
The Next-Generation of Combination Cancer Immunotherapy: Epigenetic Immunomodulators Transmogrify Immune Training to Enhance Immunotherapy

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Targets of Immunotherapy
1.2. Limitations of Immunotherapy
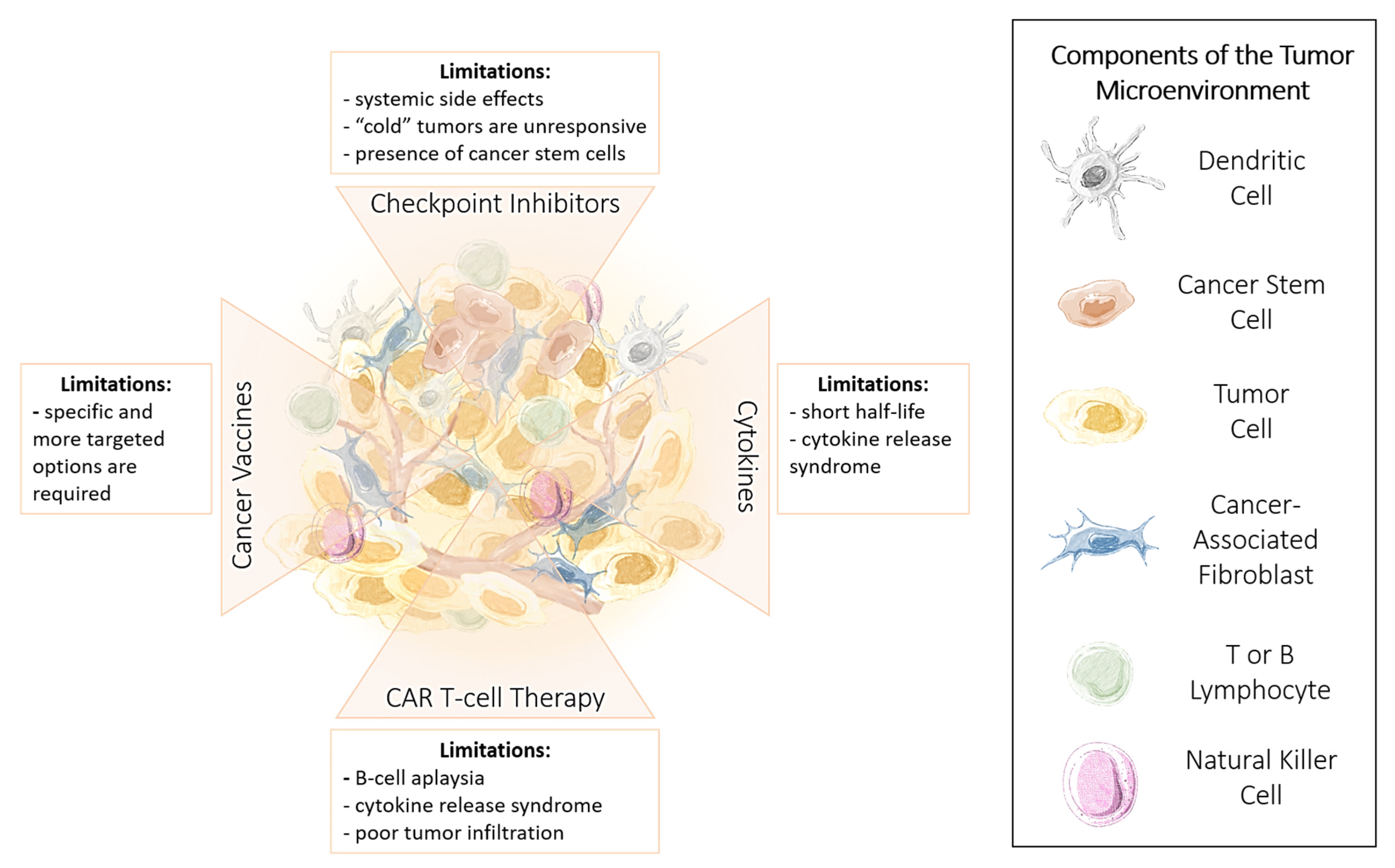
1.3. Advantages and Limitations of Cancer Immunotherapy
1.4. Immune Checkpoint Inhibitors
Limitations and Challenges of ICI Therapy
1.5. Cytokines
1.6. Chimeric Antigen Receptor (CAR) T Cells
1.7. Cancer Vaccines
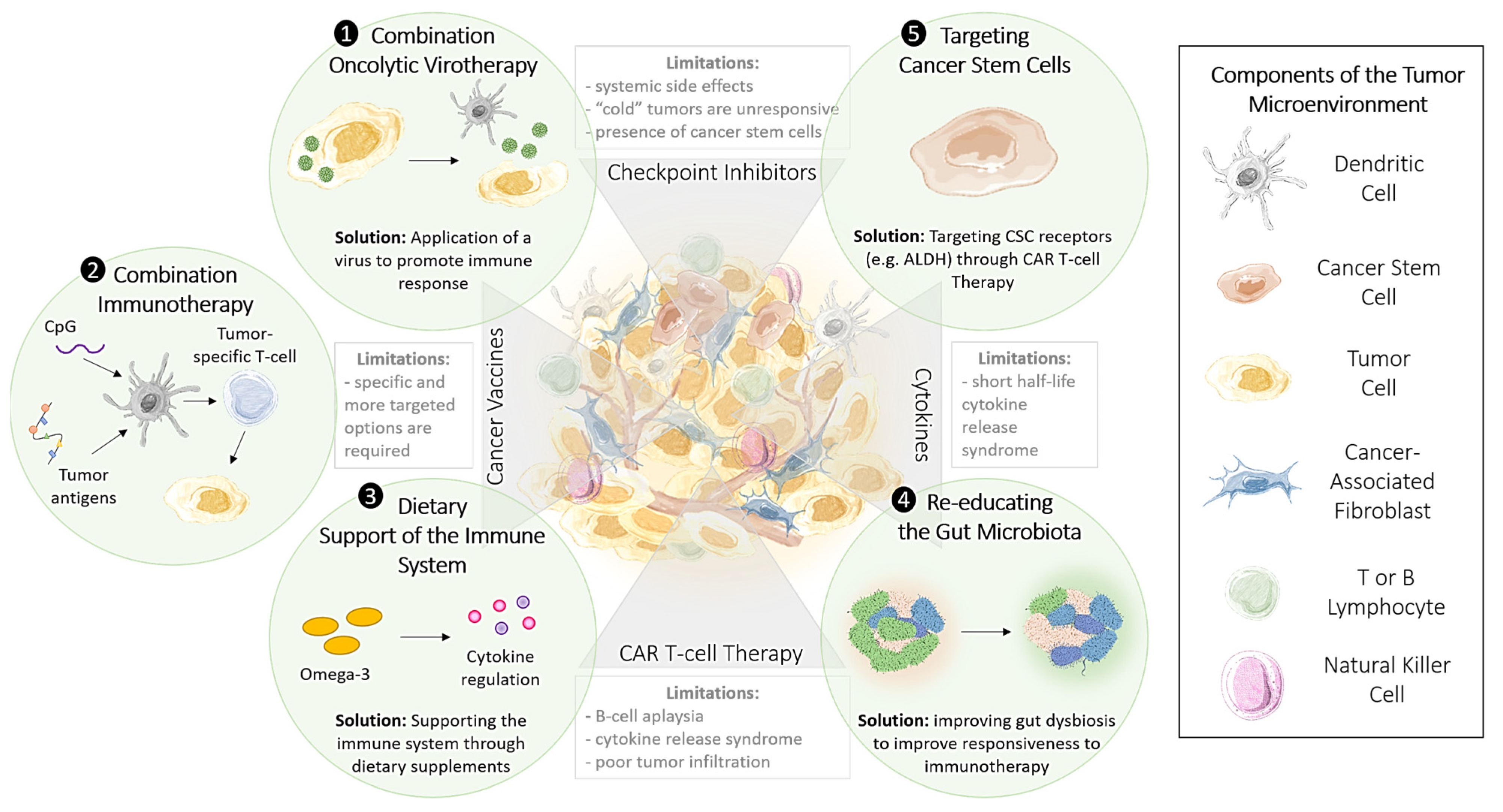
1.8. Methods to Improve the Effectiveness of Immunotherapy
1.9. Targeting Cancer Stem Cells to Enhance the Effectiveness of Immunotherapy
2. Training Immunity with Epigenetic Immunomodulators
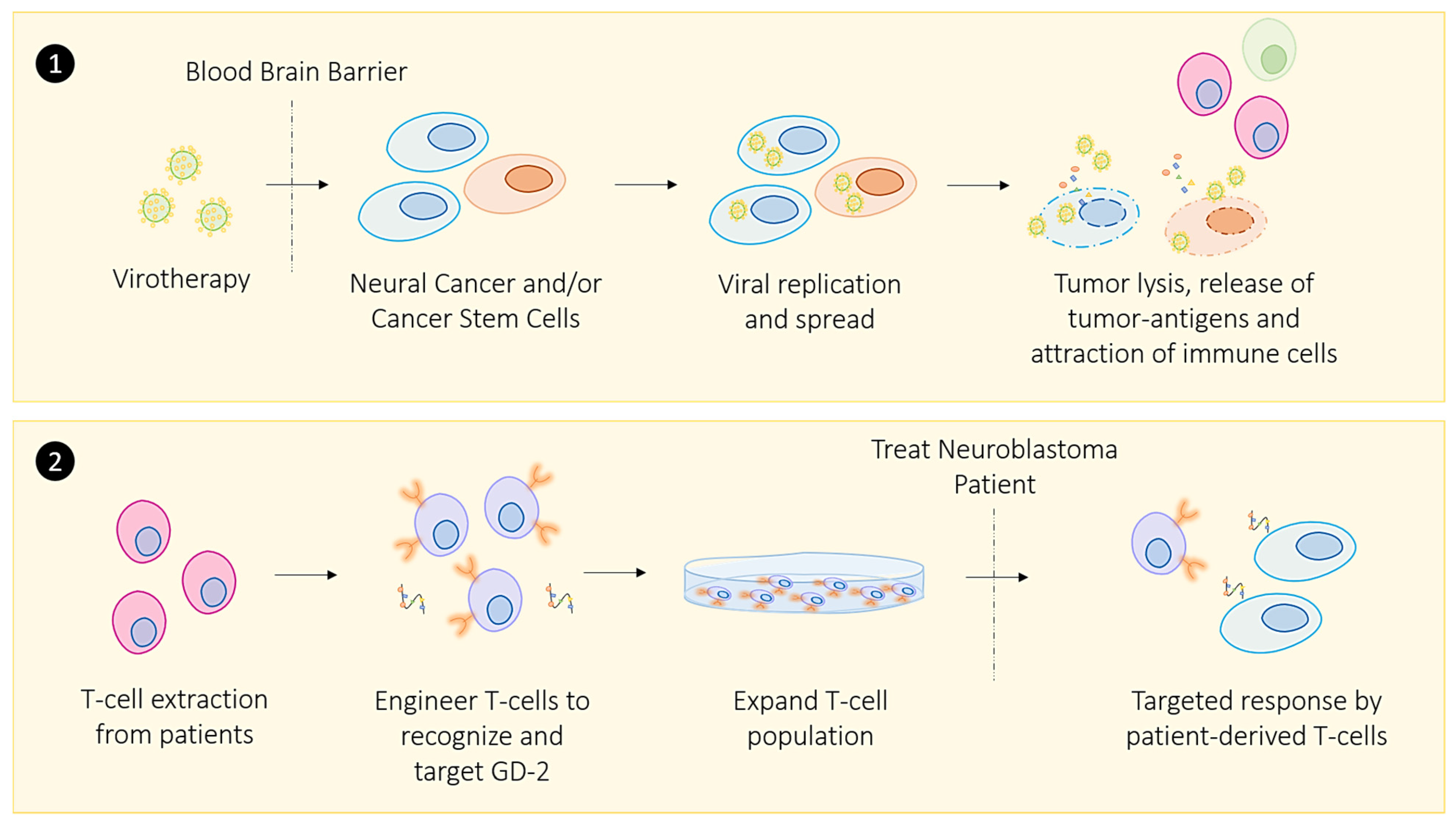
2.1. Oncolytic Viral Immune Training
2.2. Zika Viral Immune Training
2.3. COVID-19 Viral Immune Training in Convalescing Patients
2.4. Bacterial Immune Training
3. Immunomodulators Targeting Epigenetic Regulators
3.1. CpG Nucleotides: Combination Options to Increase the Effectiveness of Checkpoint Inhibitors
3.2. Restoring Epigenetic Reprogramming with Immunotherapy to Improve Therapeutic Responsiveness
3.3. Exercise-Induced Epigenetic Modifications in Genes
3.4. Targeting Cancer Stem Cells Through Telomerase-Targeted Immunotherapy
4. Harnessing the Gut Microbiota to Enhance the Immune Response
4.1. Diet and Nutrient Influence Epigenetic Modifications
4.2. Re-Educating the Gut Microbiota to Modulate the Immune Response
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef] [Green Version]
- De Angelis, M.L.; Francescangeli, F.; La Torre, F.; Zeuner, A. Stem Cell Plasticity and Dormancy in the Development of Cancer Therapy Resistance. Front. Oncol. 2019, 9, 626. [Google Scholar] [CrossRef] [Green Version]
- Gorodetska, I.; Kozeretska, I.; Dubrovska, A. BRCA Genes: The Role in Genome Stability, Cancer Stemness and Therapy Resistance. J. Cancer 2019, 10, 2109–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, C.U.; Shohet, J.M. Neuroblastoma: Molecular pathogenesis and therapy. Annu. Rev. Med. 2015, 66, 49–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betters, E.; Liu, Y.; Kjaeldgaard, A.; Sundström, E.; García-Castro, M.I. Analysis of early human neural crest development. Dev. Biol. 2010, 344, 578–592. [Google Scholar] [CrossRef]
- Cohn, S.L.; Pearson, A.D.J.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The international neuroblastoma risk group (INRG) classification system: An INRG task force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef] [PubMed]
- De Bernardi, B.; Mosseri, V.; Rubie, H.; Castel, V.; Foot, A.; Ladenstein, R.; Laureys, G.; Beck-Popovic, M.; De Lacerda, A.F.; Pearson, A.D.J.; et al. Treatment of localised resectable neuroblastoma. Results of the LNESG1 study by the SIOP Europe Neuroblastoma Group. Br. J. Cancer 2008, 99, 1027–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maris, J.M. Recent Advances in Neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [Green Version]
- Smith, V.; Foster, J. High-Risk Neuroblastoma Treatment Review. Children 2018, 5, 114. [Google Scholar] [CrossRef] [Green Version]
- Khalil, D.N.; Smith, E.L.; Brentjens, R.J.; Wolchok, J.D. The future of cancer treatment: Immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 273–290. [Google Scholar] [CrossRef] [Green Version]
- Dholaria, B.; Hammond, W.; Shreders, A.; Lou, Y. Emerging therapeutic agents for lung cancer. J. Hematol. Oncol. 2016, 9, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Shao, C.; Shi, Y.; Han, W. Lessons learned from the blockade of immune checkpoints in cancer immuno-therapy. J. Hematol. Oncol. 2018, 11, 31. [Google Scholar] [CrossRef] [PubMed]
- Biteghe, F.A.N.; Mungra, N.; Chalomie, N.E.T.; Ndong, J.D.L.C.; Vignaux, G.; Padayachee, E.; Naran, K.; Barth, S. Advances in epidermal growth factor receptor specific immunotherapy: Lessons to be learned from armed antibodies. Oncotarget 2020, 11, 3531–3557. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, L.C.; Shah, A.A.; Bingham, C.O. Cancer immunotherapy-induced rheumatic diseases emerge as new clinical entities. RMD Open 2016, 2, e000321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, S.C.M.; Leighl, N.B. Hyperprogressive disease with immunotherapy: New directions. J. Thorac. Dis. 2019, 11, S1877–S1880. [Google Scholar] [CrossRef]
- Angelicola, S.; Ruzzi, F.; Landuzzi, L.; Scalambra, L.; Gelsomino, F.; Ardizzoni, A.; Nanni, P.; Lollini, P.-L.; Palladini, A. IFN-γ and CD38 in Hyperprogressive Cancer Development. Cancers 2021, 13, 309. [Google Scholar] [CrossRef] [PubMed]
- Camelliti, S.; Le Noci, V.; Bianchi, F.; Moscheni, C.; Arnaboldi, F.; Gagliano, N.; Balsari, A.; Garassino, M.C.; Tagliabue, E.; Sfondrini, L.; et al. Mechanisms of hyperprogressive disease after immune checkpoint inhibitor therapy: What we (don’t) know. J. Exp. Clin. Cancer Res. 2020, 39, 1–20. [Google Scholar] [CrossRef]
- Song, W.; Musetti, S.; Huang, L. Nanomaterials for cancer immunotherapy. Biomaterials 2017, 148, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Sambi, M.; Bagheri, L.; Szewczuk, M.R. Current Challenges in Cancer Immunotherapy: Multimodal Approaches to Improve Efficacy and Patient Response Rates. J. Oncol. 2019, 2019, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alatrash, G.; Jakher, H.; Stafford, P.D.; Mittendorf, E.A. Cancer immunotherapies, their safety and toxicity. Expert Opin. Drug Saf. 2013, 12, 631–645. [Google Scholar] [CrossRef]
- Vareki, S.M.; Garrigós, C.; Duran, I. Biomarkers of response to PD-1/PD-L1 inhibition. Crit. Rev. Oncol. Hematol. 2017, 116, 116–124. [Google Scholar] [CrossRef]
- Zugazagoitia, J.; Guedes, C.; Ponce, S.; Ferrer, I.; Molina-Pinelo, S.; Paz-Ares, L. Current Challenges in Cancer Treatment. Clin. Ther. 2016, 38, 1551–1566. [Google Scholar] [CrossRef] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Taube, J.M.; Galon, J.; Sholl, L.M.; Rodig, S.J.; Cottrell, T.R.; Giraldo, N.A.; Baras, A.S.; Patel, S.S.; Anders, R.A.; Rimm, D.L.; et al. Implications of the tumor immune microenvironment for staging and therapeutics. Mod. Pathol. 2018, 31, 214–234. [Google Scholar] [CrossRef]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nat. Cell Biol. 2005, 439, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.; Kuball, J.; Voelkl, S.; Wiendl, H.; Becker, B.; Walter, B.; Majdic, O.; Gajewski, T.F.; Theobald, M.; Andreesen, R. Blockade of PD-L1 (B7-H1) augments human tumor-specific T cell responses in vitro. Int. J. Cancer 2006, 119, 317–327. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cottrell, T.; Taube, J.M. PD-L1 and Emerging Biomarkers in PD-1/PD-L1 Blockade Therapy. Cancer J. 2018, 24, 41. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Giraldo, N.A.; Nguyen, P.; Engle, E.L.; Kaunitz, G.J.; Cottrell, T.R.; Berry, S.; Green, B.; Soni, A.; Cuda, J.D.; Stein, J.E.; et al. Multidimensional, quantitative assessment of PD-1/PD-L1 expression in patients with Merkel cell carcinoma and association with response to pembrolizumab. J. Immunother. Cancer 2018, 6, 99. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153. [Google Scholar] [CrossRef]
- Kraft, S.; Fernandez-Figueras, M.-T.; Richarz, N.A.; Flaherty, K.T.; Hoang, M.P. PDL1 expression in desmoplastic melanoma is associated with tumor aggressiveness and progression. J. Am. Acad. Dermatol. 2017, 77, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Cimino-Mathews, A.; Thompson, E.; Taube, J.M.; Ye, X.; Lu, Y.; Meeker, A.; Xu, H.; Sharma, R.; Lecksell, K.; Cornish, T.; et al. PD-L1 (B7-H1) expression and the immune tumor microenvironment in primary and metastatic breast carcinomas. Hum. Pathol. 2016, 47, 52–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalian, S.L. Targeting immune checkpoints in cancer therapy. JAMA 2017, 318, 1647–1648. [Google Scholar] [CrossRef]
- Webb, E.S.; Liu, P.; Baleeiro, R.; Lemoine, N.R.; Yuan, M.; Wang, Y.-H. Immune checkpoint inhibitors in cancer therapy. J. Biomed. Res. 2017, 32, 317–326. [Google Scholar] [CrossRef]
- Vargas, F.A.; Furness, A.J.S.; Litchfield, K.; Joshi, K.; Rosenthal, R.; Ghorani, E.; Solomon, I.; Lesko, M.H.; Ruef, N.; Roddie, C.; et al. Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell 2018, 33, 649–663.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Tang, F.; Liu, M.; Su, J.; Zhang, Y.; Wu, W.; Devenport, M.; Lazarski, C.A.; Zhang, P.; Wang, X.; et al. A reappraisal of CTLA-4 checkpoint blockade in cancer immunotherapy. Cell Res. 2018, 28, 416–432. [Google Scholar] [CrossRef] [Green Version]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti–CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef]
- Contardi, E.; Palmisano, G.L.; Tazzari, P.L.; Martelli, A.M.; Falà, F.; Fabbi, M.; Kato, T.; Lucarelli, E.; Donati, D.M.; Polito, L.; et al. CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. Int. J. Cancer 2005, 117, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Pistillo, M.P.; Tazzari, P.L.; Palmisano, G.L.; Pierri, I.; Bolognesi, A.; Ferlito, F.; Capanni, P.; Polito, L.; Ratta, M.; Pileri, S.; et al. CTLA-4 is not restricted to the lymphoid cell lineage and can function as a target molecule for apoptosis induction of leukemic cells. Blood 2002, 101, 202–209. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; Warshauer, J.T.; Bluestone, J.A. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat. Med. 2017, 23, 540–547. [Google Scholar] [CrossRef] [Green Version]
- Friedman, C.F.; Proverbs-Singh, T.A.; Postow, M.A. Treatment of the immune-related adverse effects of immune checkpoint inhibitors: A review. JAMA Oncol. 2016, 2, 1346–1353. [Google Scholar] [CrossRef]
- Naidoo, J.; Wang, X.; Woo, K.M.; Iyriboz, T.; Halpenny, D.; Cunningham, J.; Chaft, J.E.; Segal, N.H.; Callahan, M.K.; Lesokhin, A.M.; et al. Pneumonitis in Patients Treated with Anti–Programmed Death-1/Programmed Death Ligand 1 Therapy. J. Clin. Oncol. 2017, 35, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Byun, D.J.; Wolchok, J.D.; Rosenberg, L.M.; Girotra, M. Cancer immunotherapy—Immune checkpoint blockade and associated endocrinopathies. Nat. Rev. Endocrinol. 2017, 13, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef]
- Restifo, N.P.; Smyth, M.J.; Snyder, A. Acquired resistance to immunotherapy and future challenges. Nat. Rev. Cancer 2016, 16, 121. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Coulie, P.G.; Eynde, B.J.V.D.; Agostinis, P. Integrating Next-Generation Dendritic Cell Vaccines into the Current Cancer Immunotherapy Landscape. Trends Immunol. 2017, 38, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Dillman, R.O. Is there a role for therapeutic cancer vaccines in the age of checkpoint inhibitors? Hum. Vaccines Immunother-Apeutics 2017, 13, 528–532. [Google Scholar] [CrossRef] [Green Version]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taube, J.M.; Anders, R.A.; Young, G.D.; Xu, H.; Sharma, R.; McMiller, T.L.; Chen, S.; Klein, A.P.; Pardoll, D.M.; Topalian, S.L.; et al. Colocalization of Inflammatory Response with B7-H1 Expression in Human Melanocytic Lesions Supports an Adaptive Resistance Mechanism of Immune Escape. Sci. Transl. Med. 2012, 4, 127ra37. [Google Scholar] [CrossRef] [Green Version]
- Fleming, V.; Hu, X.; Weber, R.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front. Immunol. 2018, 9, 398. [Google Scholar] [CrossRef]
- Russo, G.L.; Moro, M.; Sommariva, M.; Cancila, V.; Boeri, M.; Centonze, G.; Ferro, S.; Ganzinelli, M.; Gasparini, P.; Huber, V.; et al. Antibody–Fc/FcR Interaction on Macrophages as a Mechanism for Hyperprogressive Disease in Non–small Cell Lung Cancer Subsequent to PD-1/PD-L1 Blockade. Clin. Cancer Res. 2019, 25, 989–999. [Google Scholar] [CrossRef] [Green Version]
- Mokhtari, R.B.; Qorri, B.; Baluch, N.; Sparaneo, A.; Fabrizio, F.P.; Muscarella, L.A.; Tyker, A.; Kumar, S.; Cheng, H.-L.M.; Szewczuk, M.R.; et al. Next-generation multimodality of nutrigenomic cancer therapy: Sulforaphane in combination with ac-etazolamide actively target bronchial carcinoid cancer in disabling the PI3K/Akt/mTOR survival pathway and inducing apoptosis. Oncotarget 2021, 12. [Google Scholar] [CrossRef]
- Nasir, A.; Bullo, M.M.H.; Ahmed, Z.; Imtiaz, A.; Yaqoob, E.; Jadoon, M.; Ahmed, H.; Afreen, A.; Yaqoob, S. Nutrigenomics: Epigenetics and cancer prevention: A comprehensive review. Crit. Rev. Food Sci. Nutr. 2014, 60, 1375–1387. [Google Scholar] [CrossRef]
- Green, M.; Monti, S.; Rodig, S.J.; Juszczyński, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010, 116, 3268–3277. [Google Scholar] [CrossRef] [Green Version]
- Lastwika, K.J.; Wilson, W.; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT–mTOR Pathway in Non–Small Cell Lung Cancer. Cancer Res. 2015, 76, 227–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, M.T.; Anderson, K.S.; Lenkiewicz, E.; Andreozzi, M.; Cunliffe, H.E.; Klassen, C.L.; Dueck, A.C.; McCullough, A.E.; Reddy, S.K.; Ramanathan, R.K.; et al. Genomic amplification of 9p24.1 targeting JAK2, PD-L1, and PD-L2 is enriched in high-risk triple negative breast cancer. Oncotarget 2015, 6, 26483–26493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanik, E.L.; Kaunitz, G.J.; Cottrell, T.R.; Succaria, F.; McMiller, T.L.; Ascierto, M.L.; Esandrio, J.; Xu, H.; Ogurtsova, A.; Cornish, T. Association of HIV status with local immune response to anal squamous cell carcinoma: Implications for immunotherapy. JAMA Oncol. 2017, 3, 974–978. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, G.; Franzke, A.; Buer, J.; Beil, W.; Probst–Kepper, M.; Wittke, F.; Övermann, K.; Lassmann, S.; Hoffmann, R.; Kirchner, H. Pharmacokinetics of recombinant human interleukin–2 in advanced renal cell carcinoma patients following subcutaneous application. Br. J. Clin. Pharmacol. 1998, 46, 5–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katze, M.G.; He, Y.; Gale, M. Viruses and interferon: A fight for supremacy. Nat. Rev. Immunol. 2002, 2, 675–687. [Google Scholar] [CrossRef]
- Sun, T.; Yang, Y.; Luo, X.; Cheng, Y.; Zhang, M.; Wang, K.; Ge, C. Inhibition of tumor angiogenesis by interferon-γ by sup-pression of tumor-associated macrophage differentiation. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2014, 21, 227–235. [Google Scholar]
- He, T.; Tang, C.; Xu, S.; Moyana, T.; Xiang, J. Interferon gamma stimulates cellular maturation of dendritic cell line DC2.4 leading to induction of efficient cytotoxic T cell responses and antitumor immunity. Cell. Mol. Immunol. 2007, 4, 105–111. [Google Scholar] [PubMed]
- Müller, L.; Aigner, P.; Stoiber, D. Type I Interferons and Natural Killer Cell Regulation in Cancer. Front. Immunol. 2017, 8, 304. [Google Scholar] [CrossRef] [Green Version]
- Enomoto, H.; Tao, L.; Eguchi, R.; Sato, A.; Honda, M.; Kaneko, S.; Iwata, Y.; Nishikawa, H.; Imanishi, H.; Iijima, H.; et al. The in vivo antitumor effects of type I-interferon against hepatocellular carcinoma: The suppression of tumor cell growth and angiogenesis. Sci. Rep. 2017, 7, 12189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Sasson, S.Z.; Hu-Li, J.; Quiel, J.; Cauchetaux, S.; Ratner, M.; Shapira, I.; Dinarello, C.A.; Paul, W.E. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 7119–7124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinchieri, G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef]
- Itoh, K.; Hirohata, S. The role of IL-10 in human B cell activation, proliferation, and differentiation. J. Immunol. 1995, 154, 4341–4350. [Google Scholar]
- Yan, W.-L.; Shen, K.-Y.; Tien, C.-Y.; Chen, Y.-A.; Liu, S.-J. Recent progress in GM-CSF-based cancer immunotherapy. Immunotherapy 2017, 9, 347–360. [Google Scholar] [CrossRef]
- Milling, L.; Zhang, Y.; Irvine, D.J. Delivering safer immunotherapies for cancer. Adv. Drug Deliv. Rev. 2017, 114, 79–101. [Google Scholar] [CrossRef]
- Lim, W.A.; June, C.H. The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017, 168, 724–740. [Google Scholar] [CrossRef] [Green Version]
- Davila, M.L.; Brentjens, R.J. CD19-Targeted CAR T cells as novel cancer immunotherapy for relapsed or refractory B-cell acute lymphoblastic leukemia. Clin. Adv. Hematol. Oncol. 2016, 14, 802–808. [Google Scholar] [PubMed]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Fitzgerald, J.C.; Weiss, S.; Maude, S.L.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; Shaw, P.; Berg, R.A.; June, C.H.; Porter, D.L.; et al. Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy for Acute Lymphoblastic Leukemia. Crit. Care Med. 2017, 45, e124–e131. [Google Scholar] [CrossRef]
- Berg, J.H.V.D.; Gomez-Eerland, R.; van de Wiel, B.; Hulshoff, L.; Broek, D.V.D.; Bins, A.; Tan, H.L.; Harper, J.V.; Hassan, N.J.; Jakobsen, B.K.; et al. Case Report of a Fatal Serious Adverse Event Upon Administration of T Cells Transduced With a MART-1-specific T-cell Receptor. Mol. Ther. 2015, 23, 1541–1550. [Google Scholar] [CrossRef] [Green Version]
- Graham, C.; Hewitson, R.; Pagliuca, A.; Benjamin, R. Cancer immunotherapy with CAR-T cells—Behold the future. Clin. Med. 2018, 18, 324–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oved, J.H.; Barrett, D.M.; Teachey, D.T. Cellular therapy: Immune-related complications. Immunol. Rev. 2019, 290, 114–126. [Google Scholar] [CrossRef]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global Manufacturing of CAR T Cell Therapy. Mol. Ther. Methods Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barros, L.; Paixão, E.; Valli, A.; Naozuka, G.; Fassoni, A.; Almeida, R. CARTmath—A Mathematical Model of CAR-T Immunotherapy in Preclinical Studies of Hematological Cancers. Cancers 2021, 13, 2941. [Google Scholar] [CrossRef] [PubMed]
- Cunto-Amesty, G.; Monzavi-Karbassi, B.; Luo, P.; Jousheghany, F.; Kieber-Emmons, T. Strategies in cancer vaccines development. Int. J. Parasitol. 2003, 33, 597–613. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Ren, Y.; Yu, X.; Qian, F.; Bian, B.-S.-J.; Xiao, H.-L.; Wang, W.-G.; Xu, S.-L.; Yang, J.; Cui, W.; et al. ALDH1A1 defines invasive cancer stem-like cells and predicts poor prognosis in patients with esophageal squamous cell carcinoma. Mod. Pathol. 2013, 27, 775–783. [Google Scholar] [CrossRef] [Green Version]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer stem cells (CSCs): Metabolic strategies for their identification and eradica-tion. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef] [Green Version]
- Jagust, P.; De Luxán-Delgado, B.; Parejo-Alonso, B.; Sancho, P. Metabolism-Based Therapeutic Strategies Targeting Cancer Stem Cells. Front. Pharmacol. 2019, 10, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawood, S.; Austin, L.; Cristofanilli, M. Cancer stem cells: Implications for cancer therapy. Oncolology 2014, 28, 28. [Google Scholar]
- Kreso, A.; Dick, J.E. Evolution of the Cancer Stem Cell Model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Rofstad, E.K. Cancer stem cells (CSCs), cervical CSCs and targeted therapies. Oncotarget 2017, 8, 35351–35367. [Google Scholar] [CrossRef] [Green Version]
- Boesch, M.; Sopper, S.; Zeimet, A.G.; Reimer, D.; Gastl, G.; Ludewig, B.; Wolf, D. Heterogeneity of Cancer Stem Cells: Rationale for Targeting the Stem Cell Niche. Biochim. Biophys. Acta (BBA) Bioenerg. 2016, 1866, 276–289. [Google Scholar] [CrossRef] [Green Version]
- Franco, S.S.; Szczesna, K.; Iliou, M.S.; Al-Qahtani, M.; Mobasheri, A.; Kobolák, J.; Dinnyés, A. In vitro models of cancer stem cells and clinical applications. BMC Cancer 2016, 16, 23–49. [Google Scholar] [CrossRef] [Green Version]
- Das, B.; Pal, B.; Bhuyan, R.; Li, H.; Sarma, A.; Gayan, S.; Talukdar, J.; Sandhya, S.; Bhuyan, S.; Gogoi, G.; et al. MYC Regulates the HIF2α Stemness Pathway via Nanog and Sox2 to Maintain Self-Renewal in Cancer Stem Cells versus Non-Stem Cancer Cells. Cancer Res. 2019, 79, 4015–4025. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Velazquez, M.A.; Yu, Z.; Jiao, X.; Pestell, R.G. Cancer stem cells and the cell cycle: Targeting the drive behind breast cancer. Expert Rev. Anticancer. Ther. 2009, 9, 275–279. [Google Scholar] [CrossRef] [Green Version]
- Zeuner, A.; Francescangeli, F.; Contavalli, P.; Zapparelli, G.; Apuzzo, T.; Eramo, A.; Baiocchi, M.; De Angelis, M.; Biffoni, M.; Sette, G. Elimination of quiescent/slow-proliferating cancer stem cells by Bcl-X L inhibition in non-small cell lung cancer. Cell Death Differ. 2014, 21, 1877–1888. [Google Scholar] [CrossRef]
- Mertins, S.D. Cancer stem cells: A systems biology view of their role in prognosis and therapy. Anti-Cancer Drugs 2014, 25, 353. [Google Scholar] [CrossRef]
- Hassan, K.A.; Wang, L.; Korkaya, H.; Chen, G.; Maillard, I.; Etherton-Beer, C.; Kalemkerian, G.P.; Wicha, M.S. Notch Pathway Activity Identifies Cells with Cancer Stem Cell–like Properties and Correlates with Worse Survival in Lung Adenocarcinoma. Clin. Cancer Res. 2013, 19, 1972–1980. [Google Scholar] [CrossRef] [Green Version]
- Beck, B.; Blanpain, C. Unravelling cancer stem cell potential. Nat. Rev. Cancer 2013, 13, 727–738. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immuno-deficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Medema, J.P.; Vermeulen, L. Microenvironmental regulation of stem cells in intestinal homeostasis and cancer. Nat. Cell Biol. 2011, 474, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Shestopalov, I.A.; Zon, L.I. The right neighbour. Nature 2012, 481, 453–454. [Google Scholar] [CrossRef]
- Badrinath, N.; Yoo, S.Y. Recent Advances in Cancer Stem Cell-Targeted Immunotherapy. Cancers 2019, 11, 310. [Google Scholar] [CrossRef] [Green Version]
- Dirks, P.B. Brain tumor stem cells: The cancer stem cell hypothesis writ large. Mol. Oncol. 2010, 4, 420–430. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Iden-tification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Kleffel, S.; Schatton, T. Tumor Dormancy and Cancer Stem Cells: Two Sides of the Same Coin? In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2013; Volume 734, pp. 145–179. [Google Scholar]
- Flesken-Nikitin, A.; Hwang, C.-I.; Cheng, C.-Y.; Michurina, T.V.; Enikolopov, G.; Nikitin, A.Y. Ovarian surface epithelium at the junction area contains a cancer-prone stem cell niche. Nat. Cell Biol. 2013, 495, 241–245. [Google Scholar] [CrossRef]
- Giancotti, F.G. Mechanisms Governing Metastatic Dormancy and Reactivation. Cell 2013, 155, 750–764. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Rich, J.N. Hypoxia and Hypoxia Inducible Factors in Cancer Stem Cell Maintenance. In Current Topics in Microbiology and Immunology; Springer: Heidelberg, Germany, 2010; Volume 345, pp. 21–30. [Google Scholar]
- Miyake, K.; Mickley, L.; Litman, T.; Zhan, Z.; Robey, R.; Cristensen, B.; Brangi, M.; Greenberger, L.; Dean, M.; Fojo, T.; et al. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: Demonstration of homology to ABC transport genes. Cancer Res. 1999, 59, 8–13. [Google Scholar]
- Nunes, T.; Hamdan, D.; Leboeuf, C.; El Bouchtaoui, M.; Gapihan, G.; Nguyen, T.T.; Meles, S.; Angeli, E.; Ratajczak, P.; Lu, H.; et al. Targeting Cancer Stem Cells to Overcome Chemoresistance. Int. J. Mol. Sci. 2018, 19, 4036. [Google Scholar] [CrossRef] [Green Version]
- Campbell, L.L.; Polyak, K. Breast Tumor Heterogeneity: Cancer Stem Cells or Clonal Evolution? Cell Cycle 2007, 6, 2332–2338. [Google Scholar] [CrossRef] [Green Version]
- Wakamatsu, Y.; Sakamoto, N.; Oo, H.Z.; Naito, Y.; Uraoka, N.; Anami, K.; Sentani, K.; Oue, N.; Yasui, W. Expression of cancer stem cell markers ALDH1, CD44 and CD133 in primary tumor and lymph node metastasis of gastric cancer. Pathol. Int. 2011, 62, 112–119. [Google Scholar] [CrossRef]
- Gaur, P.; Sceusi, E.L.; Samuel, S.; Xia, L.; Fan, F.; Zhou, Y.; Lu, J.; Tozzi, F.; Lopez-Berestein, G.; Vivas-Mejia, P.; et al. Identification of Cancer Stem Cells in Human Gastrointestinal Carcinoid and Neuroendocrine Tumors. Gastroenterology 2011, 141, 1728–1737. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Liu, C.; Yuan, J.; Xiao, X.; Tang, N.; Hao, J.; Wang, H.; Bian, X.; Deng, Y.; Ding, Y. CD133+ single cell-derived progenies of colorectal cancer cell line SW480 with different invasive and metastatic potential. Clin. Exp. Metastasis 2010, 27, 517–527. [Google Scholar] [CrossRef]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Migrating cancer stem cells—an integrated concept of malignant tumour progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar] [CrossRef]
- Ortensi, B.; Setti, M.; Osti, D.; Pelicci, G. Cancer stem cell contribution to glioblastoma invasiveness. Stem Cell Res. Ther. 2013, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Raza, A.; Franklin, M.J.; Dudek, A.Z. Pericytes and vessel maturation during tumor angiogenesis and metastasis. Am. J. Hematol. 2010, 85, 593–598. [Google Scholar] [CrossRef]
- Neman, J.; Termini, J.; Wilczynski, S.; Vaidehi, N.; Choy, C.; Kowolik, C.M.; Li, H.; Hambrecht, A.C.; Roberts, E.; Jandial, R. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc. Natl. Acad. Sci. USA 2014, 111, 984–989. [Google Scholar] [CrossRef] [Green Version]
- Pal, B.; Das, B. In vitro Culture of Naïve Human Bone Marrow Mesenchymal Stem Cells: A Stemness Based Approach. Front. Cell Dev. Biol. 2017, 5, 69. [Google Scholar] [CrossRef] [Green Version]
- Pathak, L.; Gayan, S.; Pal, B.; Talukdar, J.; Bhuyan, S.; Sandhya, S.; Yeger, H.; Baishya, D.; Das, B. Coronavirus activates an altruistic stem cell mediated defense mechanism that reactivates dormant tuberculosis: Implications in COVID-19 pandemic. Am. J. Pathol. 2021, 191, 1255–1268. [Google Scholar] [CrossRef]
- Das, B.; Bayat-Mokhtari, R.; Tsui, M.; Lotfi, S.; Tsuchida, R.; Felsher, D.W.; Yeger, H. HIF-2α Suppresses p53 to Enhance the Stemness and Regenerative Potential of Human Embryonic Stem Cells. Stem Cells 2012, 30, 1685–1695. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Huang, Y.-H.; Chen, J.-L. Understanding and targeting cancer stem cells: Therapeutic implications and challenges. Acta Pharmacol. Sin. 2013, 34, 732–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells—What challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef] [Green Version]
- Besançon, R.; Valsesia-Wittmann, S.; Puisieux, A.; De Fromentel, C.C.; Maguer-Satta, V. Cancer Stem Cells: The Emerging Challenge of Drug Targeting. Curr. Med. Chem. 2009, 16, 394–416. [Google Scholar] [CrossRef] [PubMed]
- Turdo, A.; Veschi, V.; Gaggianesi, M.; Chinnici, A.; Bianca, P.; Todaro, M.; Stassi, G. Meeting the Challenge of Targeting Cancer Stem Cells. Front. Cell Dev. Biol. 2019, 7, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruttel, V.S.; Wischhusen, J.; Wischhusen, J. Cancer Stem Cell Immunology: Key to Understanding Tumorigenesis and Tumor Immune Escape? Front. Immunol. 2014, 5, 360. [Google Scholar] [CrossRef] [Green Version]
- Hamaï, A.; Meslin, F.; Benlalam, H.; Jalil, A.; Mehrpour, M.; Faure, F.; Lecluse, Y.; Vielh, P.; Avril, M.-F.; Robert, C.; et al. ICAM-1 Has a Critical Role in the Regulation of Metastatic Melanoma Tumor Susceptibility to CTL Lysis by Interfering with PI3K/AKT Pathway. Cancer Res. 2008, 68, 9854–9864. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.; Wiesner, S.; Xiao, J.; Ericson, K.; Chen, W.; Hall, W.A.; Low, W.C.; Ohlfest, J.R. Expression of MHC I and NK ligands on human CD133+ glioma cells: Possible targets of immunotherapy. J. Neuro-Oncol. 2006, 83, 121–131. [Google Scholar] [CrossRef]
- Schatton, T.; Frank, M.H. Antitumor Immunity and Cancer Stem Cells. Ann. N. Y. Acad. Sci. 2009, 1176, 154–169. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, M.; Wu, P.; Chen, C.; Xu, Z.P.; Gu, W. Increased PD-L1 expression in breast and colon cancer stem cells. Clin. Exp. Pharmacol. Physiol. 2017, 44, 602–604. [Google Scholar] [CrossRef]
- Todaro, M.; Alea, M.P.; di Stefano, A.B.; Cammareri, P.; Vermeulen, L.; Iovino, F.; Tripodo, C.; Russo, A.; Gulotta, G.; Medema, J.P.; et al. Colon Cancer Stem Cells Dictate Tumor Growth and Resist Cell Death by Production of Interleukin. Cell Stem Cell 2007, 1, 389–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Tomaso, T.; Mazzoleni, S.; Wang, E.; Sovena, G.; Clavenna, D.; Franzin, A.; Mortini, P.; Ferrone, S.; Doglioni, C.; Marincola, F.M.; et al. Immunobiological Characterization of Cancer Stem Cells Isolated from Glioblastoma Patients. Clin. Cancer Res. 2010, 16, 800–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.P.; Singh, R.; Gupta, O.P.; Gupta, S.; Bhatt, M.L.B. Immunotherapy: Newer Therapeutic Armamentarium against Cancer Stem Cells. J. Oncol. 2020, 2020, 1–15. [Google Scholar] [CrossRef]
- Luo, H.; Zeng, C.; Fang, C.; Seeruttun, S.R.; Lv, L.; Wang, W. A New Strategy Using ALDHhigh-CD8+T Cells to Inhibit Tumorigenesis. PLoS ONE 2014, 9, e103193. [Google Scholar] [CrossRef] [Green Version]
- Jena, B.; Dotti, G.; Cooper, L.J.N. Redirecting T-cell specificity by introducing a tumor-specific chimeric antigen receptor. Blood 2010, 116, 1035–1044. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Joosten, L.A.B.; Latz, E.; Mills, K.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Brommer, B.; Tian, X.; Krishnan, A.; Meer, M.; Wang, C.; Vera, D.L.; Zeng, Q.; Yu, D.; Bonkowski, M.S.; et al. Reprogramming to recover youthful epigenetic information and restore vision. Nat. Cell Biol. 2020, 588, 124–129. [Google Scholar] [CrossRef]
- Reddy, V.S.; Palika, R.; Ismail, A.; Pullakhandam, R.; Reddy, G.B. Nutrigenomics: Opportunities & challenges for public health nutrition. Indian J. Med. Res. 2018, 148, 632–641. [Google Scholar] [CrossRef]
- Marelli, G.; Howells, A.; Lemoine, N.R.; Wang, Y. Oncolytic Viral Therapy and the Immune System: A Double-Edged Sword Against Cancer. Front. Immunol. 2018, 9, 866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, L.; Peng, K.-W. The emerging role of oncolytic virus therapy against cancer. Chin. Clin. Oncol. 2018, 7, 16. [Google Scholar] [CrossRef]
- Li, L.; Liu, S.; Han, D.; Tang, B.; Ma, J. Delivery and Biosafety of Oncolytic Virotherapy. Front. Oncol. 2020, 10, 475. [Google Scholar] [CrossRef] [Green Version]
- Su, K.Y.; Balasubramaniam, V.R.M.T. Zika Virus as Oncolytic Therapy for Brain Cancer: Myth or Reality? Front. Microbiol. 2019, 10, 2715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doniņa, S.; Strele, I.; Proboka, G.; Auziņš, J.; Alberts, P.; Jonsson, B.; Venskus, D.; Muceniece, A. Adapted ECHO-7 virus Rigvir immunotherapy (oncolytic virotherapy) prolongs survival in melanoma patients after surgical excision of the tumour in a retrospective study. Melanoma Res. 2015, 25, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Ries, S.; Korn, W.M. ONYX-015: Mechanisms of action and clinical potential of a replication-selective adenovirus. Br. J. Cancer 2002, 86, 5–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Z.-J.; Chang, J.-H.; Zhang, L.; Jiang, W.-Q.; Guan, Z.-Z.; Liu, J.-W.; Zhang, Y.; Hu, X.-H.; Wu, G.-H.; Wang, H.-Q.; et al. [Phase III randomized clinical trial of intratumoral injection of E1B gene-deleted adenovirus (H101) combined with cisplatin-based chemotherapy in treating squamous cell cancer of head and neck or esophagus]. Ai Zheng = Aizheng = Chin. J. Cancer 2004, 23, 1666–1670. [Google Scholar]
- Yanagi, Y.; Takeda, M.; Ohno, S. Measles virus: Cellular receptors, tropism and pathogenesis. J. Gen. Virol. 2006, 87, 2767–2779. [Google Scholar] [CrossRef]
- Schirrmacher, V. Fifty Years of Clinical Application of Newcastle Disease Virus: Time to Celebrate! Biomedicines 2016, 4, 16. [Google Scholar] [CrossRef] [Green Version]
- Vellinga, J.; Van Der Heijdt, S.; Hoeben, R. The adenovirus capsid: Major progress in minor proteins. J. Gen. Virol. 2005, 86, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Sivanandam, V.; LaRocca, C.J.; Chen, N.G.; Fong, Y.; Warner, S.G. Oncolytic Viruses and Immune Checkpoint Inhibition: The Best of Both Worlds. Mol. Ther. Oncolytics 2019, 13, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Carteaux, G.; Maquart, M.; Bedet, A.; Contou, D.; Brugières, P.; Fourati, S.; Cleret de Langavant, L.; De Broucker, T.; Brun-Buisson, C.; Leparc-Goffart, I.; et al. Zika Virus Associated with Meningoencephalitis. N. Engl. J. Med. 2016, 374, 1595–1596. [Google Scholar] [CrossRef]
- Wells, M.F.; Salick, M.R.; Wiskow, O.; Ho, D.J.; Worringer, K.; Ihry, R.J.; Kommineni, S.; Bilican, B.; Klim, J.R.; Hill, E.J.; et al. Genetic Ablation of AXL Does Not Protect Human Neural Progenitor Cells and Cerebral Organoids from Zika Virus Infection. Cell Stem Cell 2016, 19, 703–708. [Google Scholar] [CrossRef] [Green Version]
- Onorati, M.; Li, Z.; Liu, F.; Sousa, A.M.M.; Nakagawa, N.; Li, M.; Dell’Anno, M.T.; Gulden, F.O.; Pochareddy, S.; Tebbenkamp, A.T.N.; et al. Zika Virus Disrupts Phospho-TBK1 Localization and Mitosis in Human Neuroepithelial Stem Cells and Radial Glia. Cell Rep. 2016, 16, 2576–2592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavali, P.L.; Stojic, L.; Meredith, L.W.; Joseph, N.; Nahorski, M.S.; Sanford, T.J.; Sweeney, T.R.; Krishna, B.A.; Hosmillo, M.; Firth, A.E.; et al. Neurodevelopmental protein Musashi-1 interacts with the Zika genome and promotes viral replication. Science 2017, 357, 83–88. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Gorman, M.J.; McKenzie, L.; Chai, J.N.; Hubert, C.G.; Prager, B.C.; Fernandez, E.; Richner, J.; Zhang, R.; Shan, C.; et al. Zika virus has oncolytic activity against glioblastoma stem cells. J. Exp. Med. 2017, 214, 2843–2857. [Google Scholar] [CrossRef] [Green Version]
- Macedo, N.; Miller, D.M.; Haq, R.; Kaufman, H.L. Clinical landscape of oncolytic virus research in 2020. J. Immunother. Cancer 2020, 8, e001486. [Google Scholar] [CrossRef]
- Liang, W.; Guan, W.; Chen, R.; Wang, W.; Li, J.; Xu, K.; Li, C.; Ai, Q.; Lu, W.; Liang, H.; et al. Cancer patients in SARS-CoV-2 infection: A nationwide analysis in China. Lancet Oncol. 2020, 21, 335–337. [Google Scholar] [CrossRef]
- Derosa, L.; Melenotte, C.; Griscelli, F.; Gachot, B.; Marabelle, A.; Kroemer, G.; Zitvogel, L. The immuno-oncological challenge of COVID-19. Nat. Cancer 2020, 1, 946–964. [Google Scholar] [CrossRef]
- Yazdanpanah, F.; Hamblin, M.R.; Rezaei, N. The immune system and COVID-19: Friend or foe? Life Sci. 2020, 256, 117900. [Google Scholar] [CrossRef]
- Rokni, M.; Ghasemi, V.; Tavakoli, Z. Immune responses and pathogenesis of SARS-CoV-2 during an outbreak in Iran: Comparison withSARSandMERS. Rev. Med. Virol. 2020, 30, 2107. [Google Scholar] [CrossRef] [Green Version]
- Prompetchara, E.; Ketloy, C.; Palaga, T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pac. J. Allergy Immunol. 2020, 38, 1–9. [Google Scholar] [CrossRef]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Soria, G.; Ofri-Shahak, M.; Haas, I.; Yaal-Hahoshen, N.; Leider-Trejo, L.; Leibovich-Rivkin, T.; Weitzenfeld, P.; Meshel, T.; Shabtai, E.; Gutman, M.; et al. Inflammatory mediators in breast cancer: Coordinated expression of TNFα & IL-1β with CCL2 & CCL5 and effects on epithelial-to-mesenchymal transition. BMC Cancer 2011, 11, 130–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Challenor, S.; Tucker, D. SARS-CoV-2-induced remission of Hodgkin lymphoma. Br. J. Haemaatol 2021, 192, 415. [Google Scholar] [CrossRef]
- Pettenati, C.; Ingersoll, M.A. Mechanisms of BCG immunotherapy and its outlook for bladder cancer. Nat. Rev. Urol. 2018, 15, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Özcan, Y.; Çağlar, F.; Celik, S.; Demir, A.B.; Erçetin, A.P.; Altun, Z.; Aktas, S. The role of cancer stem cells in immunotherapy for bladder cancer: An in vitro study. Urol. Oncol. Semin. Orig. Investig. 2020, 38, 476–487. [Google Scholar] [CrossRef]
- Das, B.; Tsuchida, R.; Malkin, D.; Koren, G.; Baruchel, S.; Yeger, H. Hypoxia Enhances Tumor Stemness by Increasing the Invasive and Tumorigenic Side Population Fraction. Stem Cells 2008, 26, 1818–1830. [Google Scholar] [CrossRef]
- Bhuyan, S.; Pal, B.; Li, H.; Bhuyan, R.; Pathak, L.; Gayan, S.; Baishya, D.; Das, B. Abstract 5539: Mesenchymal stem cell altruism mediates a novel HMGB1/NKG2D immune response in oral cancer. Immunology 2020, 80, 5539. [Google Scholar] [CrossRef]
- Karlitepe, A.; Özalp, Ö.; Avci, C.B. New approaches for cancer immunotherapy. Tumor Biol. 2015, 36, 4075–4078. [Google Scholar] [CrossRef] [PubMed]
- Linch, S.; McNamara, M.J.; Redmond, W.L. OX40 Agonists and Combination Immunotherapy: Putting the Pedal to the Metal. Front. Oncol. 2015, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Sagiv-Barfi, I.; Czerwinski, D.K.; Levy, S.; Alam, I.S.; Mayer, A.T.; Gambhir, S.S.; Levy, R. Eradication of spontaneous malignancy by local immunotherapy. Sci. Transl. Med. 2018, 10, eaan4488. [Google Scholar] [CrossRef] [Green Version]
- Houot, R.; Levy, R. T-cell modulation combined with intratumoral CpG cures lymphoma in a mouse model without the need for chemotherapy. Blood 2009, 113, 3546–3552. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Poltorak, A.; Wei, Y.; Beutler, B. Three novel mammalian toll-like receptors: Gene structure, expression, and evolution. Eur. Cytokine Netw. 2000, 11, 362–371. [Google Scholar]
- Li, J.; Song, W.; Czerwinski, D.K.; Varghese, B.; Uematsu, S.; Akira, S.; Krieg, A.M.; Levy, R. Lymphoma Immunotherapy with CpG Oligodeoxynucleotides Requires TLR9 Either in the Host or in the Tumor Itself. J. Immunol. 2007, 179, 2493–2500. [Google Scholar] [CrossRef] [Green Version]
- Baxter, A.G.; Hodgkin, P. Activation rules: The two-signal theories of immune activation. Nat. Rev. Immunol. 2002, 2, 439–446. [Google Scholar] [CrossRef]
- Lane, P. Role of Ox40 Signals in Coordinating Cd4 T Cell Selection, Migration, and Cytokine Differentiation in T Helper (Th)1 and Th2 Cells. J. Exp. Med. 2000, 191, 201–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petty, J.K.; He, K.; Corless, C.L.; Vetto, J.T.; Weinberg, A.D. Survival in human colorectal cancer correlates with expression of the T-cell costimulatory molecule OX-40 (CD134). Am. J. Surg. 2002, 183, 512–518. [Google Scholar] [CrossRef]
- Valzasina, B.; Guiducci, C.; Dislich, H.; Killeen, N.; Weinberg, A.D.; Colombo, M.P. Triggering of OX40 (CD134) on CD4+CD25+ T cells blocks their inhibitory activity: A novel regulatory role for OX40 and its comparison with GITR. Blood 2005, 105, 2845–2851. [Google Scholar] [CrossRef] [Green Version]
- Ruby, C.E.; Yates, M.A.; Hirschhorn-Cymerman, D.; Chlebeck, P.; Wolchok, J.D.; Houghton, A.N.; Offner, H.; Weinberg, A.D. Cutting Edge: OX40 Agonists Can Drive Regulatory T Cell Expansion if the Cytokine Milieu Is Right. J. Immunol. 2009, 183, 4853–4857. [Google Scholar] [CrossRef] [Green Version]
- Tomasi, T.B.; Magner, W.J.; Khan, A.N.H. Epigenetic regulation of immune escape genes in cancer. Cancer Immunol. Immunother. 2006, 55, 1159–1184. [Google Scholar] [CrossRef]
- Cao, J.; Yan, Q. Cancer Epigenetics, Tumor Immunity, and Immunotherapy. Trends Cancer 2020, 6, 580–592. [Google Scholar] [CrossRef]
- Topper, M.J.; Vaz, M.; Marrone, K.A.; Brahmer, J.R.; Baylin, S.B. The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Zahnow, C.A.; Ahuja, N.; Baylin, S.B. Combining Epigenetic and Immunotherapy to Combat Cancer. Cancer Res. 2016, 76, 1683–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigalotti, L.; Fratta, E.; Coral, S.; Maio, M. Epigenetic drugs as immunomodulators for combination therapies in solid tumors. Pharmacol. Ther. 2014, 142, 339–350. [Google Scholar] [CrossRef]
- James, S.R.; A Link, P.; Karpf, A.R. Epigenetic regulation of X-linked cancer/germline antigen genes by DNMT1 and DNMT3b. Oncogene 2006, 25, 6975–6985. [Google Scholar] [CrossRef] [Green Version]
- Héninger, E.; Krueger, T.E.G.; Lang, J.M.; Heninger, E. Augmenting Antitumor Immune Responses with Epigenetic Modifying Agents. Front. Immunol. 2015, 6, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wischnewski, F.; Pantel, K.; Schwarzenbach, H. Promoter Demethylation and Histone Acetylation Mediate Gene Expression of MAGE-A1, -A2, -A3, and -A12 in Human Cancer Cells. Mol. Cancer Res. 2006, 4, 339–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvic, M.; Talpur, R.; Ni, X.; Zhang, C.; Hazarika, P.; Kelly, C.; Chiao, J.H.; Reilly, J.F.; Ricker, J.L.; Richon, V.M.; et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2006, 109, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-C.; Li, H.; Van Neste, L.; Cai, Y.; Robert, C.; Rassool, F.V.; Shin, J.J.; Harbom, K.M.; Beaty, R.; Pappou, E.; et al. Transient Low Doses of DNA-Demethylating Agents Exert Durable Antitumor Effects on Hematological and Epithelial Tumor Cells. Cancer Cell 2012, 21, 430–446. [Google Scholar] [CrossRef] [Green Version]
- Lisiero, D.N.; Soto, H.; Everson, R.G.; Liau, L.M.; Prins, R.M. The histone deacetylase inhibitor, LBH589, promotes the systemic cytokine and effector responses of adoptively transferred CD8+ T cells. J. Immunother. Cancer 2014, 2, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voisin, S.; Eynon, N.; Yan, X.; Bishop, D. Exercise training and DNA methylation in humans. Acta Physiol. 2015, 213, 39–59. [Google Scholar] [CrossRef]
- Choi, S.-W.; Friso, S. Epigenetics: A New Bridge between Nutrition and Health. Adv. Nutr. 2010, 1, 8–16. [Google Scholar] [CrossRef]
- Barron-Cabrera, E.; Ramos-Lopez, O.; González-Becerra, K.; Riezu-Boj, J.I.; Milagro, F.; Martínez-López, E.; Martínez, J.A. Epigenetic Modifications as Outcomes of Exercise Interventions Related to Specific Metabolic Alterations: A Systematic Review. Lifestyle Genom. 2019, 12, 25–44. [Google Scholar] [CrossRef] [PubMed]
- Holmen Olofsson, G.; Jensen, A.W.P.; Idorn, M.; thor Straten, P. Exercise Oncology and Immuno-Oncology; A (Future) Dy-namic Duo. Int. J. Mol. Sci. 2020, 21, 3816. [Google Scholar] [CrossRef]
- Nieman, D.C.; Wentz, L.M. The compelling link between physical activity and the body’s defense system. J. Sport Health Sci. 2019, 8, 201–217. [Google Scholar] [CrossRef]
- De Gonzalo-Calvo, D.; Davalos, A.; Montero, A.; García-González, Á.; Tyshkovska, I.; González-Medina, A.; Soares, S.; Camblor, P.M.; Casas-Agustench, P.; Rabadán, M.; et al. Circulating inflammatory miRNA signature in response to different doses of aerobic exercise. J. Appl. Physiol. 2015, 119, 124–134. [Google Scholar] [CrossRef] [Green Version]
- Maciejowski, J.; De Lange, J.M.T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Mizukoshi, E.; Kaneko, S. Telomerase-Targeted Cancer Immunotherapy. Int. J. Mol. Sci. 2019, 20, 1823. [Google Scholar] [CrossRef] [Green Version]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT Promoter Mutations in Familial and Sporadic Melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.-P.; Chen, W.; Schwarer, A.P.; Li, H. Telomerase in cancer immunotherapy. Biochim. Biophys. Acta (BBA) Rev. Cancer 2010, 1805, 35–42. [Google Scholar] [CrossRef]
- Nakamura, T.; Morin, G.; Chapman, K.B.; Weinrich, S.L.; Andrews, W.H.; Lingner, J.; Harley, C.B.; Cech, T.R. Telomerase Catalytic Subunit Homologs from Fission Yeast and Human. Science 1997, 277, 955–959. [Google Scholar] [CrossRef]
- Kumagai, M.; Mizukoshi, E.; Tamai, T.; Kitahara, M.; Yamashita, T.; Arai, K.; Terashima, T.; Iida, N.; Fushimi, K.; Kaneko, S. Immune response to human telomerase reverse transcriptase-derived helper T cell epitopes in hepatocellular carcinoma patients. Liver Int. 2018, 38, 1635–1645. [Google Scholar] [CrossRef]
- Staff, C.; Mozaffari, F.; Frodin, J.-E.; Mellstedt, H.; Liljefors, M.G. Telomerase (GV1001) vaccination together with gemcitabine in advanced pancreatic cancer patients. Int. J. Oncol. 2014, 45, 1293–1303. [Google Scholar] [CrossRef] [Green Version]
- Park, J.K.; Kim, Y.; Kim, H.; Jeon, J.; Kim, T.W.; Park, J.-H.; Hwnag, Y.-I.; Lee, W.J.; Kang, J.S. The anti-fibrotic effect of GV1001 combined with gemcitabine on treatment of pancreatic ductal adenocarcinoma. Oncotarget 2016, 7, 75081–75093. [Google Scholar] [CrossRef] [Green Version]
- Brunsvig, P.F.; Aamdal, S.; Gjertsen, M.K.; Kvalheim, G.; Markowski-Grimsrud, C.J.; Sve, I.; Dyrhaug, M.; Trachsel, S.; Møller, M.; Eriksen, J.A.; et al. Telomerase peptide vaccination: A phase I/II study in patients with non-small cell lung cancer. Cancer Immunol. Immunother. 2006, 55, 1553–1564. [Google Scholar] [CrossRef] [PubMed]
- Fenoglio, D.; Parodi, A.; Lavieri, R.; Kalli, F.; Ferrera, F.; Tagliamacco, A.; Guastalla, A.; Lamperti, M.G.; Giacomini, M.; Filaci, G. Immunogenicity of GX301 cancer vaccine: Four (telomerase peptides) are better than one. Hum. Vaccines Immunother. 2015, 11, 838–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lilleby, W.; Gaudernack, G.; Brunsvig, P.F.; Vlatkovic, L.; Schulz, M.; Mills, K.; Hole, K.H.; Inderberg, E.M. Phase I/IIa clinical trial of a novel hTERT peptide vaccine in men with metastatic hormone-naive prostate cancer. Cancer Immunol. Immunother. 2017, 66, 891–901. [Google Scholar] [CrossRef]
- Kotsakis, A.; Papadimitraki, E.; Vetsika, E.K.; Aggouraki, D.; Dermitzaki, E.K.; Hatzidaki, D.; Kentepozidis, N.; Mavroudis, D.; Georgoulias, V. A phase II trial evaluating the clinical and immunologic response of HLA-A2+ non-small cell lung cancer patients vaccinated with an hTERT cryptic peptide. Lung Cancer 2014, 86, 59–66. [Google Scholar] [CrossRef]
- Zhang, Y.; Kutateladze, T.G. Diet and the epigenome. Nat. Commun. 2018, 9, 3375. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hodes, G.; Zhang, H.; Zhang, S.; Zhao, W.; Golden, S.A.; Bi, W.; Menard, C.; Kana, V.; Leboeuf, M.; et al. Epigenetic modulation of inflammation and synaptic plasticity promotes resilience against stress in mice. Nat. Commun. 2018, 9, 477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Zhang, D.; Wang, Y.; Perez-Neut, M.; Han, Z.; Zheng, Y.G.; Hao, Q.; Zhao, Y. Lysine benzoylation is a histone mark regulated by SIRT2. Nat. Commun. 2018, 9, 3374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Tsujimoto, K.; Hashimoto, K.; Kawahori, K.; Hanzawa, N.; Hamaguchi, M.; Seki, T.; Nawa, M.; Ehara, T.; Kitamura, Y.; et al. Epigenetic modulation of Fgf21 in the perinatal mouse liver ameliorates diet-induced obesity in adulthood. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.; Suarez-Alvarez, B.; Lopez-Larrea, C. Therapeutic Epigenetic Reprogramming of Trained Immunity in Myeloid Cells. Trends Immunol. 2019, 40, 66–80. [Google Scholar] [CrossRef] [PubMed]
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2017, 359, 582–587. [Google Scholar] [CrossRef] [Green Version]
- Tominaga, S.; Kuroishi, T. An ecological study on diet/nutrition and cancer in Japan. Int. J. Cancer 1997, 71, 2–6. [Google Scholar] [CrossRef]
- Soldati, L.; Di Renzo, L.; Jirillo, E.; Ascierto, P.A.; Marincola, F.M.; De Lorenzo, A. The influence of diet on anti-cancer immune responsiveness. J. Transl. Med. 2018, 16, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ponzo, V.; Pellegrini, M.; D’Eusebio, C.; Bioletto, F.; Goitre, I.; Buscemi, S.; Frea, S.; Ghigo, E.; Bo, S. Mediterranean Diet and SARS-COV-2 Infection: Is There Any Association? A Proof-of-Concept Study. Nutrition 2021, 13, 1721. [Google Scholar] [CrossRef]
- Grudzien, M.; Rapak, A. Effect of Natural Compounds on NK Cell Activation. J. Immunol. Res. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Erkurt, M.A.; Aydogdu, I.; Dikilitaş, M.; Kuku, I.; Kaya, E.; Bayraktar, N.; Ozhan, O.; Ozkan, I.; Sönmez, A. Effects of Cyanocobalamin on Immunity in Patients with Pernicious Anemia. Med. Princ. Pract. 2008, 17, 131–135. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Quesada, C.; López-Biedma, A.; Toledo, E.; Gaforio, J.J. Squalene Stimulates a Key Innate Immune Cell to Foster Wound Healing and Tissue Repair. Evid. Based Complement. Altern. Med. 2018, 2018, 1–9. [Google Scholar] [CrossRef]
- Desbien, A.L.; Reed, S.J.; Bailor, H.R.; Cauwelaert, N.D.; Laurance, J.D.; Orr, M.; Fox, C.B.; Carter, D.; Duthie, M. Squalene emulsion potentiates the adjuvant activity of the TLR4 agonist, GLA, via inflammatory caspases, IL-18, and IFN-γ. Eur. J. Immunol. 2014, 45, 407–417. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, Y.; Tian, K.; Chen, X.; Zhang, R.; Mu, X.; Wu, Y.; Wang, D.; Wang, S.; Liu, F.; et al. Apigenin suppresses PD-L1 expression in melanoma and host dendritic cells to elicit synergistic therapeutic effects. J. Exp. Clin. Cancer Res. 2018, 37, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Casas, R.; Sacanella, E.; Estruch, R. The immune protective effect of the Mediterranean diet against chronic low-grade in-flammatory diseases. Endocr. Metab. Immune Disord. Drug Targets 2014, 14, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; Fulton, R.S. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207. [Google Scholar]
- Ding, T.; Schloss, P.D. Dynamics and associations of microbial community types across the human body. Nat. Cell Biol. 2014, 509, 357–360. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopala, S.V.; Vashee, S.; Oldfield, L.M.; Suzuki, Y.; Venter, J.C.; Telenti, A.; Nelson, K.E. The Human Microbiome and Cancer. Cancer Prev. Res. 2017, 10, 226–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braga, M.; Wischmeyer, P.E.; Drover, J.; Heyland, D.K. Clinical Evidence for Pharmaconutrition in Major Elective Surgery. J. Parenter. Enter. Nutr. 2013, 37, 66S–72S. [Google Scholar] [CrossRef]
- Bachmeier, B.E.; Mohrenz, I.V.; Mirisola, V.; Schleicher, E.; Romeo, F.; Höhneke, C.; Jochum, M.; Nerlich, A.G.; Pfeffer, U. Curcumin downregulates the inflammatory cytokines CXCL1 and -2 in breast cancer cells via NFκB. Carcinogenesis 2007, 29, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.-Y.; Yang, N.-S.; Lin, T.-J. Phytochemicals Approach for Developing Cancer Immunotherapeutics. Front. Pharmacol. 2017, 8, 386. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-M.; Wang, P.-H.; Chen, S.-S.; Wen, C.-C.; Chen, Y.-H.; Yang, W.-C.; Yang, N.-S. Shikonin induces immunogenic cell death in tumor cells and enhances dendritic cell-based cancer vaccine. Cancer Immunol. Immunother. 2012, 61, 1989–2002. [Google Scholar] [CrossRef] [PubMed]
- Ayeka, P.A. Potential of Mushroom Compounds as Immunomodulators in Cancer Immunotherapy: A Review. Evid. Based Complement. Altern. Med. 2018, 2018, 1–9. [Google Scholar] [CrossRef]
- Armideo, R.E.; Callahan, R.C.; Madonia, R.L. Immunotherapy for High-Risk Neuroblastoma: Management of Side Effects and Complications. J. Adv. Pract. Oncol. 2017, 8, 44–55. [Google Scholar] [CrossRef]
- Richards, R.M.; Sotillo, E.; Majzner, R.G. CAR T Cell Therapy for Neuroblastoma. Front. Immunol. 2018, 9, 2380. [Google Scholar] [CrossRef] [Green Version]
- Jabbari, P.; Hanaei, S.; Rezaei, N. State of the art in immunotherapy of neuroblastoma. Immunotherapy 2019, 11, 831–850. [Google Scholar] [CrossRef]
- Tilak, T.; Sherawat, S.; Agarwala, S.; Gupta, R.; Vishnubhatla, S.; Bakhshi, S. Circulating T-Regulatory Cells in Neuroblastoma: A Pilot Prospective Study. Pediatr. Hematol. Oncol. 2014, 31, 717–722. [Google Scholar] [CrossRef]
- Kock, A.; Bergqvist, F.; Steinmetz, J.; Elfman, L.H.M.; Korotkova, M.; Johnsen, J.I.; Jakobsson, P.-J.; Kogner, P.; Larsson, K. Establishment of an in vitro 3D model for neuroblastoma enables preclinical investigation of combined tumor-stroma drug targeting. Fed. Am. Soc. Exp. Biol. 2020, 34, 11101–11114. [Google Scholar] [CrossRef]
- Chaicharoenaudomrung, N.; Kunhorm, P.; Noisa, P. Three-dimensional cell culture systems as an in vitro platform for cancer and stem cell modeling. World J. Stem Cells 2019, 11, 1065–1083. [Google Scholar] [CrossRef]
- Corallo, D.; Frabetti, S.; Candini, O.; Gregianin, E.; Dominici, M.; Fischer, H.; Aveic, S. Emerging Neuroblastoma 3D In Vitro Models for Pre-Clinical Assessments. Front. Immunol. 2020, 11, 3110. [Google Scholar] [CrossRef]
- Nolan, J.; Frawley, T.; Tighe, J.; Soh, H.; Curtin, C.; Piskareva, O.; Nolan, J.; Frawley, T.; Tighe, J.; Soh, H.; et al. Preclinical models for neuroblastoma: Advances and challenges. Cancer Lett. 2020, 474, 53–62. [Google Scholar] [CrossRef]
- Joshi, S. Targeting the Tumor Microenvironment in Neuroblastoma: Recent Advances and Future Directions. Cancers 2020, 12, 2057. [Google Scholar] [CrossRef]
- Courau, T.; Bonnereau, J.; Chicoteau, J.; Bottois, H.; Remark, R.; Miranda, L.A.; Toubert, A.; Blery, M.; Aparicio, T.; Allez, M.; et al. Cocultures of human colorectal tumor spheroids with immune cells reveal the therapeutic potential of MICA/B and NKG2A targeting for cancer treatment. J. Immunother. Cancer 2019, 7, 74. [Google Scholar] [CrossRef] [Green Version]
- Saraiva, D.P.; Matias, A.T.; Braga, S.; Jacinto, A.; Cabral, M.G. Establishment of a 3D Co-culture With MDA-MB-231 Breast Cancer Cell Line and Patient-Derived Immune Cells for Application in the Development of Immunotherapies. Front. Oncol. 2020, 10, 1543. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mokhtari, R.B.; Sambi, M.; Qorri, B.; Baluch, N.; Ashayeri, N.; Kumar, S.; Cheng, H.-L.M.; Yeger, H.; Das, B.; Szewczuk, M.R. The Next-Generation of Combination Cancer Immunotherapy: Epigenetic Immunomodulators Transmogrify Immune Training to Enhance Immunotherapy. Cancers 2021, 13, 3596. https://doi.org/10.3390/cancers13143596
Mokhtari RB, Sambi M, Qorri B, Baluch N, Ashayeri N, Kumar S, Cheng H-LM, Yeger H, Das B, Szewczuk MR. The Next-Generation of Combination Cancer Immunotherapy: Epigenetic Immunomodulators Transmogrify Immune Training to Enhance Immunotherapy. Cancers. 2021; 13(14):3596. https://doi.org/10.3390/cancers13143596
Chicago/Turabian StyleMokhtari, Reza Bayat, Manpreet Sambi, Bessi Qorri, Narges Baluch, Neda Ashayeri, Sushil Kumar, Hai-Ling Margaret Cheng, Herman Yeger, Bikul Das, and Myron R. Szewczuk. 2021. "The Next-Generation of Combination Cancer Immunotherapy: Epigenetic Immunomodulators Transmogrify Immune Training to Enhance Immunotherapy" Cancers 13, no. 14: 3596. https://doi.org/10.3390/cancers13143596
APA StyleMokhtari, R. B., Sambi, M., Qorri, B., Baluch, N., Ashayeri, N., Kumar, S., Cheng, H. -L. M., Yeger, H., Das, B., & Szewczuk, M. R. (2021). The Next-Generation of Combination Cancer Immunotherapy: Epigenetic Immunomodulators Transmogrify Immune Training to Enhance Immunotherapy. Cancers, 13(14), 3596. https://doi.org/10.3390/cancers13143596