Scholar] [CrossRef] [Green Version]
- Bellacosa, A.; Kumar, C.C.; Di Cristofano, A.; Testa, J.R. Activation of AKT Kinases in Cancer: Implications for Therapeutic Targeting. Adv. Cancer Res. 2005, 94, 29–86. [Google Scholar] [CrossRef]
- Banerji, U.; Dean, E.J.; Pérez-Fidalgo, J.A.; Batist, G.; Bedard, P.L.; You, B.; Westin, S.N.; Kabos, P.; Garrett, M.D.; Tall, M.; et al. A Phase I Open-Label Study to Identify a Dosing Regimen of the Pan-AKT Inhibitor AZD5363 for Evaluation in Solid Tumors and inPIK3CA-Mutated Breast and Gynecologic Cancers. Clin. Cancer Res. 2017, 24, 2050–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bono, J.S.; De Giorgi, U.; Rodrigues, D.N.; Massard, C.; Bracarda, S.; Font, A.; Arija, J.A.A.; Shih, K.C.; Radavoi, G.D.; Xu, N.; et al. Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clin. Cancer Res. 2019, 25, 928–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mundi, P.; Sachdev, J.; McCourt, C.; Kalinsky, K. AKT in cancer: New molecular insights and advances in drug development. Br. J. Clin. Pharmacol. 2016, 82, 943–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landel, I.; Quambusch, L.; Depta, L.; Rauh, D. Spotlight on AKT: Current Therapeutic Challenges. ACS Med. Chem. Lett. 2020, 11, 225–227. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.; Lin, N.U.; Maurer, M.A.; Chen, H.; Mahvash, A.; Sahin, A.; Akcakanat, A.; Li, Y.; Abramson, V.; Litton, J.; et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 2019, 21, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, S.; Vasilevski, N.; Serra, V.; Rodon, J.; Eichhorn, P. Mechanisms of Resistance to PI3K Inhibitors in Cancer: Adaptive Responses, Drug Tolerance and Cellular Plasticity. Cancers 2021, 13, 1538. [Google Scholar] [CrossRef]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Weisner, J.; Landel, I.; Reintjes, C.; Uhlenbrock, N.; Trajkovic-Arsic, M.; Dienstbier, N.; Hardick, J.; Ladigan, S.; Lindemann, M.; Smith, S.; et al. Preclinical Efficacy of Covalent-Allosteric AKT Inhibitor Borussertib in Combination with Trametinib in KRAS-mutant Pancreatic and Colorectal Cancer. Cancer Res. 2019, 79, 2367–2378. [Google Scholar] [CrossRef] [Green Version]
- Dey, N.; De, P.; Leyland-Jones, B. PI3K-AKT-mTOR inhibitors in breast cancers: From tumor cell signaling to clinical trials. Pharmacol. Ther. 2017, 175, 91–106. [Google Scholar] [CrossRef]
- Gnant, M.; Baselga, J.; Rugo, H.S.; Noguchi, S.; Burris, H.A.; Piccart, M.; Hortobagyi, G.N.; Eakle, J.; Mukai, H.; Iwata, H.; et al. Effect of Everolimus on Bone Marker Levels and Progressive Disease in Bone in BOLERO-2. J. Natl. Cancer Inst. 2013, 105, 654–663. [Google Scholar] [CrossRef] [Green Version]
- Hasskarl, J. Everolimus. Methods Mol. Biol. 2018, 211, 101–123. [Google Scholar]
- Bachelot, T.; Bourgier, C.; Cropet, C.; Ray-Coquard, I.; Ferrero, J.-M.; Freyer, G.; Abadie-Lacourtoisie, S.; Eymard, J.-C.; Debled, M.; Spaëth, D.; et al. Randomized Phase II Trial of Everolimus in Combination With Tamoxifen in Patients With Hormone Receptor–Positive, Human Epidermal Growth Factor Receptor 2–Negative Metastatic Breast Cancer With Prior Exposure to Aromatase Inhibitors: A GINECO Study. J. Clin. Oncol. 2012, 30, 2718–2724. [Google Scholar] [CrossRef]
- Royce, M.; Bachelot, T.; Villanueva, C.; Özgüroglu, M.; Azevedo, S.J.; Cruz, F.M.; Debled, M.; Hegg, R.; Toyama, T.; Falkson, C.; et al. Everolimus Plus Endocrine Therapy for Postmenopausal Women With Estrogen Receptor–Positive, Human Epidermal Growth Factor Receptor 2–Negative Advanced Breast Cancer. JAMA Oncol. 2018, 4, 977. [Google Scholar] [CrossRef]
- Petrossian, K.; Nguyen, D.; Lo, C.; Kanaya, N.; Somlo, G.; Cui, Y.X.; Huang, C.-S.; Chen, S. Use of dual mTOR inhibitor MLN0128 against everolimus-resistant breast cancer. Breast Cancer Res. Treat. 2018, 170, 499–506. [Google Scholar] [CrossRef]
- Lim, B.; Potter, D.A.; Salkeni, M.A.; Silverman, P.; Haddad, T.C.; Forget, F.; Awada, A.; Canon, J.-L.; Danso, M.; Lortholary, A.; et al. Sapanisertib Plus Exemestane or Fulvestrant in Women with Hormone Receptor–Positive/HER2-Negative Advanced or Metastatic Breast Cancer. Clin. Cancer Res. 2021, 27, 3329–3338. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Zaiss, M.; Harper-Wynne, C.; Ferreira, M.; Dubey, S.; Chan, S.; Makris, A.; Nemsadze, G.; Brunt, A.M.; Kuemmel, S.; et al. Fulvestrant Plus Vistusertib vs Fulvestrant Plus Everolimus vs Fulvestrant Alone for Women With Hormone Receptor–Positive Metastatic Breast Cancer. JAMA Oncol. 2019, 5, 1556–1563. [Google Scholar] [CrossRef]
- Brana, I.; Lorusso, P.; Baselga, J.; Heath, E.I.; Patnaik, A.; Gendreau, S.; Laird, A.; Papadopoulos, K. A phase I dose-escalation study of the safety, pharmacokinetics (PK), and pharmacodynamics of XL765 (SAR245409), a PI3K/TORC1/TORC2 inhibitor administered orally to patients (pts) with advanced malignancies. J. Clin. Oncol. 2010, 28, 3030. [Google Scholar] [CrossRef]
- Funakoshi, T.; Latif, A.; Galsky, M.D. Risk of hematologic toxicities in patients with solid tumors treated with everolimus: A systematic review and meta-analysis. Crit. Rev. Oncol. 2013, 88, 30–41. [Google Scholar] [CrossRef]
- Tabernero, J.; Rojo, F.; Calvo, E.; Burris, H.; Judson, I.; Hazell, K.; Martinelli, E.; Cajal, S.R.Y.; Jones, S.; Vidal, L.; et al. Dose- and Schedule-Dependent Inhibition of the Mammalian Target of Rapamycin Pathway With Everolimus: A Phase I Tumor Pharmacodynamic Study in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2008, 26, 1603–1610. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.H.; Perisic, O.; Ried, C.; Stephens, L.; Williams, R. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nat. Cell Biol. 1999, 402, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rudge, D.G.; Koos, J.; Vaidialingam, B.; Yang, H.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nat. Cell Biol. 2013, 497, 217–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell 2005, 8, 179–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Reilly, K.E.; Rojo, F.; She, Q.-B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR Inhibition Induces Upstream Receptor Tyrosine Kinase Signaling and Activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wainberg, Z.A.; Shapiro, G.; Curigliano, G.; Leong, S.; Kristeleit, R.S.; Maqueda, M.A.; Britten, C.D.; Milella, M.; Middleton, M.R.; Olszanski, A.J.; et al. Phase I study of the PI3K/mTOR inhibitor PF-05212384 in combination with other antitumor agents. J. Clin. Oncol. 2015, 33, 2590. [Google Scholar] [CrossRef]
- Forero-Torres, A.; Han, H.; Dees, E.C.; Wesolowski, R.; Bardia, A.; Kabos, P.; Layman, R.M.; Lu, J.M.; Kern, K.A.; Perea, R.; et al. Phase Ib study of gedatolisib in combination with palbociclib and endocrine therapy (ET) in women with estrogen receptor (ER) positive (+) metastatic breast cancer (MBC) (B2151009). J. Clin. Oncol. 2018, 36, 1040. [Google Scholar] [CrossRef]
- Franco, I.; Margaria, J.P.J.; De Santis, M.C.; A Ranghino, A.; Monteyne, D.; Chiaravalli, M.; Pema, M.M.; Campa, C.C.; E Ratto, E.; Gulluni, F.; et al. Phosphoinositide 3-Kinase-C2α Regulates Polycystin-2 Ciliary Entry and Protects against Kidney Cyst Formation. J. Am. Soc. Nephrol. 2015, 27, 1135–1144. [Google Scholar] [CrossRef] [Green Version]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef]
- Braccini, L.; Ciraolo, E.; Campa, C.C.; Perino, A.; Longo, D.L.; Tibolla, G.; Pregnolato, M.; Cao, Y.; Tassone, B.; Damilano, F.; et al. PI3K-C2γ is a Rab5 effector selectively controlling endosomal Akt2 activation downstream of insulin signalling. Nat. Commun. 2015, 6, 7400. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, K.; Yoshida, K.; Cui, H.; Wakayama, T.; Takuwa, N.; Okamoto, Y.; Du, W.; Qi, X.; Asanuma, K.; Sugihara, K.; et al. Endothelial PI3K-C2α, a class II PI3K, has an essential role in angiogenesis and vascular barrier function. Nat. Med. 2012, 18, 1560–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valet, C.; Chicanne, G.; Severac, C.; Chaussade, C.; Whitehead, M.A.; Cabou, C.; Gratacap, M.-P.; Gaits-Iacovoni, F.; Vanhaesebroeck, B.; Payrastre, B.; et al. Essential role of class II PI3K-C2α in platelet membrane morphology. Blood 2015, 126, 1128–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, I.; Gulluni, F.; Campa, C.C.; Costa, C.; Margaria, J.P.; Ciraolo, E.; Martini, M.; Monteyne, D.; De Luca, E.; Germena, G.; et al. PI3K Class II α Controls Spatially Restricted Endosomal PtdIns3P and Rab11 Activation to Promote Primary Cilium Function. Dev. Cell 2014, 28, 647–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marat, A.L.; Wallroth, A.; Lo, W.-T.; Müller, R.; Norata, G.D.; Falasca, M.; Schultz, C.; Haucke, V. mTORC1 activity repression by late endosomal phosphatidylinositol 3,4-bisphosphate. Science 2017, 356, 968–972. [Google Scholar] [CrossRef] [PubMed]
- Campa, C.C.; Margaria, J.P.; Derle, A.; Del Giudice, M.; De Santis, M.C.; Gozzelino, L.; Copperi, F.; Bosia, C.; Hirsch, E. Rab11 activity and PtdIns(3)P turnover removes recycling cargo from endosomes. Nat. Chem. Biol. 2018, 14, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Ciraolo, E.; Gulluni, F.; Hirsch, E. Methods to Measure the Enzymatic Activity of PI3Ks. Methods Enzymol. 2014, 543, 115–140. [Google Scholar] [CrossRef]
- Gozzelino, L.; De Santis, M.C.; Gulluni, F.; Hirsch, E.; Martini, M. PI(3,4)P2 Signaling in Cancer and Metabolism. Front. Oncol. 2020, 10, 360. [Google Scholar] [CrossRef] [Green Version]
- Posor, Y.; Eichhorn-Gruenig, M.; Puchkov, D.; Schöneberg, J.; Ullrich, A.; Lampe, A.; Müller, R.; Zarbakhsh, S.; Gulluni, F.; Hirsch, E.; et al. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nat. Cell Biol. 2013, 499, 233–237. [Google Scholar] [CrossRef]
- Wang, H.; Lo, W.-T.; Žagar, A.V.; Gulluni, F.; Lehmann, M.; Scapozza, L.; Haucke, V.; Vadas, O. Autoregulation of Class II Alpha PI3K Activity by Its Lipid-Binding PX-C2 Domain Module. Mol. Cell 2018, 71, 343–351.e4. [Google Scholar] [CrossRef] [Green Version]
- Virbasius, J.V.; Guilherme, A.; Czech, M.P. Mouse p170 is a novel phosphatidylinositol 3-kinase containing a C2 domain. J. Biol. Chem. 1996, 271, 13304–13307. [Google Scholar] [CrossRef] [Green Version]
- Domin, J.; Pages, F.; Volinia, S.; Rittenhouse, S.E.; Zvelebil, M.J.; Stein, R.C.; Waterfield, M.D. Cloning of a human phosphoinositide 3-kinase with a C2 domain that displays reduced sensitivity to the inhibitor wortmannin. Biochem. J. 1997, 326, 139–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martini, M.; Ciraolo, E.; Gulluni, F.; Hirsch, E. Targeting PI3K in Cancer: Any Good News? Front. Oncol. 2013, 3, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Wulfkuhle, J.; Zhang, H.; Gu, P.; Yang, Y.; Deng, J.; Margolick, J.B.; Liotta, L.A.; Petricoin, E.; Zhang, Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. USA 2007, 104, 16158–16163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulluni, F.; Martini, M.; De Santis, M.C.; Campa, C.C.; Ghigo, A.; Margaria, J.P.; Ciraolo, E.; Franco, I.; Ala, U.; Annaratone, L.; et al. Mitotic Spindle Assembly and Genomic Stability in Breast Cancer Require PI3K-C2α Scaffolding Function. Cancer Cell 2017, 32, 444–459.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chikh, A.; Ferro, R.; Abbott, J.J.; Piñeiro, R.; Buus, R.; Iezzi, M.; Ricci, F.; Bergamaschi, D.; Ostano, P.; Chiorino, G.; et al. Class II phosphoinositide 3-kinase C2β regulates a novel signaling pathway involved in breast cancer progression. Oncotarget 2016, 7, 18325–18345. [Google Scholar] [CrossRef] [Green Version]
- Domin, J.; Harper, L.; Aubyn, D.; Wheeler, M.; Florey, O.; Haskard, D.; Yuan, M.; Zicha, D. The class II phosphoinositide 3-kinase PI3K-C2β regulates cell migration by a PtdIns(3)P dependent mechanism. J. Cell. Physiol. 2005, 205, 452–462. [Google Scholar] [CrossRef]
- Katso, R.M.; Pardo, O.; Palamidessi, A.; Franz, C.M.; Marinov, M.; De Laurentiis, A.; Downward, J.; Scita, G.; Ridley, A.J.; Waterfield, M.D.; et al. Phosphoinositide 3-Kinase C2β Regulates Cytoskeletal Organization and Cell Migration via Rac-dependent Mechanisms. Mol. Biol. Cell 2006, 17, 3729–3744. [Google Scholar] [CrossRef]
- Maffucci, T.; Cooke, F.T.; Foster, F.M.; Traer, C.J.; Fry, M.; Falasca, M. Class II phosphoinositide 3-kinase defines a novel signaling pathway in cell migration. J. Cell Biol. 2005, 169, 789–799. [Google Scholar] [CrossRef]
- Falasca, M.; Hamilton, J.R.; Selvadurai, M.; Sundaram, K.; Adamska, A.; Thompson, P.E. Class II Phosphoinositide 3-Kinases as Novel Drug Targets. J. Med. Chem. 2016, 60, 47–65. [Google Scholar] [CrossRef]
- Knight, Z.; Gonzalez, B.; Feldman, M.E.; Zunder, E.R.; Goldenberg, D.D.; Williams, O.; Loewith, R.; Stokoe, D.; Balla, A.; Toth, B.; et al. A Pharmacological Map of the PI3-K Family Defines a Role for p110α in Insulin Signaling. Cell 2006, 125, 733–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, D.; Dan, S.; Yamazaki, K.; Yamori, T. Inhibition profiles of phosphatidylinositol 3-kinase inhibitors against PI3K superfamily and human cancer cell line panel JFCR. Eur. J. Cancer 2010, 46, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Boller, D.; Doepfner, K.T.; De Laurentiis, A.; Guerreiro, A.S.; Marinov, M.; Shalaby, T.; Depledge, P.; Robson, A.; Saghir, N.; Hayakawa, M.; et al. Targeting PI3KC2beta impairs proliferation and survival in acute leukemia, brain tumours and neuroendocrine tumours. Anticancer Res 2012, 32, 3015–3027. [Google Scholar] [PubMed]
- Selvadurai, M.V.; Moon, M.J.; Mountford, S.J.; Ma, X.; Zheng, Z.; Jennings, I.G.; Setiabakti, N.M.; Iman, R.P.; Brazilek, R.J.; Abidin, N.A.Z.; et al. Disrupting the platelet internal membrane via PI3KC2α inhibition impairs thrombosis independently of canonical platelet activation. Sci. Transl. Med. 2020, 12, eaar8430. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Prever, L.; Hirsch, E.; Gulluni, F. Targeting PI3K/AKT/mTOR Signaling Pathway in Breast Cancer. Cancers 2021, 13, 3517. https://doi.org/10.3390/cancers13143517
Li H, Prever L, Hirsch E, Gulluni F. Targeting PI3K/AKT/mTOR Signaling Pathway in Breast Cancer. Cancers. 2021; 13(14):3517. https://doi.org/10.3390/cancers13143517
Chicago/Turabian StyleLi, Huayi, Lorenzo Prever, Emilio Hirsch, and Federico Gulluni. 2021. "Targeting PI3K/AKT/mTOR Signaling Pathway in Breast Cancer" Cancers 13, no. 14: 3517. https://doi.org/10.3390/cancers13143517
APA StyleLi, H., Prever, L., Hirsch, E., & Gulluni, F. (2021). Targeting PI3K/AKT/mTOR Signaling Pathway in Breast Cancer. Cancers, 13(14), 3517. https://doi.org/10.3390/cancers13143517