
Differences in Extracellular Vesicle Protein Cargo Are Dependent on Head and Neck Squamous Cell Carcinoma Cell of Origin and Human Papillomavirus Status

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
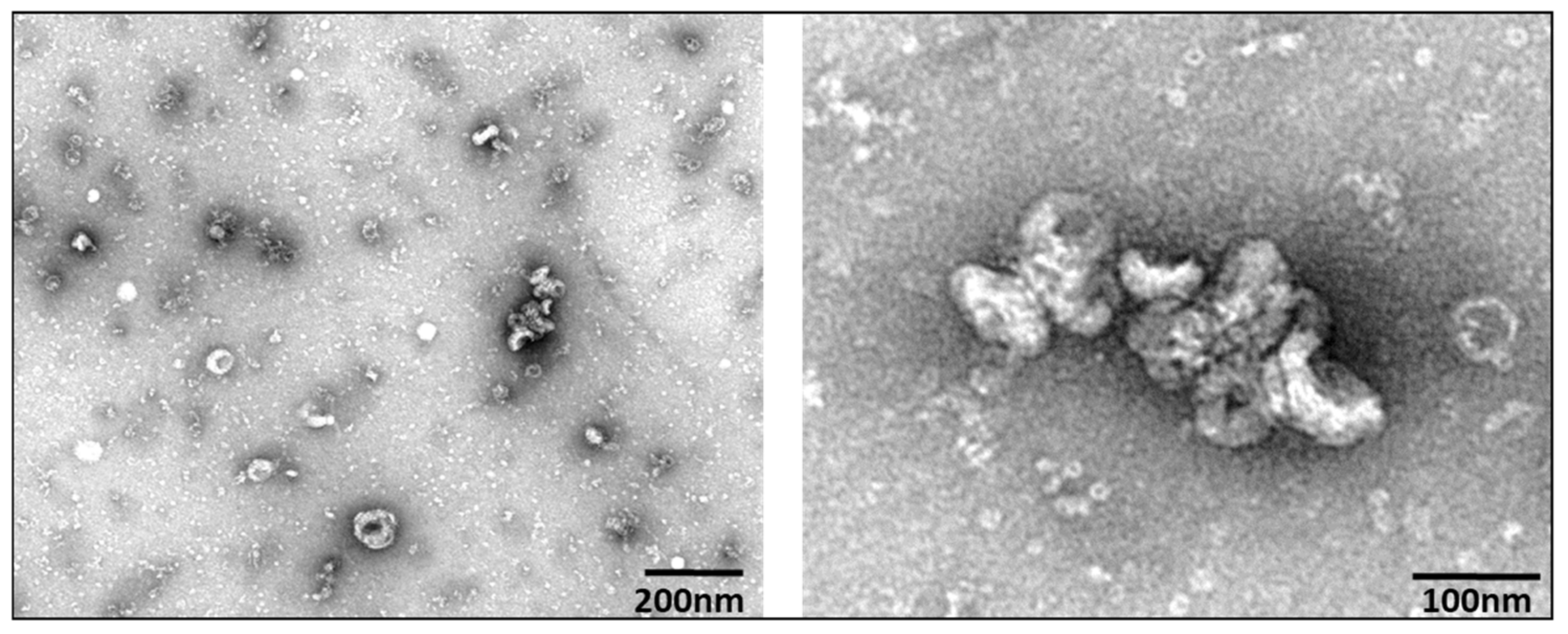
3.1. Extracellular Vesicle Characterization
3.2. Western Confirmation of EV Markers
3.3. Proteomic Analysis by Mass Spectrometry
3.4. Western Analysis of Candidate Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.; Khwaja, S.S.; Baker, C.M.; Gay, H.A.; Thorstad, W.L.; Daly, M.D.; Lewis, J.S.; Wang, X. Prognostic micro RNA signatures derived from The Cancer Genome Atlas for head and neck squamous cell carcinomas. Cancer Med. 2016, 5, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Prim. 2020, 6, 1–22. [Google Scholar] [CrossRef]
- Leemans, C.R.; Braakhuis, B.J.M.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2010, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Applebaum, K.M.; Furniss, C.S.; Zeka, A.; Posner, M.R.; Smith, J.F.; Bryan, J.; Eisen, E.A.; Peters, E.S.; McClean, M.D.; Kelsey, K.T. Lack of Association of Alcohol and Tobacco with HPV16-Associated Head and Neck Cancer. J. Natl. Cancer Inst. 2007, 99, 1801–1810. [Google Scholar] [CrossRef] [Green Version]
- Gillison, M.L.; D’Souza, G.; Westra, W.; Sugar, E.; Xiao, W.; Begum, S.; Viscidi, R. Distinct Risk Factor Profiles for Human Papillomavirus Type 16–Positive and Human Papillomavirus Type 16–Negative Head and Neck Cancers. J. Natl. Cancer Inst. 2008, 100, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Morris, L.G.; Sikora, A.G.; Patel, S.G.; Hayes, R.B.; Ganly, I. Second Primary Cancers after an Index Head and Neck Cancer: Subsite-Specific Trends in the Era of Human Papillomavirus–Associated Oropharyngeal Cancer. J. Clin. Oncol. 2011, 29, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quon, H.; Cohen, M.A.; Montone, K.T.; Ziober, A.F.; Wang, L.P.; Weinstein, G.S.; O’Malley, B.W. Transoral robotic surgery and adjuvant therapy for oropharyngeal carcinomas and the influence of p16INK4a on treatment outcomes. Laryngoscope 2013, 123, 635–640. [Google Scholar] [CrossRef]
- Haughey, B.H.; Hinni, M.L.; Salassa, J.R.; Hayden, R.E.; Grant, D.G.; Rich, J.T.; Milov, S.; Lewis, J.S.; Krishna, M. Transoral laser microsurgery as primary treatment for advanced-stage oropharyngeal cancer: A united states multicenter study. Head Neck 2011, 33, 1683–1694. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteside, T.L. The potential of tumor-derived exosomes for noninvasive cancer monitoring. Expert Rev. Mol. Diagn. 2015, 15, 1293–1310. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Rai, A.J.; DeCastro, G.J.; Zeringer, E.; Barta, T.; Magdaleno, S.; Setterquist, R.; Vlassov, A.V. An optimized procedure for exosome isolation and analysis using serum samples: Application to cancer biomarker discovery. Methods 2015, 87, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Tumor-Derived Exosomes and Their Role in Cancer Progression. Adv. Clin. Chem. 2016, 74, 103–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, S.; Malinowska, K.; Zöller, M. Exosomal Tumor MicroRNA Modulates Premetastatic Organ Cells. Neoplasia 2013, 15, 281-IN31. [Google Scholar] [CrossRef] [Green Version]
- Hannafon, B.N.; Ding, W.-Q. Intercellular Communication by Exosome-Derived microRNAs in Cancer. Int. J. Mol. Sci. 2013, 14, 14240–14269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhondt, B.; Rousseau, Q.; De Wever, O.; Hendrix, A. Function of extracellular vesicle-associated miRNAs in metastasis. Cell Tissue Res. 2016, 365, 621–641. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Investig. 2016, 126, 1208–1215. [Google Scholar] [CrossRef]
- Koga, K.; Matsumoto, K.; Akiyoshi, T.; Kubo, M.; Yamanaka, N.; Tasaki, A.; Nakashima, H.; Nakamura, M.; Kuroki, S.; Tanaka, M.; et al. Purification, characterization and biological significance of tumor-derived exosomes. Anticancer. Res. 2005, 25, 3703–3707. [Google Scholar]
- Jin, Y.; Chen, K.; Wang, Z.; Wang, Y.; Liu, J.; Lin, L.; Shao, Y.; Gao, L.; Yin, H.; Cui, C.; et al. DNA in serum extracellular vesicles is stable under different storage conditions. BMC Cancer 2016, 16, 753. [Google Scholar] [CrossRef] [Green Version]
- Ralhan, R.; Masui, O.; DeSouza, L.V.; Matta, A.; Macha, M.; Siu, K.W.M. Identification of proteins secreted by head and neck cancer cell lines using LC-MS/MS: Strategy for discovery of candidate serological biomarkers. J. Proteom. 2011, 11, 2363–2376. [Google Scholar] [CrossRef]
- Dowling, P.; Wormald, R.; Meleady, P.; Henry, M.; Curran, A.; Clynes, M. Analysis of the saliva proteome from patients with head and neck squamous cell carcinoma reveals differences in abundance levels of proteins associated with tumour progression and metastasis. J. Proteom. 2008, 71, 168–175. [Google Scholar] [CrossRef]
- Park, N.-H.; Min, B.-M.; Li, S.-L.; Huang, M.Z.; Doniger, J. Immortalization of normal human oral keratinocytes with type 16 human papillomavirus. Carcinogenesis 1991, 12, 1627–1631. [Google Scholar] [CrossRef] [PubMed]
- Castilho, R.M.; Squarize, C.; Leelahavanichkul, K.; Zheng, Y.; Bugge, T.; Gutkind, J.S. Rac1 Is Required for Epithelial Stem Cell Function during Dermal and Oral Mucosal Wound Healing but Not for Tissue Homeostasis in Mice. PLoS ONE 2010, 5, e10503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and Characterization of Exosomes from Cell Culture Supernatants and Biological Fluids. Curr. Protoc. Cell Biol. 2006, 30, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Rider, M.A.; Hurwitz, S.N.; Meckes, D.G., Jr. ExtraPEG: A Polyethylene Glycol-Based Method for Enrichment of Extracellular Vesicles. Sci. Rep. 2016, 6, 23978. [Google Scholar] [CrossRef]
- Tank, E.M.; Figueroa-Romero, C.; Hinder, L.M.; Bedi, K.; Archbold, H.C.; Li, X.; Weskamp, K.; Safren, N.; Paez-Colasante, X.; Pacut, C.; et al. Abnormal RNA stability in amyotrophic lateral sclerosis. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- McAlister, G.C.; Nusinow, D.; Jedrychowski, M.P.; Wühr, M.; Huttlin, E.; Erickson, B.; Rad, R.; Haas, W.; Gygi, S.P. MultiNotch MS3 Enables Accurate, Sensitive, and Multiplexed Detection of Differential Expression across Cancer Cell Line Proteomes. Anal. Chem. 2014, 86, 7150–7158. [Google Scholar] [CrossRef] [PubMed]
- Martins, T.; Catita, J.; Rosa, I.M.; Silva, O.A.; Henriques, A.G. Exosome isolation from distinct biofluids using precipitation and column-based approaches. PLoS ONE 2018, 13, e0198820. [Google Scholar] [CrossRef] [Green Version]
- Lässer, C.; Eldh, M.; Lötvall, J. Isolation and Characterization of RNA-Containing Exosomes. J. Vis. Exp. 2012, 2012, e3037. [Google Scholar] [CrossRef]
- Hartjes, T.A.; Mytnyk, S.; Jenster, G.W.; Van Steijn, V.; Van Royen, M.E. Extracellular Vesicle Quantification and Characterization: Common Methods and Emerging Approaches. Bioengineering 2019, 6, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midwood, K.S.; Hussenet, T.; Langlois, B.; Orend, G. Advances in tenascin-C biology. Cell Mol. Life Sci. 2011, 68, 3175–3199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midwood, K.S.; Orend, G. The role of tenascin-C in tissue injury and tumorigenesis. J. Cell Commun. Signal. 2009, 3, 287–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, N.; Maenaka, K. The Roles of Matricellular Proteins in Oncogenic Virus-Induced Cancers and Their Potential Utilities as Therapeutic Targets. Int. J. Mol. Sci. 2017, 18, 2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udalova, I.; Ruhmann, M.; Thomson, S.J.; Midwood, K.S. Expression and Immune Function of Tenascin-C. Crit. Rev. Immunol. 2011, 31, 115–145. [Google Scholar] [CrossRef] [PubMed]
- Lyons, A.; Jones, J. Cell adhesion molecules, the extracellular matrix and oral squamous carcinoma. Int. J. Oral Maxillofac. Surg. 2007, 36, 671–679. [Google Scholar] [CrossRef]
- Oskarsson, T.; Acharyya, S.; Zhang, X.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massague, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874. [Google Scholar] [CrossRef] [Green Version]
- Mirzaei, R.; Sarkar, S.; Dzikowski, L.; Rawji, K.S.; Khan, L.; Faissner, A.; Bose, P.; Yong, V.W. Brain tumor-initiating cells export tenascin-C associated with exosomes to suppress T cell activity. OncoImmunology 2018, 7, e1478647. [Google Scholar] [CrossRef] [Green Version]
- Nissen, N.I.; Karsdal, M.; Willumsen, N. Collagens and Cancer associated fibroblasts in the reactive stroma and its relation to Cancer biology. J. Exp. Clin. Cancer Res. 2019, 38, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Brown, Y.; Hua, S.; Tanwar, P.S. Extracellular matrix-mediated regulation of cancer stem cells and chemoresistance. Int. J. Biochem. Cell Biol. 2019, 109, 90–104. [Google Scholar] [CrossRef]
- Sundquist, E.; Kauppila, J.H.; Veijola, J.; Mroueh, R.; Lehenkari, P.; Laitinen, S.; Risteli, J.; Soini, Y.; Kosma, V.-M.; Sawazaki-Calone, I.; et al. Tenascin-C and fibronectin expression divide early stage tongue cancer into low- and high-risk groups. Br. J. Cancer 2017, 116, 640–648. [Google Scholar] [CrossRef] [Green Version]
- Lowy, C.M.; Oskarsson, T. Tenascin C in metastasis: A view from the invasive front. Cell Adhes. Migr. 2015, 9, 112–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauzenberger, D.; Olivier, P.; Gundersen, D.; Ruegg, C. Tenascin-C inhibits beta1 integrin-dependent T lymphocyte adhesion to fibronectin through the binding of its fnIII 1-5 repeats to fibronectin. Eur. J. Immunol. 1999, 29, 1435–1447. [Google Scholar] [CrossRef]
- Joo, J.; Omae, Y.; Hitomi, Y.; Park, B.; Shin, H.-J.; Yoon, K.-A.; Sawai, H.; Tsuiji, M.; Hayashi, T.; Kong, S.-Y.; et al. The association of integration patterns of human papilloma virus and single nucleotide polymorphisms on immune- or DNA repair-related genes in cervical cancer patients. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lacina, L.; Plzák, J.; Kodet, O.; Szabo, P.; Chovanec, M.; Dvořánková, B.; Smetana, K., Jr. Cancer Microenvironment: What Can We Learn from the Stem Cell Niche. Int. J. Mol. Sci. 2015, 16, 24094–24110. [Google Scholar] [CrossRef]
- Tiitta, O.; Happonen, R.-P.; Virtanen, I.; Luomanen, M. Distribution of tenascin in oral premalignant lesions and squamous cell carcinoma. J. Oral. Pathol. Med. 1994, 23, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Atula, T.; Hedström, J.; Finne, P.; Leivo, I.; Markkanen-Leppänen, M.; Haglund, C. Tenascin-C expression and its prognostic significance in oral and pharyngeal squamous cell carcinoma. Anticancer. Res. 2003, 23, 3051–3056. [Google Scholar] [PubMed]
- Xiong, A.; Yang, Z.; Shen, Y.; Zhou, J.; Shen, Q. Transcription Factor STAT3 as a Novel Molecular Target for Cancer Prevention. Cancers 2014, 6, 926–957. [Google Scholar] [CrossRef] [Green Version]
- Grandis, J.R.; Drenning, S.D.; Chakraborty, A.; Zhou, M.Y.; Zeng, Q.; Pitt, A.S.; Tweardy, D.J. Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor- mediated cell growth In vitro. J. Clin. Investig. 1998, 102, 1385–1392. [Google Scholar] [CrossRef] [Green Version]
- Geiger, J.L.; Grandis, J.R.; Bauman, J.E. The STAT3 pathway as a therapeutic target in head and neck cancer: Barriers and innovations. Oral. Oncol. 2016, 56, 84–92. [Google Scholar] [CrossRef] [Green Version]
- Morgan, E.; Wasson, C.W.; Hanson, L.; Kealy, D.; Pentland, I.; McGuire, V.; Scarpini, C.; Coleman, N.; Arthur, J.S.C.; Parish, J.L.; et al. STAT3 activation by E6 is essential for the differentiation-dependent HPV18 life cycle. PLOS Pathog. 2018, 14, e1006975. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.L.; Macdonald, A. Autocrine STAT3 activation in HPV positive cervical cancer through a virus-driven Rac1—NFκB—IL-6 signalling axis. PLOS Pathog. 2019, 15, e1007835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez, A.A.R.; Van Renne, N.; Baumert, T.F.; Lupberger, J. Viral manipulation of STAT3: Evade, exploit, and injure. PLOS Pathog. 2018, 14, e1006839. [Google Scholar] [CrossRef] [Green Version]
- Silva, T.G.; Crispim, J.C.O.; Miranda, F.A.; Hassumi, M.K.; De Mello, J.M.Y.; Simões, R.T.; Souto, F.; Soares, E.G.; Donadi, E.A.; Soares, C.P. Expression of the nonclassical HLA-G and HLA-E molecules in laryngeal lesions as biomarkers of tumor invasiveness. Histol. Histopathol. 2011, 26, 1487–1497. [Google Scholar] [PubMed]
- Kochan, G.; Escors-Murugarren, D.; Breckpot, K.; Guerrero-Setas, D. Role of non-classical MHC class I molecules in cancer immunosuppression. OncoImmunology 2013, 2, e26491. [Google Scholar] [CrossRef] [Green Version]
- Foroni, I.; Bettencourt, B.F.; Santos, M.; Lima, M.; Bruges-Armas, J. HLA-E, HLA-F and HLA-G—The Non-Classical Side of the MHC Cluster. In HLA and Associated Important Diseases; Xi, Y., Ed.; IntechOpen: London, UK, 2014. [Google Scholar]
- Wischhusen, J.; Waschbisch, A.; Wiendl, H. Immune-refractory cancers and their little helpers—An extended role for immunetolerogenic MHC molecules HLA-G and HLA-E? Semin. Cancer Biol. 2007, 17, 459–468. [Google Scholar] [CrossRef]
- D’Souza, M.P.; Adams, E.; Altman, J.D.; Birnbaum, M.; Boggiano, C.; Casorati, G.; Chien, Y.-H.; Conley, A.; Eckle, S.B.G.; Früh, K.; et al. Casting a wider net: Immunosurveillance by nonclassical MHC molecules. PLOS Pathog. 2019, 15, e1007567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosono, S.; Kawase, T.; Matsuo, K.; Watanabe, M.; Kajiyama, H.; Hirose, K.; Suzuki, T.; Kidokoro, K.; Ito, H.; Nakanishi, T.; et al. HLA-A Alleles and the Risk of Cervical Squamous Cell Carcinoma in Japanese Women. J. Epidemiol. 2010, 20, 295–301. [Google Scholar] [CrossRef] [Green Version]
- Ferns, D.M.; Heeren, A.M.; Samuels, S.; Bleeker, M.C.G.; De Gruijl, T.D.; Kenter, G.G.; Jordanova, E.S. Classical and non-classical HLA class I aberrations in primary cervical squamous- and adenocarcinomas and paired lymph node metastases. J. Immunother. Cancer 2016, 4, 78. [Google Scholar] [CrossRef] [Green Version]
- Gensterblum-Miller, E.; Brenner, J.C. Protecting Tumors by Preventing Human Papilloma Virus Antigen Presentation: Insights from Emerging Bioinformatics Algorithms. Cancers 2019, 11, 1543. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.-W.; Yang, Y.-C.; Lin, H.-F.; Lin, M.-F.; Cheng, Y.-W.; Chu, C.-C.; Tsao, Y.-P.; Chen, S.-L. Cytotoxic T-Lymphocyte Responses to Human Papillomavirus Type 16 E5 and E7 Proteins and HLA-A*0201-Restricted T-Cell Peptides in Cervical Cancer Patients. J. Virol. 2007, 81, 2869–2879. [Google Scholar] [CrossRef] [Green Version]
- Paaso, A.; Jaakola, A.; Syrjänen, S.; Louvanto, K. From HPV Infection to Lesion Progression: The Role of HLA Alleles and Host Immunity. Acta Cytol. 2019, 63, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and Genetic Properties of Tumors Associated with Local Immune Cytolytic Activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Eura, M.; Katsura, F.; Oiso, M.; Obata, A.; Nakano, K.; Masuyama, K.; Ishikawa, T. Frequency of HLA-A alleles in Japanese patients with head and neck cancer. Jpn. J. Clin. Oncol. 1999, 29, 535–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Näsman, A.; Andersson, E.; Marklund, L.; Tertipis, N.; Hammarstedt-Nordenvall, L.; Attner, P.; Nyberg, T.; Masucci, G.V.; Munck-Wikland, E.; Ramqvist, T.; et al. HLA Class I and II Expression in Oropharyngeal Squamous Cell Carcinoma in Relation to Tumor HPV Status and Clinical Outcome. PLoS ONE 2013, 8, e77025. [Google Scholar] [CrossRef] [Green Version]
- Tisch, M.; Kyrberg, H.; Weidauer, H.; Mytilineos, J.; Conradt, C.; Opelz, G.; Maier, H. Human Leukocyte Antigens and Prognosis in Patients With Head and Neck Cancer: Results of a Prospective Follow-up Study. Laryngoscope 2002, 112, 651–657. [Google Scholar] [CrossRef]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743.e13. [Google Scholar] [CrossRef] [Green Version]
- Network, T.C.G.A. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nat. Cell Biol. 2015, 517, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Müller, S.; Qian, G.; Xu, J.; Kim, S.; Chen, Z.; Jiang, N.; Wang, D.; Zhang, H.; Saba, N.F.; et al. Human papillomavirus 16 oncoprotein regulates the translocation of β-catenin via the activation of epidermal growth factor receptor. Cancer 2015, 121, 214–225. [Google Scholar] [CrossRef]
- Padhi, S.; Saha, A.; Kar, M.; Ghosh, C.; Adhya, A.; Baisakh, M.; Mohapatra, N.; Venkatesan, S.; Hande, M.P.; Banerjee, B. Clinico-Pathological Correlation of β-Catenin and Telomere Dysfunction in Head and Neck Squamous Cell Carcinoma Patients. J. Cancer 2015, 6, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Alamoud, K.; Kukuruzinska, M. Emerging Insights into Wnt/β-catenin Signaling in Head and Neck Cancer. J. Dent. Res. 2018, 97, 665–673. [Google Scholar] [CrossRef]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Zimmermann, M.; Tinhofer, I.; Kaufmann, A.M.; Albers, A.E. Epithelial-to-mesenchymal transition and cancer stem(-like) cells in head and neck squamous cell carcinoma. Cancer Lett. 2013, 338, 47–56. [Google Scholar] [CrossRef]
- Medici, D.; Hay, E.D.; Goodenough, D.A. Cooperation between Snail and LEF-1 Transcription Factors Is Essential for TGF-β1-induced Epithelial-Mesenchymal Transition. Mol. Biol. Cell 2006, 17, 1871–1879. [Google Scholar] [CrossRef] [Green Version]
- Chien, M.-H.; Chou, L.S.-S.; Chung, T.-T.; Lin, C.-H.; Chou, M.-Y.; Weng, M.-S.; Yang, S.-F.; Chen, M.-K. Effects of E-cadherin (CDH1) gene promoter polymorphisms on the risk and clinicopathologic development of oral cancer. Head Neck 2011, 34, 405–411. [Google Scholar] [CrossRef]
- Takeichi, M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science 1991, 251, 1451–1455. [Google Scholar] [CrossRef]
- Loh, C.-Y.; Chai, J.; Tang, T.; Wong, W.; Sethi, G.; Shanmugam, M.; Chong, P.; Looi, C. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [Green Version]
- Yao, D.; Dai, C.; Peng, S. Mechanism of the Mesenchymal–Epithelial Transition and Its Relationship with Metastatic Tumor Formation. Mol. Cancer Res. 2011, 9, 1608–1620. [Google Scholar] [CrossRef] [Green Version]
- Mitra, A.; Mishra, L.; Li, S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget 2015, 6, 10697–10711. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Filho, M.S.; Nör, J.E. The biology of head and neck cancer stem cells. Oral Oncol. 2012, 48, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, L.; Jayakar, S.K.; Ow, T.J.; Segall, J.E. Mechanisms of Invasion in Head and Neck Cancer. Arch. Pathol. Lab. Med. 2015, 139, 1334–1348. [Google Scholar] [CrossRef] [Green Version]
- Ihler, F.; Gratz, R.; Wolff, H.A.; Weiss, B.G.; Bertlich, M.; Kitz, J.; Salinas, G.; Rave-Fränk, M.; Canis, M. Epithelial-Mesenchymal Transition during Metastasis of HPV-Negative Pharyngeal Squamous Cell Carcinoma. BioMed Res. Int. 2018, 2018, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Wells, A.; Yates, C.; Shepard, C.R. E-cadherin as an indicator of mesenchymal to epithelial reverting transitions during the metastatic seeding of disseminated carcinomas. Clin. Exp. Metastasis 2008, 25, 621–628. [Google Scholar] [CrossRef] [Green Version]
- Yazdani, J.; Ghavimi, M.A.; Hagh, E.J.; Ahmadpour, F. The Role of E-Cadherin as a Prognostic Biomarker in Head and Neck Squamous Carcinoma: A Systematic Review and Meta-Analysis. Mol. Diagn. Ther. 2018, 22, 523–535. [Google Scholar] [CrossRef]
- Diniz-Freitas, M.; García-Caballero, T.; Antúnez-López, J.; Gándara-Rey, J.M.; Garcia-Garcia, A. Reduced E-cadherin expression is an indicator of unfavourable prognosis in oral squamous cell carcinoma. Oral Oncol. 2006, 42, 190–200. [Google Scholar] [CrossRef]
- Cioce, M.; Fazio, V. EphA2 and EGFR: Friends in Life, Partners in Crime. Can EphA2 Be a Predictive Biomarker of Response to Anti-EGFR Agents? Cancers 2021, 13, 700. [Google Scholar] [CrossRef]
- Pasquale, E.B. Eph-Ephrin Bidirectional Signaling in Physiology and Disease. Cell 2008, 133, 38–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soler, M.M.G.; Gehring, M.P.; Lechtenberg, B.C.; Zapata-Mercado, E.; Hristova, K.; Pasquale, E.B. Engineering nanomolar peptide ligands that differentially modulate EphA2 receptor signaling. J. Biol. Chem. 2019, 294, 8791–8805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateo-Lozano, S.; Bazzocco, S.; Rodrigues, P.; Mazzolini, R.; Andretta, E.; Dopeso, H.; Fernàndez, Y.; Del Llano, E.; Bilic, J.; Lopez, L.S.; et al. Loss of the EPH receptor B6 contributes to colorectal cancer metastasis. Sci. Rep. 2017, 7, 43702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barquilla, A.; Pasquale, E.B. Eph Receptors and Ephrins: Therapeutic Opportunities. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 465–487. [Google Scholar] [CrossRef] [Green Version]
- Barile, E.; Wang, S.; Das, S.K.; Noberini, R.; Dahl, R.; Stebbins, J.L.; Pasquale, E.B.; Fisher, P.B.; Pellecchia, M. Design, Synthesis and Bioevaluation of an EphA2 Receptor-Based Targeted Delivery System. ChemMedChem 2014, 9, 1403–1412. [Google Scholar] [CrossRef]
- Riedl, S.J.; Pasquale, S.J.R.A.E.B. Targeting the Eph System with Peptides and Peptide Conjugates. Curr. Drug Targets 2015, 16, 1031–1047. [Google Scholar] [CrossRef]
- Boyd, A.W.; Bartlett, P.F.; Lackmann, M. Therapeutic targeting of EPH receptors and their ligands. Nat. Rev. Drug Discov. 2014, 13, 39–62. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Y.; Wang, H.-B.; Zhang, A.; Chen, M.-L.; Fang, Z.-X.; Dong, X.-D.; Li, S.-B.; Du, Y.; Xiong, D.; et al. Ephrin receptor A2 is an epithelial cell receptor for Epstein–Barr virus entry. Nat. Microbiol. 2018, 3, 164–171. [Google Scholar] [CrossRef]
- Saloura, V.; Izumchenko, E.; Zuo, Z.; Bao, R.; Korzinkin, M.; Ozerov, I.; Zhavoronkov, A.; Sidransky, D.; Bedi, A.; Hoque, M.O.; et al. Immune profiles in primary squamous cell carcinoma of the head and neck. Oral Oncol. 2019, 96, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lin, P.; Sun, B.; Zhang, S.; Cai, W.; Han, C.; Li, L.; Lu, H.; Zhao, X. Epithelial-Mesenchymal Transition Regulated by EphA2 Contributes to Vasculogenic Mimicry Formation of Head and Neck Squamous Cell Carcinoma. BioMed Res. Int. 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodie, T.M.; Tosevski, V.; Medova, M. OMIP-045: Characterizing human head and neck tumors and cancer cell lines with mass cytometry. Cytom. Part A 2018, 93, 406–410. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Yang, X.; Si, H.; Saleh, A.D.; Xiao, W.; Coupar, J.; Gollin, S.M.; Ferris, R.L.; Issaeva, N.; Yarbrough, W.G.; et al. Genomic and Transcriptomic Characterization Links Cell Lines with Aggressive Head and Neck Cancers. Cell Rep. 2018, 25, 1332–1345.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nisa, L.; Francica, P.; Giger, R.; Medo, M.; Elicin, O.; Friese-Hamim, M.; Wilm, C.; Stroh, C.; Bojaxhiu, B.; Quintin, A.; et al. Targeting the MET Receptor Tyrosine Kinase as a Strategy for Radiosensitization in Locoregionally Advanced Head and Neck Squamous Cell Carcinoma. Mol. Cancer Ther. 2019, 19, 614–626. [Google Scholar] [CrossRef]
- Shang, X.; Lin, X.; Howell, S.B. Claudin-4 controls the receptor tyrosine kinase EphA2 pro-oncogenic switch through beta-catenin. Cell Commun. Signal. 2014, 12, 59. [Google Scholar] [CrossRef] [Green Version]
- Zantek, N.; Azimi, M.; Fedor-Chaiken, M.; Wang, B.; Brackenbury, R.; Kinch, M.S. E-cadherin regulates the function of the EphA2 receptor tyrosine kinase. Cell Growth Differ. Mol. Boil. J. Am. Assoc. Cancer Res. 1999, 10, 629–638. [Google Scholar]
- Li, Z.; Liao, J.; Yang, Z.; Choi, E.Y.; Lapidus, R.G.; Liu, X.; Cullen, K.J.; Dan, H. Co-targeting EGFR and IKKβ/NF-κB signalling pathways in head and neck squamous cell carcinoma: A potential novel therapy for head and neck squamous cell cancer. Br. J. Cancer 2019, 120, 306–316. [Google Scholar] [CrossRef] [Green Version]
- Broek, R.V.; Mohan, S.; Eytan, D.; Chen, Z.; Van Waes, C. The PI3K/Akt/mTOR axis in head and neck cancer: Functions, aberrations, cross-talk, and therapies. Oral Dis. 2013, 21, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhang, S.; MacLennan, G.T.; Eble, J.N.; López-Beltrán, A.; Yang, X.J.; Pan, C.-X.; Zhou, H.; Montironi, R.; Cheng, L. Epidermal Growth Factor Receptor Protein Expression and Gene Amplification in Small Cell Carcinoma of the Urinary Bladder. Clin. Cancer Res. 2007, 13, 953–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milanezi, F.; Carvalho, S.; Schmitt, F.C. EGFR/HER2 in breast cancer: A biological approach for molecular diagnosis and therapy. Expert Rev. Mol. Diagn. 2008, 8, 417–434. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, I.; Vorsteher, N.; Bühler, H.; Evers, K.; Sehouli, J.; Schaller, G.; Kümmel, S. The prognostic significance of human epidermal growth factor receptor correlations in squamous cell cervical carcinoma. Anticancer. Res. 2007, 27, 959–963. [Google Scholar] [PubMed]
- Voelzke, W.R.; Petty, W.J.; Lesser, G.J. Targeting the Epidermal Growth Factor Receptor in High-Grade Astrocytomas. Curr. Treat. Options Oncol. 2008, 9, 23–31. [Google Scholar] [CrossRef]
- Tu, J.; Yu, Y.; Liu, W.; Chen, S. Significance of human epidermal growth factor receptor 2 expression in colorectal cancer. Exp. Ther. Med. 2015, 9, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Chen, L.; Sheng, L.; Nordgren, H.; Wester, K.; Carlsson, J. EGFR, HER2 and HER3 expression in esophageal primary tumours and corresponding metastases. Int. J. Oncol. 2007, 31, 493–499. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.A.; Lee, H.S.; Lee, H.E.; Jeon, Y.K.; Yang, H.K.; Kim, W.H. EGFR in gastric carcinomas: Prognostic significance of protein overexpression and high gene copy number. Histopathology 2008, 52, 738–746. [Google Scholar] [CrossRef]
- Tang, X.; Varella-Garcia, M.; Xavier, A.C.; Massarelli, E.; Ozburn, N.C.; Moran, C.A.; Wistuba, I.I. Epidermal Growth Factor Receptor Abnormalities in the Pathogenesis and Progression of Lung Adenocarcinomas. Cancer Prev. Res. 2008, 1, 192–200. [Google Scholar] [CrossRef] [Green Version]
- Lafky, J.M.; Wilken, J.A.; Baron, A.; Maihle, N.J. Clinical implications of the ErbB/epidermal growth factor (EGF) receptor family and its ligands in ovarian cancer. Biochim. et Biophys. Acta (BBA) Bioenerg. 2008, 1785, 232–265. [Google Scholar] [CrossRef]
- Nicholson, R.; Gee, J.; Harper, M. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37, 9–15. [Google Scholar] [CrossRef]
- Bs, M.E.S.; Ferris, R.L.; Ferrone, S.; Grandis, J.R. Epidermal growth factor receptor targeted therapy of squamous cell carcinoma of the head and neck. Head Neck 2010, 32, 1412–1421. [Google Scholar] [CrossRef]
- Rodriguez, I.; Finbow, M.E.; Alonso, A. Binding of human papillomavirus 16 E5 to the 16 kDa subunit c (proteolipid) of the vacuolar H+-ATPase can be dissociated from the E5-mediated epidermal growth factor receptor overactivation. Oncogene 2000, 19, 3727–3732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, S.M.G. Requirement of Epidermal Growth Factor Receptor for Hyperplasia Induced by E5, a High-Risk Human Papillomavirus Oncogene. Cancer Res. 2005, 65, 6534–6542. [Google Scholar] [CrossRef] [Green Version]
- Wetherill, L.; Holmes, K.K.; Verow, M.; Müller, M.; Howell, G.; Harris, M.; Fishwick, C.; Stonehouse, N.; Foster, R.; Blair, G.; et al. High-Risk Human Papillomavirus E5 Oncoprotein Displays Channel-Forming Activity Sensitive to Small-Molecule Inhibitors. J. Virol. 2012, 86, 5341–5351. [Google Scholar] [CrossRef] [Green Version]
- Wasson, C.W.; Morgan, E.L.; Müller, M.; Ross, R.L.; Hartley, M.; Roberts, S.; Macdonald, A. Human papillomavirus type 18 E5 oncogene supports cell cycle progression and impairs epithelial differentiation by modulating growth factor receptor signalling during the virus life cycle. Oncotarget 2017, 8, 103581–103600. [Google Scholar] [CrossRef] [Green Version]
- Cassell, A.; Grandis, J.R. Investigational EGFR-targeted therapy in head and neck squamous cell carcinoma. Expert Opin. Investig. Drugs 2010, 19, 709–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harari, P.M.; Wheeler, D.L.; Grandis, J.R. Molecular Target Approaches in Head and Neck Cancer: Epidermal Growth Factor Receptor and Beyond. Semin. Radiat. Oncol. 2009, 19, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Werner, S.; Keller, L.; Pantel, K. Epithelial keratins: Biology and implications as diagnostic markers for liquid biopsies. Mol. Asp. Med. 2020, 72, 100817. [Google Scholar] [CrossRef]
- Karantza, V. Keratins in health and cancer: More than mere epithelial cell markers. Oncogene 2010, 30, 127–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagi, T.; Watanabe, M.; Hata, H.; Kitamura, S.; Imafuku, K.; Yanagi, H.; Homma, A.; Wang, L.; Takahashi, H.; Shimizu, H.; et al. Loss of TRIM29 Alters Keratin Distribution to Promote Cell Invasion in Squamous Cell Carcinoma. Cancer Res. 2018, 78, 6795–6806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.K.; Yin, Y.; Chae, H.S.; Cho, S.-G. Opposing Regulation of Cancer Properties via KRT19-Mediated Differential Modulation of Wnt/β-Catenin/Notch Signaling in Breast and Colon Cancers. Cancers 2019, 11, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, J.-H.; Oh, S.; Lee, K.-M.; Yang, W.; Nam, K.S.; Moon, H.-G.; Noh, D.-Y.; Kim, C.G.; Park, G.; Park, J.B.; et al. Cytokeratin19 induced by HER2/ERK binds and stabilizes HER2 on cell membranes. Cell Death Differ. 2015, 22, 665–676. [Google Scholar] [CrossRef] [Green Version]
- Ohtsuka, T.; Sakaguchi, M.; Yamamoto, H.; Tomida, S.; Takata, K.; Shien, K.; Hashida, S.; Miyata-Takata, T.; Watanabe, M.; Suzawa, K.; et al. Interaction of cytokeratin 19 head domain and HER2 in the cytoplasm leads to activation of HER2-Erk pathway. Sci. Rep. 2016, 6, 39557. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Kawano, S.; Hattori, T.; Matsubara, R.; Sakamoto, T.; Hashiguchi, Y.; Kaneko, N.; Mikami, Y.; Morioka, M.; Maruse, Y.; et al. Cytokeratin 19 as a biomarker of highly invasive oral squamous cell carcinoma with metastatic potential. J. Oral Maxillofac. Surg. Med. Pathol. 2020, 32, 1–7. [Google Scholar] [CrossRef]
- Chan, J.K.; Yuen, D.; Too, P.H.-M.; Sun, Y.; Willard, B.; Man, D.; Tam, C. Keratin 6a reorganization for ubiquitin–proteasomal processing is a direct antimicrobial response. J. Cell Biol. 2017, 217, 731–744. [Google Scholar] [CrossRef] [Green Version]
- Gu, L.-H.; Coulombe, P.A. Keratin function in skin epithelia: A broadening palette with surprising shades. Curr. Opin. Cell Biol. 2007, 19, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Gu, X.; Moore-Medlin, T.N.; Nathan, C.-A.; Hutt-Fletcher, L.M. Oral dysplasia and squamous cell carcinoma: Correlation between increased expression of CD21, Epstein-Barr virus and CK19. Oral Oncol. 2012, 48, 836–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, T.; Sugihara, K.; Hirayama, Y.; Hirano, M.; Tanuma, J.-I.; Semba, I. Immunohistological evaluation of Ki-67, p63, CK19 and p53 expression in oral epithelial dysplasias. J. Oral Pathol. Med. 2006, 35, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Alsharif, S.; Fallatah, A.; Chung, B.M. Intermediate Filaments as Effectors of Cancer Development and Metastasis: A Focus on Keratins, Vimentin, and Nestin. Cells 2019, 8, 497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Wong, P.; Coulombe, P.A. A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nat. Cell Biol. 2006, 441, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Ikenberg, K.; Apel, B.; Schumann, D.; Huber, G.; Studer, G.; Rordorf, T.; Riesterer, O.; Rössle, M.; Korol, D.; et al. Expression of CK19 is an independent predictor of negative outcome for patients with squamous cell carcinoma of the tongue. Oncotarget 2016, 7, 76151–76158. [Google Scholar] [CrossRef]
- Zhong, L.-P.; Chen, W.-T.; Zhang, C.-P.; Zhang, Z.-Y. Increased CK19 expression correlated with pathologic differentiation grade and prognosis in oral squamous cell carcinoma patients. Oral Surgery Oral Med. Oral Pathol. Oral Radiol. Endodontology 2007, 104, 377–384. [Google Scholar] [CrossRef]
- Ram Prassad, V.V.; Nirmala, N.R.; Kotian, M.S. Immunohistochemical evaluation of expression of cytokeratin 19 in different histological grades of leukoplakia and oral squamous cell carcinoma. Indian J. Dent. Res. 2005, 16, 6–11. [Google Scholar]
- Tada, H.; Takahashi, H.; Kuwabara-Yokobori, Y.; Shino, M.; Chikamatsu, K. Molecular profiling of circulating tumor cells predicts clinical outcome in head and neck squamous cell carcinoma. Oral Oncol. 2020, 102, 104558. [Google Scholar] [CrossRef]
- Ravindranath, N.M.; Shuler, C. Cell-surface density of complement restriction factors (CD46, CD55, and CD59): Oral squamous cell carcinoma versus other solid tumors. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontology 2007, 103, 231–239. [Google Scholar] [CrossRef]
- Uhlén, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [Green Version]
- Roumenina, L.T.; Daugan, M.V.; Petitprez, F.; Sautès-Fridman, C.; Fridman, W.H. Context-dependent roles of complement in cancer. Nat. Rev. Cancer 2019, 19, 698–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dakubo, G.D. Cancer Biomarkers in Body Fluids: Biomarkers in Proximal Fluids; Springer Nature: Cham, Switzerland, 2019; p. 296. [Google Scholar]
- Csősz, É.; Lábiscsák, P.; Kalló, G.; Márkus, B.; Emri, M.; Szabó, A.; Tar, I.; Tőzsér, J.; Kiss, C.; Márton, I. Proteomics investigation of OSCC-specific salivary biomarkers in a Hungarian population highlights the importance of identification of population-tailored biomarkers. PLoS ONE 2017, 12, e0177282. [Google Scholar] [CrossRef] [Green Version]
- Clayton, A.; Harris, C.L.; Court, J.; Mason, M.D.; Morgan, P. Antigen-presenting cell exosomes are protected from complement-mediated lysis by expression of CD55 and CD59. Eur. J. Immunol. 2003, 33, 522–531. [Google Scholar] [CrossRef]
- Ludwig, S.; Sharma, P.; Theodoraki, M.-N.; Pietrowska, M.; Yerneni, S.S.; Lang, S.; Ferrone, S.; Whiteside, T.L. Molecular and Functional Profiles of Exosomes From HPV(+) and HPV(−) Head and Neck Cancer Cell Lines. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Tumor Site | Tumor Stage | HPV Status | Patient Sex | Patient Age | Smoker? | Follow-Up |
|---|---|---|---|---|---|---|---|
| UM-SCC-38 | Tonsil | T2N2aM0 | Negative | Male | 60 | Yes | Patient died from disease one year after diagnosis |
| UM-SCC-47 | Lateral Tongue | T3N1M0 | HPV16 | Male | 53 | Yes | Patient died from disease within a year of diagnosis |
| UM-SCC-118 | Lateral Tongue | T4aN2M0 | Negative | Female | 23 | No | Patient died from disease within a year of diagnosis |
| UM-SCC-104 | Floor of Mouth | T4N2bM0 | HPV16 | Male | 56 | Yes | Patient died from disease within a year of diagnosis |
| UM-SCC-17a | Larynx | T1N0M0 | Negative | Female | 47 | Yes | Patient alive with no evidence of disease 14+ years after diagnosis |
| UM-SCC-105 | Larynx | T4N0M0 | HPV18 | Male | 51 | No | Patient alive with no evidence of disease 5+ years after diagnosis |
| UM-SCC-92 | Tongue | T2N0M0 | Negative | Female | 38 | No | Patient alive with no evidence of disease 4+ years after diagnosis |
| UPCI:SCC152 | Recurrent Hypopharynx | T2N1M0 | HPV16 | Male | 47 | Yes | Patient died from disease 4 years after diagnosis |
| Antibody | Clone | Supplier | Catalog # | Lot | Secondary |
|---|---|---|---|---|---|
| Annexin V | N/A | Novus | NB100-1930 | 4E15L33570 | Rabbit |
| βCatenin | D10AB | Cell Signaling | 8480 | 5 | Rabbit |
| Calnexin | N/A | Novus | NB100-1965ss | D-2 | Rabbit |
| CD59 | N/A | R & D Systems | AF1987 | KIF011911A | Goat |
| CD9 | C-4 | Santa Cruz | SC-13118 | E1719 | Mouse |
| Cyclin D1 | H-295 | Santa Cruz | sc-753 | G2315 | Rabbit |
| Cytokeratin 19 | A53-B/A2 | Santa Cruz | SC6278 | D1618 | Mouse |
| E-Cadherin | 180215 | R & D Systems | MAB1838 | JAT0219051 | Mouse |
| EGFR | N/A | Origene | TA312545 | 20100915076 | Rabbit |
| EPHA2 | C-3 | Santa Cruz | SC-398832 | D2519 | Mouse |
| HLA-A | EP1395Y | Abcam | ab52922 | GR25873 | Rabbit |
| HPV16 E7 | N/A | Santa Cruz | SC-65711 | I1216 | Mouse |
| p16 | N/A | Ventana | 725-4793 | E04025 | Mouse |
| p53 | DO-1 | Neomarkers | MS-187-p | 187p1201E | Mouse |
| Rb | 1F8 | Neomarkers | MS-107-p1 | 107p1607F | Mouse |
| STAT3 | 124H6 | Cell Signaling | 9139 | 7 | Mouse |
| Tenascin-C | EPR4219 | Abcam | ab108930 | GR3209212-7 | Rabbit |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goudsmit, C.; da Veiga Leprevost, F.; Basrur, V.; Peters, L.; Nesvizhskii, A.; Walline, H. Differences in Extracellular Vesicle Protein Cargo Are Dependent on Head and Neck Squamous Cell Carcinoma Cell of Origin and Human Papillomavirus Status. Cancers 2021, 13, 3714. https://doi.org/10.3390/cancers13153714
Goudsmit C, da Veiga Leprevost F, Basrur V, Peters L, Nesvizhskii A, Walline H. Differences in Extracellular Vesicle Protein Cargo Are Dependent on Head and Neck Squamous Cell Carcinoma Cell of Origin and Human Papillomavirus Status. Cancers. 2021; 13(15):3714. https://doi.org/10.3390/cancers13153714
Chicago/Turabian StyleGoudsmit, Christine, Felipe da Veiga Leprevost, Venkatesha Basrur, Lila Peters, Alexey Nesvizhskii, and Heather Walline. 2021. "Differences in Extracellular Vesicle Protein Cargo Are Dependent on Head and Neck Squamous Cell Carcinoma Cell of Origin and Human Papillomavirus Status" Cancers 13, no. 15: 3714. https://doi.org/10.3390/cancers13153714
APA StyleGoudsmit, C., da Veiga Leprevost, F., Basrur, V., Peters, L., Nesvizhskii, A., & Walline, H. (2021). Differences in Extracellular Vesicle Protein Cargo Are Dependent on Head and Neck Squamous Cell Carcinoma Cell of Origin and Human Papillomavirus Status. Cancers, 13(15), 3714. https://doi.org/10.3390/cancers13153714