
Claisened Hexafluoro Inhibits Metastatic Spreading of Amoeboid Melanoma Cells

 , , , ,
, , , ,  ,
,  , ,
, ,  and
and 
Abstract
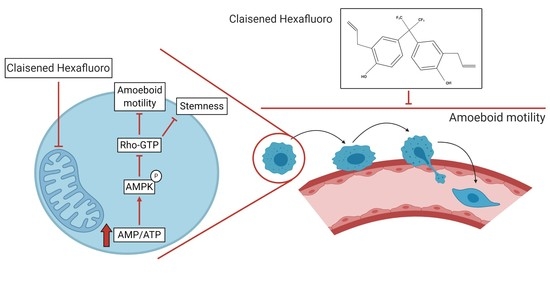
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Materials
2.2. Cell Viability Assay
2.3. Western Blotting
2.4. RhoA Activity Assay
2.5. Invasion Assay
2.6. 3D Invasion Assay
2.7. Cell Migration in Three-Dimensional Collagen Matrices
2.8. Adhesion Assay
2.9. Trans-Endothelial Migration Assay
2.10. Flow Cytometric Analysis
2.11. Confocal Image Acquisition and Analysis
2.12. RT-PCR
2.13. Melanosphere Formation
2.14. Seahorse XFe96 Metabolic Assays
2.15. Lactate Quantification Assay
2.16. AMP/ATP Quantification Assay
2.17. Lung Colonization Assay
2.18. Statistical Analysis
3. Results
3.1. CH Inhibits Amoeboid Motility and Invasive Ability of Melanoma Cells
3.2. CH Decreases Adhesion Abilities and Trans-Endothelium Migration of Amoeboid Melanoma Cells
3.3. CH Inhibits the Stemness Features of A375M6 Cells
3.4. CH Inhibits the Mitochondrial Activity and ATP Production of Melanoma Cells
3.5. CH Affects AMPK Signaling
3.6. CH Decreases the In Vivo Lung Metastasis Formation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef]
- Friedl, P.; Zänker, K.S.; Bröcker, E.B. Cell migration strategies in 3-D extracellular matrix: Differences in morphology, cell matrix interactions, and integrin function. Microsc. Res. Tech. 1998, 43, 369–378. [Google Scholar] [CrossRef]
- Renkawitz, J.; Schumann, K.; Weber, M.; Lämmermann, T.; Pflicke, H.; Piel, M.; Polleux, J.; Spatz, J.P.; Sixt, M. Adaptive force transmission in amoeboid cell migration. Nat. Cell Biol. 2009, 11, 1438–1443. [Google Scholar] [CrossRef] [PubMed]
- Polte, T.R.; Eichler, G.S.; Wang, N.; Ingber, D.E. Extracellular matrix controls myosin light chain phosphorylation and cell contractility through modulation of cell shape and cytoskeletal prestress. Am. J. Physiol. Cell Physiol. 2004, 286. [Google Scholar] [CrossRef] [Green Version]
- Leader, W.M.; Stopak, D.; Harris, A.K. Increased contractile strenght and tightened adhesions to the substratum result from reverse transformation of CHO cells by dibutyryl cyclic adenosine monophosphate. J. Cell Sci. 1983, 64, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Moreno, V.; Gadea, G.; Ahn, J.; Paterson, H.; Marra, P.; Pinner, S.; Sahai, E.; Marshall, C.J. Rac Activation and Inactivation Control Plasticity of Tumor Cell Movement. Cell 2008, 135, 510–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolyada, A.Y.; Riley, K.N.; Herman, I.M. Rho GTPase signaling modulates cell shape and contractile phenotype in an isoactin-specific manner. Am. J. Physiol. Cell Physiol. 2003, 285. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Wolf, K. Plasticity of cell migration: A multiscale tuning model. J. Cell Biol. 2010, 188, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Pelham, R.J.; Wang, Y.L. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc. Natl. Acad. Sci. USA 1997, 94, 13661–13665. [Google Scholar] [CrossRef] [Green Version]
- Young, W.C.; Herman, I.M. Extracellular matrix modulation of endothelial cell shape and motility following injury in vitro. J. Cell Sci. 1985, 73, 19–32. [Google Scholar] [CrossRef]
- Taddei, M.L.; Giannoni, E.; Morandi, A.; Ippolito, L.; Ramazzotti, M.; Callari, M.; Gandellini, P.; Chiarugi, P. Mesenchymal to amoeboid transition is associated with stem-like features of melanoma cells. Cell Commun. Signal. 2014, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Borgmann, S.; Bröcker, E.B. Amoeboid leukocyte crawling through extracellular matrix: Lessons from the Dictyostelium paradigm of cell movement. J. Leukoc. Biol. 2001, 70, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Lämmermann, T.; Sixt, M. Mechanical modes of “amoeboid” cell migration. Curr. Opin. Cell Biol. 2009, 21, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Hegerfeldt, Y.; Tusch, M. Collective cell migration in morphogenesis and cancer. Int. J. Dev. Biol. 2004, 48, 441–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, D.; Kurisu, S.; Takenawa, T. Regulation of cancer cell motility through actin reorganization. Cancer Sci. 2005, 96, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Worthylake, R.A.; Lemoine, S.; Watson, J.M.; Burridge, K. RhoA is required for monocyte tail retraction during transendothelial migration. J. Cell Biol. 2001, 154, 147–160. [Google Scholar] [CrossRef] [Green Version]
- Michielin, O.; Atkins, M.B.; Koon, H.B.; Dummer, R.; Ascierto, P.A. Evolving impact of long-term survival results on metastatic melanoma treatment. J. Immunother. Cancer 2020, 8, 948. [Google Scholar] [CrossRef] [PubMed]
- Parri, M.; Taddei, M.L.; Bianchini, F.; Calorini, L.; Chiarugi, P. EphA2 reexpression prompts invasion of melanoma cells shifting from mesenchymal to amoeboid-like motility style. Cancer Res. 2009, 69, 2072–2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, S.; te Boekhorst, V.; Odenthal, J.; Bianchi, R.; van Helvert, S.; Ikenberg, K.; Ilina, O.; Stoma, S.; Xandry, J.; Jiang, L.; et al. Hypoxia Induces a HIF-1-Dependent Transition from Collective-to-Amoeboid Dissemination in Epithelial Cancer Cells. Curr. Biol. 2017, 27, 392–400. [Google Scholar] [CrossRef] [Green Version]
- Liou, K.T.; Shen, Y.C.; Chen, C.F.; Tsao, C.M.; Tsai, S.K. Honokiol protects rat brain from focal cerebral ischemia-reperfusion injury by inhibiting neutrophil infiltration and reactive oxygen species production. Brain Res. 2003, 992, 159–166. [Google Scholar] [CrossRef]
- Tsai, S.K.; Huang, C.H.; Huang, S.S.; Hung, L.M.; Hong, C.Y. Antiarrhythmic effect of magnolol and honokiol during acute phase of coronary occlusion in anesthetized rats: Influence of L-NAME and aspirin. Pharmacology 1999, 59, 227–233. [Google Scholar] [CrossRef]
- Zhao, C.; Liu, Z.Q. Comparison of antioxidant abilities of magnolol and honokiol to scavenge radicals and to protect DNA. Biochimie 2011, 93, 1755–1760. [Google Scholar] [CrossRef]
- Talarek, S.; Listos, J.; Barreca, D.; Tellone, E.; Sureda, A.; Nabavi, S.F.; Braidy, N.; Nabavi, S.M. Neuroprotective effects of honokiol: From chemistry to medicine. BioFactors 2017, 43, 760–769. [Google Scholar] [CrossRef]
- Bai, X.; Cerimele, F.; Ushio-Fukai, M.; Waqas, M.; Campbell, P.M.; Govindarajan, B.; Der, C.J.; Battle, T.; Frank, D.A.; Ye, K.; et al. Honokiol, a small molecular weight natural product, inhibits angiogenesis in vitro and tumor growth in vivo. J. Biol. Chem. 2003, 278, 35501–35507. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Chen, L.J.; Liu, L.; Chen, X.; Chen, P.; Yang, G.L.; Hou, W.L.; Tang, M.H.; Zhang, F.; Wang, X.H.; et al. Liposomal honokiol, a potent anti-angiogenesis agent, in combination with radiotherapy produces a synergistic antitumor efficacy without increasing toxicity. Exp. Mol. Med. 2008, 40, 617–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, B.; Lee, Y.; Ku, Y.; Bae, K.; Chung, C.P. Antimicrobial activity of magnolol and honokiol against periodontopathic microorganisms. Planta Med. 1998, 64, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.Y.; Tsai, C.C.; Chen, C.P.; Huang, J.S.; Lin, C.C. Antimicrobial activity of honokiol and magnolol isolated from Magnolia officinalis. Phyther. Res. 2001, 15, 139–141. [Google Scholar] [CrossRef]
- Park, J.; Lee, J.; Jung, E.; Park, Y.; Kim, K.; Park, B.; Jung, K.; Park, E.; Kim, J.; Park, D. In vitro antibacterial and anti-inflammatory effects of honokiol and magnolol against Propionibacterium sp. Eur. J. Pharmacol. 2004, 496, 189–195. [Google Scholar] [CrossRef]
- Qiang, L.Q.; Wang, C.P.; Wang, F.M.; Pan, Y.; Yi, L.T.; Zhang, X.; Kong, L.D. Combined administration of the mixture of honokiol and magnolol and ginger oil evokes antidepressant-like synergism in rats. Arch. Pharm. Res. 2009, 32, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Yi, L.T.; Pan, Y.; Wang, X.; Li, Y.C.; Li, J.M.; Wang, C.P.; Kong, L.D. Antidepressant-like effects of the mixture of honokiol and magnolol from the barks of Magnolia officinalis in stressed rodents. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2008, 32, 715–725. [Google Scholar] [CrossRef]
- Eastham, L.L.; Howard, C.M.; Balachandran, P.; Pasco, D.S.; Claudio, P.P. Eating Green: Shining Light on the Use of Dietary Phytochemicals as a Modern Approach in the Prevention and Treatment of Head and Neck Cancers. Curr. Top. Med. Chem. 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Guillermo-Lagae, R.; Santha, S.; Thomas, M.; Zoelle, E.; Stevens, J.; Kaushik, R.S.; Dwivedi, C. Antineoplastic effects of honokiol on melanoma. BioMed Res. Int. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.S.; Tsai, C.H.; Hsieh, M.S.; Tsai, S.C.; Jan, Y.J.; Lin, W.Y.; Lai, D.W.; Wu, S.M.; Hsing, H.Y.; Arbiser, J.L.; et al. Exploiting Honokiol-induced ER stress CHOP activation inhibits the growth and metastasis of melanoma by suppressing the MITF and β-catenin pathways. Cancer Lett. 2019, 442, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Die Wang, W.; Shang, Y.; Li, Y.; Chen, S.-Z. Honokiol inhibits breast cancer cell metastasis by blocking EMT through modulation of Snail/Slug protein translation. Acta Pharmacol. Sin. 2019, 40, 1219–1227. [Google Scholar] [CrossRef]
- Akamata, K.; Wei, J.; Bhattacharyya, M.; Cheresh, P.; Bonner, M.Y.; Arbiser, J.L.; Raparia, K.; Gupta, M.P.; Kamp, D.W.; Varga, J. SIRT3 is attenuated in systemic sclerosis skin and lungs, and its pharmacologic activation mitigates organ fibrosis. Oncotarget 2016, 7, 69321–69336. [Google Scholar] [CrossRef] [Green Version]
- Bonner, M.Y.; Karlsson, I.; Rodolfo, M.; Arnold, R.S.; Vergani, E.; Arbiser, J.L. Honokiol bis-dichloroacetate (Honokiol DCA) demonstrates activity in vemurafenib-resistant melanoma in vivo. Oncotarget 2016, 7, 12857–12868. [Google Scholar] [CrossRef]
- Laurenzana, A.; Chillà, A.; Luciani, C.; Peppicelli, S.; Biagioni, A.; Bianchini, F.; Tenedini, E.; Torre, E.; Mocali, A.; Calorini, L.; et al. uPA/uPAR system activation drives a glycolytic phenotype in melanoma cells. Int. J. Cancer 2017, 141, 1190–1200. [Google Scholar] [CrossRef] [Green Version]
- Taddei, M.L.; Parri, M.; Angelucci, A.; Bianchini, F.; Marconi, C.; Giannoni, E.; Raugei, G.; Bologna, M.; Calorini, L.; Chiarugi, P. EphA2 induces metastatic growth regulating amoeboid motility and clonogenic potential in prostate carcinoma cells. Mol. Cancer Res. 2011, 9, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Bröcker, E.B. Reconstructing leukocyte migration in 3D extracellular matrix by time-lapse videomicroscopy and computer-assisted tracking. Methods Mol. Biol. 2004, 239, 77–90. [Google Scholar] [CrossRef]
- Pietrovito, L.; Comito, G.; Parri, M.; Giannoni, E.; Chiarugi, P.; Taddei, M.L. Zoledronic Acid Inhibits the RhoA-mediated Amoeboid Motility of Prostate Cancer Cells. Curr. Cancer Drug Targets 2019, 19, 807–816. [Google Scholar] [CrossRef]
- Ahn, J.; Sanz-Moreno, V.; Marshall, C.J. The metastasis gene NEDD9 product acts through integrin β3 and Src to promote mesenchymal motility and inhibit amoeboid motility. J. Cell Sci. 2012, 125, 1814–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz-Moreno, V.; Gaggioli, C.; Yeo, M.; Albrengues, J.; Wallberg, F.; Viros, A.; Hooper, S.; Mitter, R.; Féral, C.C.; Cook, M.; et al. ROCK and JAK1 Signaling Cooperate to Control Actomyosin Contractility in Tumor Cells and Stroma. Cancer Cell 2011, 20, 229–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, E.A.; Golub, T.R.; Lander, E.S.; Hynes, R.O. Genomic analysis of metastasis reveals an essential role for RhoC. Nature 2000, 406, 532–535. [Google Scholar] [CrossRef]
- Lorentzen, H.F. Targeted therapy for malignant melanoma. Curr. Opin. Pharmacol. 2019, 46, 116–121. [Google Scholar] [CrossRef]
- Sadok, A.; McCarthy, A.; Caldwell, J.; Collins, I.; Garrett, M.D.; Yeo, M.; Hooper, S.; Sahai, E.; Kuemper, S.; Mardakheh, F.K.; et al. Rho kinase inhibitors block melanoma cell migration and inhibit metastasis. Cancer Res. 2015, 75, 2272–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udayakumar, D.; Zhang, G.; Ji, Z.; Njauw, C.N.; Mroz, P.; Tsao, H. Epha2 is a critical oncogene in melanoma. Oncogene 2011, 30, 4921–4929. [Google Scholar] [CrossRef] [Green Version]
- Giannoni, E.; Taddei, M.L.; Parri, M.; Bianchini, F.; Santosuosso, M.; Grifantini, R.; Fibbi, G.; Mazzanti, B.; Calorini, L.; Chiarugi, P. EphA2-mediated mesenchymal–amoeboid transition induced by endothelial progenitor cells enhances metastatic spread due to cancer-associated fibroblasts. J. Mol. Med. 2013, 91, 103–115. [Google Scholar] [CrossRef]
- Zhang, Y.; Weinberg, R.A. Epithelial-to-mesenchymal transition in cancer: Complexity and opportunities. Front. Med. 2018, 12, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Hernandez, I.; Maiques, O.; Kohlhammer, L.; Cantelli, G.; Perdrix-Rosell, A.; Monger, J.; Fanshawe, B.; Bridgeman, V.L.; Karagiannis, S.N.; Penin, R.M.; et al. WNT11-FZD7-DAAM1 signalling supports tumour initiating abilities and melanoma amoeboid invasion. Nat. Commun. 2020, 11, 5315. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noh, K.H.; Kim, B.W.; Song, K.H.; Cho, H.; Lee, Y.H.; Kim, J.H.; Chung, J.Y.; Kim, J.H.; Hewitt, S.M.; Seong, S.Y.; et al. Nanog signaling in cancer promotes stem-like phenotype and immune evasion. J. Clin. Investig. 2012, 122, 4077–4093. [Google Scholar] [CrossRef] [Green Version]
- Perego, M.; Tortoreto, M.; Tragni, G.; Mariani, L.; Deho, P.; Carbone, A.; Santinami, M.; Patuzzo, R.; Della Mina, P.; Villa, A.; et al. Heterogeneous phenotype of human melanoma cells with in vitro and in vivo features of tumor-initiating cells. J. Investig. Dermatol. 2010, 130, 1877–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porporato, P.E.; Payen, V.L.; Pérez-Escuredo, J.; De Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.; Lee, Y.; Wang, Y.; You, M. Honokiol targets mitochondria to halt cancer progression and metastasis. Mol. Nutr. Food Res. 2016, 60, 1383–1395. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-J.; Kuo, C.-H.; Chen, S.-H.; Lin, C.-Y.; Lee, Y.-R. Honokiol inhibits in vitro and in vivo growth of oral squamous cell carcinoma through induction of apoptosis, cell cycle arrest and autophagy. J. Cell. Mol. Med. 2018, 22, 1894–1908. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ren, X.; Shi, M.; Jiang, Z.; Wang, H.; Su, Q.; Liu, Q.; Li, G.; Jiang, G. Downregulation of STAT3 and activation of MAPK are involved in the induction of apoptosis by HNK in glioblastoma cell line U87. Oncol. Rep. 2014, 32, 2038–2046. [Google Scholar] [CrossRef]
- Zhao, B.; Qiang, L.; Joseph, J.; Kalyanaraman, B.; Viollet, B.; He, Y.Y. Mitochondrial dysfunction activates the AMPK signaling and autophagy to promote cell survival. Genes Dis. 2016, 3, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. Sensing of energy and nutrients by AMP-activated protein kinase. Am. J. Clin. Nutr. 2011, 93, 891S–896S. [Google Scholar] [CrossRef] [Green Version]
- Schaffer, B.E.; Levin, R.S.; Hertz, N.T.; Maures, T.J.; Schoof, M.L.; Hollstein, P.E.; Benayoun, B.A.; Banko, M.R.; Shaw, R.J.; Shokat, K.M.; et al. Identification of AMPK Phosphorylation Sites Reveals a Network of Proteins Involved in Cell Invasion and Facilitates Large-Scale Substrate Prediction. Cell Metab. 2015, 22, 907–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, B.; Seibel, W. Compound C/Dorsomorphin: Its use and misuse as an AMPK inhibitor. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2018; Volume 1732, pp. 195–202. [Google Scholar]
- Elia, I.; Doglioni, G.; Fendt, S.-M. Metabolic Hallmarks of Metastasis Formation. Trends Cell Biol. 2018, 28, 673–684. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, K.; Hignett, E.; Khachemoune, A. Current and emerging treatment options for metastatic melanoma: A focused review. Dermatol. Online J. 2020, 26. [Google Scholar] [CrossRef]
- Sahai, E.; Marshall, C.J. Differing modes for tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 2003, 5, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Hegerfeldt, Y.; Tusch, M.; Bröcker, E.B.; Friedl, P. Collective cell movement in primary melanoma explants: Plasticity of cell-cell interaction, β1-integrin function, and migration strategies. Cancer Res. 2002, 62, 2125–2130. [Google Scholar]
- Charras, G.T.; Hu, C.K.; Coughlin, M.; Mitchison, T.J. Reassembly of contractile actin cortex in cell blebs. J. Cell Biol. 2006, 175, 477–490. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, S.; Paterson, H.F.; Marshall, C.J. Cdc42-MRCK and Rho-ROCK signalling cooperate in myosin phosphorylation and cell invasion. Nat. Cell Biol. 2005, 7, 255–261. [Google Scholar] [CrossRef]
- Wyckoff, J.B.; Pinner, S.E.; Gschmeissner, S.; Condeelis, J.S.; Sahai, E. ROCK- and Myosin-Dependent Matrix Deformation Enables Protease-Independent Tumor-Cell Invasion In Vivo. Curr. Biol. 2006, 16, 1515–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orgazy, J.L.; Herraizy, C.; Sanz-Moreno, V. Rho GTPases modulate malignant transformation of tumor cells. Small GTPases 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Misek, S.A.; Appleton, K.M.; Dexheimer, T.S.; Lisabeth, E.M.; Lo, R.S.; Larsen, S.D.; Gallo, K.A.; Neubig, R.R. Rho-mediated signaling promotes BRAF inhibitor resistance in de-differentiated melanoma cells. Oncogene 2020, 39, 1466–1483. [Google Scholar] [CrossRef]
- Routhier, A.; Astuccio, M.; Lahey, D.; Monfredo, N.; Johnson, A.; Callahan, W.; Partington, A.; Fellows, K.; Ouellette, L.; Zhidro, S.; et al. Pharmacological inhibition of Rho-kinase signaling with Y-27632 blocks melanoma tumor growth. Oncol. Rep. 2010, 23, 861–867. [Google Scholar] [PubMed]
- Arbiser, J.L.; Bonner, M.Y.; Gilbert, L.C. Targeting the duality of cancer. NPJ Precis. Oncol. 2017, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trotta, A.P.; Gelles, J.D.; Serasinghe, M.N.; Loi, P.; Arbiser, J.L.; Chipuk, J.E. Disruption of mitochondrial electron transport chain function potentiates the pro-apoptotic effects of MAPK inhibition. J. Biol. Chem. 2017, 292, 11727–11739. [Google Scholar] [CrossRef] [Green Version]
- Zhou, A.Y.; Johnson, D.B. Combinatorial Therapies in Melanoma: MAPK Inhibitors and Beyond. Am. J. Clin. Dermatol. 2018, 19, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Ruocco, M.R.; Avagliano, A.; Granato, G.; Vigliar, E.; Masone, S.; Montagnani, S.; Arcucci, A. Metabolic flexibility in melanoma: A potential therapeutic target. Semin. Cancer Biol. 2019, 59, 187–207. [Google Scholar] [CrossRef]
- Ratnikov, B.I.; Scott, D.A.; Osterman, A.L.; Smith, J.W.; Ronai, Z.A. Metabolic rewiring in melanoma. Oncogene 2017, 36, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Corazao-Rozas, P.; Guerreschi, P.; André, F.; Gabert, P.-E.; Lancel, S.; Dekiouk, S.; Fontaine, D.; Tardivel, M.; Savina, A.; Quesnel, B.; et al. Mitochondrial oxidative phosphorylation controls cancer cell’s life and death decisions upon exposure to MAPK inhibitors. Oncotarget 2016, 7, 39473–39485. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Goñi, T.; Galobart, T.C.; Wantuch, S.; Normantaite, D.; Leach, M.O.; Whittaker, S.R.; Beloueche-Babari, M. Increased inflammatory lipid metabolism and anaplerotic mitochondrial activation follow acquired resistance to vemurafenib in BRAF-mutant melanoma cells. Br. J. Cancer 2020, 122, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Gayard, M.; Guilluy, C.; Rousselle, A.; Viollet, B.; Henrion, D.; Pacaud, P.; Loirand, G.; Rolli-Derkinderen, M. AMPK Alpha 1-induced RhoA phosphorylation mediates vasoprotective effect of estradiol. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2634–2642. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Wang, H.; Yang, Y.; Guo, S.; Zhang, W.; Liu, Y.; Yi, X.; Ma, J.; Zhao, T.; Liu, L.; et al. Down-regulated miR-23a contributes to the metastasis of cutaneous melanoma by promoting autophagy. Theranostics 2017, 7, 2231–2249. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leo, A.; Pranzini, E.; Pietrovito, L.; Pardella, E.; Parri, M.; Cirri, P.; Bruno, G.; Calvani, M.; Peppicelli, S.; Torre, E.; et al. Claisened Hexafluoro Inhibits Metastatic Spreading of Amoeboid Melanoma Cells. Cancers 2021, 13, 3551. https://doi.org/10.3390/cancers13143551
Leo A, Pranzini E, Pietrovito L, Pardella E, Parri M, Cirri P, Bruno G, Calvani M, Peppicelli S, Torre E, et al. Claisened Hexafluoro Inhibits Metastatic Spreading of Amoeboid Melanoma Cells. Cancers. 2021; 13(14):3551. https://doi.org/10.3390/cancers13143551
Chicago/Turabian StyleLeo, Angela, Erica Pranzini, Laura Pietrovito, Elisa Pardella, Matteo Parri, Paolo Cirri, Gennaro Bruno, Maura Calvani, Silvia Peppicelli, Eugenio Torre, and et al. 2021. "Claisened Hexafluoro Inhibits Metastatic Spreading of Amoeboid Melanoma Cells" Cancers 13, no. 14: 3551. https://doi.org/10.3390/cancers13143551
APA StyleLeo, A., Pranzini, E., Pietrovito, L., Pardella, E., Parri, M., Cirri, P., Bruno, G., Calvani, M., Peppicelli, S., Torre, E., Sasaki, M., Yang, L., Zhu, L., Chiarugi, P., Raugei, G., Arbiser, J. L., & Taddei, M. L. (2021). Claisened Hexafluoro Inhibits Metastatic Spreading of Amoeboid Melanoma Cells. Cancers, 13(14), 3551. https://doi.org/10.3390/cancers13143551