
Endogenous and Therapeutic Estrogens: Maestro Conductors of the Microenvironment of ER+ Breast Cancers


Abstract
:Simple Summary
Abstract
1. Introduction
2. Local Immune Environments of Clinical ER+ Breast Cancers
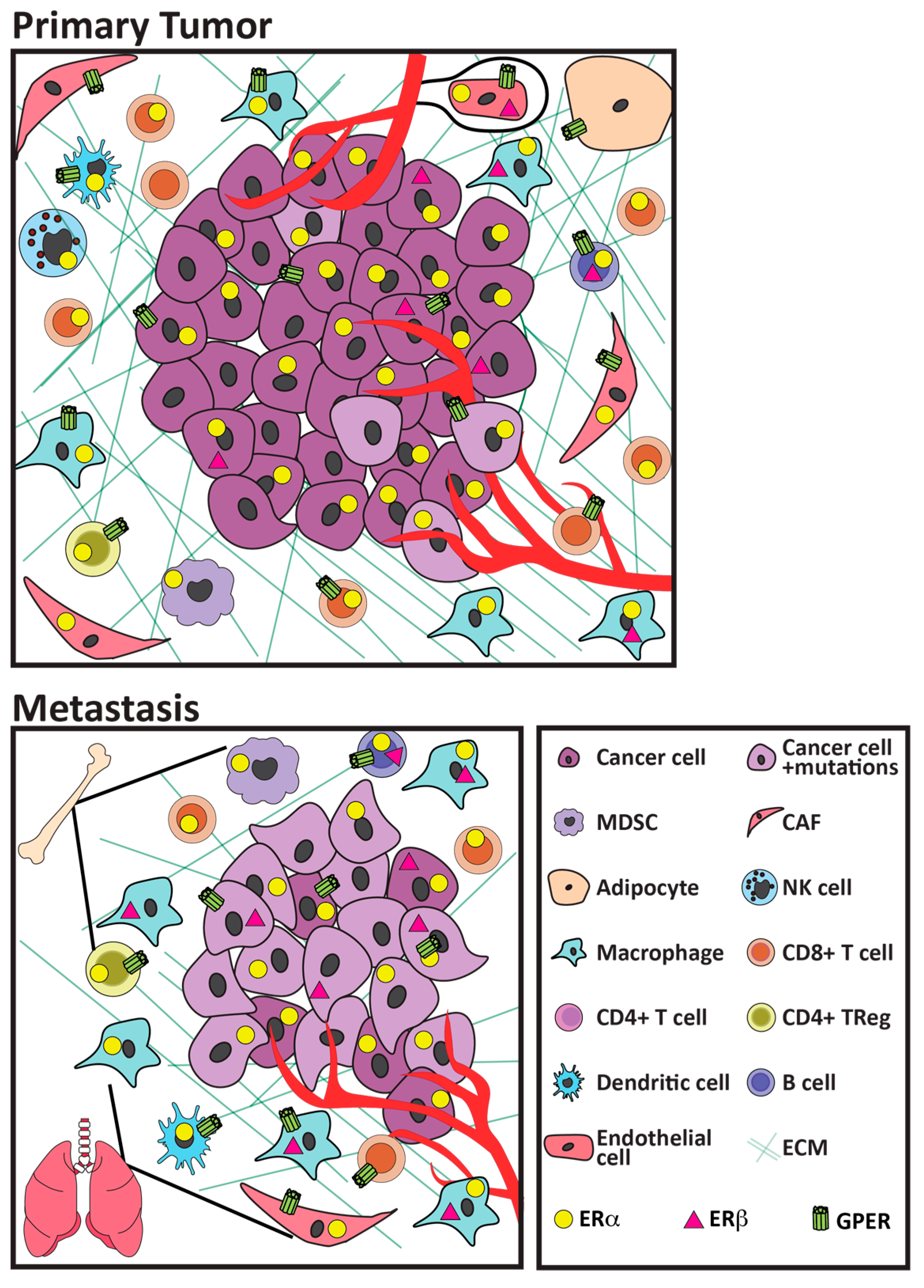
2.1. Immune Microenvironment of Primary ER+ Tumors
2.2. Influence of Somatic Mutations in Oncogenic Drivers and Anti-Estrogen Resistance
2.3. Differences between the Microenvironments of Primary and Metastatic ER+ Tumors
2.4. Bone as a Metastatic Site
2.5. Summary
3. Deposition, Composition, and Architecture of the Extracellular Matrix
4. Estrogen Receptors
4.1. ERα
4.2. ERβ
4.3. G Protein Coupled Estrogen Receptor (GPER)
4.4. Summary
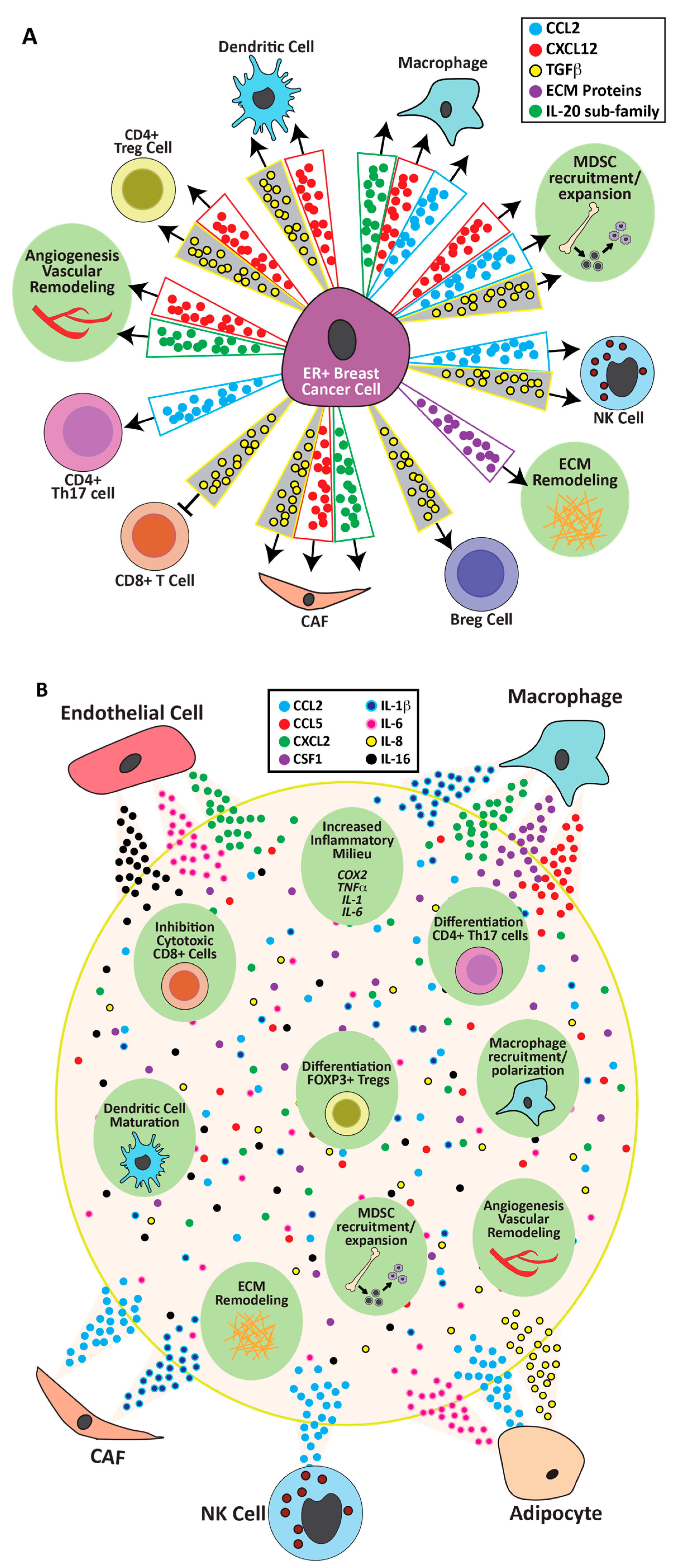
5. Cell Specific Estrogenic Regulation of Genes Which Would Modulate the Immune Microenvironment of ER+ Breast Cancers
5.1. Tumor Epithelia
5.2. Myofibroblasts/Cancer-Associated Fibroblasts (CAFs)/Immune Cells
5.3. Endothelial Cells
5.4. Multiple Cell Type Contributions to Extracellular Matrix Remodeling
5.5. Summary
6. Net Outcomes of Manipulation of Estrogen Activity In Vivo
6.1. Effects of Manipulation of Estrogen Activity in Experimental Models
6.2. Effects of Anti-Estrogen Treatments on the Microenvironment of Clinical ER+ Cancers
6.3. Summary
7. Conclusions and Important Areas of Future Study
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics. CA Cancer J. Clin. 2020, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, C.K.; Schiff, R. Mechanisms of Endocrine Resistance in Breast Cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef] [Green Version]
- Nardone, A.; de Angelis, C.; Trivedi, M.V.; Osborne, C.K.; Schiff, R. The changing role of ER in endocrine resistance. Breast 2015, 24, S60–S66. [Google Scholar] [CrossRef]
- Nagaraj, G.; Ma, C. Revisiting the estrogen receptor pathway and its role in endocrine therapy for postmenopausal women with estrogen receptor-positive metastatic breast cancer. Breast Cancer Res. Treat. 2015, 150, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Katzenellenbogen, J.A.; Mayne, C.G.; Katzenellenbogen, B.S.; Greene, G.L.; Chandarlapaty, S. Structural underpinnings of oestrogen receptor mutations in endocrine therapy resistance. Nat. Rev. Cancer 2018, 18, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.A. Parallel progression of primary tumours and metastases. Nat. Rev. Cancer 2009, 9, 302–312. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor Metastasis: Molecular Insights and Evolving Paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Pan, H.; Gray, R.; Braybrooke, J.; Davies, C.; Taylor, C.; McGale, P.; Peto, R.; Pritchard, K.I.; Bergh, J.; Dowsett, M.; et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N. Engl. J. Med. 2017, 377, 1836–1846. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Giuliano, M.; Trivedi, M.V.; Schiff, R.; Osborne, C.K. Metastasis Dormancy in Estrogen Receptor–Positive Breast Cancer. Clin. Cancer Res. 2013, 19, 6389–6397. [Google Scholar] [CrossRef] [Green Version]
- Riggio, A.I.; Varley, K.E.; Welm, A.L. The lingering mysteries of metastatic recurrence in breast cancer. Br. J. Cancer 2020, 124, 13–26. [Google Scholar] [CrossRef]
- Vesely, M.D.; Schreiber, R.D. Cancer immunoediting: Antigens, mechanisms, and implications to cancer immunotherapy. Ann. N. Y. Acad. Sci. 2013, 1284, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galon, J.; Bruni, D. Tumor Immunology and Tumor Evolution: Intertwined Histories. Immunity 2020, 52, 55–81. [Google Scholar] [CrossRef]
- Kelly, P.N. The Cancer Immunotherapy Revolution. Science 2018, 359, 1344–1345. [Google Scholar] [CrossRef] [Green Version]
- Darvin, P.; Toor, S.M.; Nair, V.S.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 1–3. [Google Scholar] [CrossRef]
- Miller, L.D.; Chou, J.A.; Black, M.; Print, C.; Chifman, J.; Alistar, A.; Putti, T.; Zhou, X.; Bedognetti, D.; Hendrickx, W.; et al. Immunogenic Subtypes of Breast Cancer Delineated by Gene Classifiers of Immune Responsiveness. Cancer Immunol. Res. 2016, 4, 600–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luen, S.; Virassamy, B.; Savas, P.; Salgado, R.; Loi, S. The genomic landscape of breast cancer and its interaction with host immunity. Breast 2016, 29, 241–250. [Google Scholar] [CrossRef]
- Dieci, M.V.; Griguolo, G.; Miglietta, F.; Guarneri, V. The immune system and hormone-receptor positive breast cancer: Is it really a dead end? Cancer Treat. Rev. 2016, 46, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H.; Domchek, S.M.; Clark, A.S. Immunotherapy for Breast Cancer: What Are We Missing? Clin. Cancer Res. 2017, 23, 2640–2646. [Google Scholar] [CrossRef] [Green Version]
- Wein, L.; Luen, S.J.; Savas, P.; Salgado, R.; Loi, S. Checkpoint blockade in the treatment of breast cancer: Current status and future directions. Br. J. Cancer 2018, 119, 4–11. [Google Scholar] [CrossRef] [Green Version]
- Straub, R.H. The Complex Role of Estrogens in Inflammation. Endocr. Rev. 2007, 28, 521–574. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Shapiro, D.J. The immune system and inflammation in breast cancer. Mol. Cell. Endocrinol. 2014, 382, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Kovats, S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell. Immunol. 2015, 294, 63–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttilla, I.K.; Adams, B.D.; White, B.A.; Ralpha, E. microRNAs, and the epithelial-mesenchymal transition in breast cancer. Trends Endocrinol. Metab. 2012, 23, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Maynadier, M.; Chambon, M.; Basile, I.; Gleizes, M.; Nirdé, P.; Gary-Bobo, M.; Garcia, M. Estrogens promote cell–cell adhesion of normal and malignant mammary cells through increased desmosome formation. Mol. Cell. Endocrinol. 2012, 364, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Simões, B.M.; Alferez, D.G.; Howell, S.; Clarke, R. The role of steroid hormones in breast cancer stem cells. Endocr. Related Cancer 2015, 22, T177–T186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bado, I.; Gugala, Z.; Fuqua, S.A.W.; Zhang, X.H.-F. Estrogen receptors in breast and bone: From virtue of remodeling to vileness of metastasis. Oncogene 2017, 36, 4527–4537. [Google Scholar] [CrossRef] [Green Version]
- Fornetti, J.; Welm, A.L.; Stewart, A.S. Understanding the Bone in Cancer Metastasis. J. Bone Miner. Res. 2018, 33, 2099–2113. [Google Scholar] [CrossRef] [Green Version]
- Somasundaram, A.; Rothenberger, N.J.; Stabile, L.P. The Impact of Estrogen in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1277, 33–52. [Google Scholar] [CrossRef] [PubMed]
- Segovia-Mendoza, M.; Morales-Montor, J. Immune Tumor Microenvironment in Breast Cancer and the Participation of Estrogen and Its Receptors in Cancer Physiopathology. Front Immunol. 2019, 10, 348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Zhou, J.; Chen, H.; Li, J.; Zhang, C.; Jiang, X.; Ni, C. The immunomodulatory effects of endocrine therapy in breast cancer. J. Exp. Clin. Cancer Res. 2021, 40, 1–16. [Google Scholar] [CrossRef]
- Stanton, S.E.; Adams, S.; Disis, M.L. Variation in the incidence and magnitude of tumor-infiltrating lymphocytes in breast cancer subtypes: A systematic review. JAMA Oncol. 2016, 2, 1354–1360. [Google Scholar] [CrossRef]
- Jackson, H.; Fischer, J.R.; Zanotelli, V.R.T.; Ali, H.R.; Mechera, R.; Soysal, S.D.; Moch, H.; Muenst, S.; Varga, Z.; Weber, W.P.; et al. The single-cell pathology landscape of breast cancer. Nat. Cell Biol. 2020, 578, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Nederlof, I.; De Bortoli, D.; Bareche, Y.; Nguyen, B.; De Maaker, M.; Hooijer, G.K.J.; Buisseret, L.; Kok, M.; Smid, M.; Van den Eynden, G.G.G.M.; et al. Comprehensive evaluation of methods to assess overall and cell-specific immune infiltrates in breast cancer. Breast Cancer Res. 2019, 21, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Perou, C.M.; Børresen-Dale, A.-L. Systems Biology and Genomics of Breast Cancer. Cold Spring Harb. Perspect. Biol. 2010, 3, a003293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, C.; METABRIC Group; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.; Speed, D.; Lynch, A.; Samarajiwa, S.; et al. he genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nat. Cell Biol. 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Budczies, J.; Bockmayr, M.; Denkert, C.; Klauschen, F.; Lennerz, J.K.; Györffy, B.; Dietel, M.; Loibl, S.; Weichert, W.; Stenzinger, A. Classical pathology and mutational load of breast cancer—Integration of two worlds. J. Pathol. Clin. Res. 2015, 1, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Phiel, K.L.; Henderson, R.A.; Adelman, S.J.; Elloso, M.M. Differential estrogen receptor gene expression in human peripheral blood mononuclear cell populations. Immunol. Lett. 2005, 97, 107–113. [Google Scholar] [CrossRef]
- Huang, M.; Li, Y.; Zhang, H.; Nan, F. Breast cancer stromal fibroblasts promote the generation of CD44+CD24- cells through SDF-1/CXCR4 interaction. J. Exp. Clin. Cancer Res. 2010, 29, 80. [Google Scholar] [CrossRef] [Green Version]
- Pierdominici, M.; Maselli, A.; Colasanti, T.; Giammarioli, A.M.; Delunardo, F.; Vacirca, D.; Sanchez, M.; Giovannetti, A.; Malorni, W.; Ortona, E. Estrogen receptor profiles in human peripheral blood lymphocytes. Immunol. Lett. 2010, 132, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Svoronos, N.; Perales-Puchalt, A.; Allegrezza, M.J.; Rutkowski, M.R.; Payne, K.; Tesone, A.J.; Nguyen, J.M.; Curiel, T.J.; Cadungog, M.G.; Singhal, S.; et al. Tumor Cell–Independent Estrogen Signaling Drives Disease Progression through Mobilization of Myeloid-Derived Suppressor Cells. Cancer Discov. 2016, 7, 72–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepermans, R.; Sharma, G.; Prossnitz, E. G Protein-Coupled Estrogen Receptor in Cancer and Stromal Cells: Functions and Novel Therapeutic Perspectives. Cells 2021, 10, 672. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Rapsomaniki, M.A.; Chevrier, S.; Anzeneder, T.; Langwieder, C.; Dykgers, A.; Rees, M.; Ramaswamy, A.; Muenst, S.; Soysal, S.D.; et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 2019, 177, 1330–1345.e18. [Google Scholar] [CrossRef] [Green Version]
- Pal, B.; Chen, Y.; Vaillant, F.; Capaldo, B.D.; Joyce, R.; Song, X.; Bryant, V.L.; Penington, J.S.; Di Stefano, L.; Ribera, N.T.; et al. A single-cell RNA expression atlas of normal, preneoplastic and tumorigenic states in the human breast. EMBO J. 2021, 40, e107333. [Google Scholar] [CrossRef]
- Denkert, C.; von Minckwitz, G.; Darb-Esfahani, S.; Lederer, B.; Heppner, B.I.; Weber, K.E.; Budczies, J.; Huober, J.; Klauschen, F.; Furlanetto, J.; et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: A pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2018, 19, 40–50. [Google Scholar] [CrossRef]
- Loi, S.; Sirtaine, N.; Piette, F.; Salgado, R.; Viale, G.; Van Eenoo, F.; Rouas, G.; Francis, P.; Crown, J.P.; Hitre, E.; et al. Prognostic and Predictive Value of Tumor-Infiltrating Lymphocytes in a Phase III Randomized Adjuvant Breast Cancer Trial in Node-Positive Breast Cancer Comparing the Addition of Docetaxel to Doxorubicin With Doxorubicin-Based Chemotherapy: BIG 02-98. J. Clin. Oncol. 2013, 31, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, C.; Bendahl, P.-O.; Ekholm, M.; Fernö, M.; Forsare, C.; Krüger, U.; Nordenskjöld, B.; Stål, O.; Rydén, L. Tumour-infiltrating lymphocytes as a prognostic and tamoxifen predictive marker in premenopausal breast cancer: Data from a randomised trial with long-term follow-up. Breast Cancer Res. 2020, 22, 1–14. [Google Scholar] [CrossRef]
- Gruosso, T.; Gigoux, M.; Manem, V.S.K.; Bertos, N.; Zuo, D.; Perlitch, I.; Saleh, S.M.I.; Zhao, H.; Souleimanova, M.; Johnson, R.M.; et al. Spatially distinct tumor immune microenvironments stratify triple-negative breast cancers. J. Clin. Investig. 2019, 129, 1785–1800. [Google Scholar] [CrossRef] [Green Version]
- Rugo, H.S.; Delord, J.-P.; Im, S.-A.; Ott, P.A.; Piha-Paul, S.; Bedard, P.; Sachdev, J.; Le Tourneau, C.; Van Brummelen, E.M.; Varga, A.; et al. Safety and Antitumor Activity of Pembrolizumab in Patients with Estrogen Receptor–Positive/Human Epidermal Growth Factor Receptor 2–Negative Advanced Breast Cancer. Clin. Cancer Res. 2018, 24, 2804–2811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirix, L.Y.; Takacs, I.; Jerusalem, G.; Nikolinakos, P.; Arkenau, H.-T.; Forero-Torres, A.; Boccia, R.; Lippman, M.E.; Somer, R.; Smakal, M.; et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: A phase 1b JAVELIN Solid Tumor study. Breast Cancer Res. Treat. 2017, 167, 671–686. [Google Scholar] [CrossRef] [Green Version]
- Waks, A.G.; Stover, D.G.; Guerriero, J.L.; Dillon, D.; Barry, W.T.; Gjini, E.; Hartl, C.; Lo, W.; Savoie, J.; Brock, J.; et al. The Immune Microenvironment in Hormone Receptor–Positive Breast Cancer Before and After Preoperative Chemotherapy. Clin. Cancer Res. 2019, 25, 4644–4655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Leun, A.M.; Thommen, D.S.; Schumacher, T.N. CD8+ T cell states in human cancer: Insights from single-cell analysis. Nat. Rev. Cancer 2020, 20, 218–232. [Google Scholar] [CrossRef]
- Hammerl, D.; Massink, M.P.G.; Smid, M.; van Deurzen, C.H.M.; Meijers-Heijboer, H.E.J.; Waisfisz, Q.; Debets, R.; Martens, J.W.M. Clonality, antigen recognition, and suppression of CD8(+) T Cells differentially affect prognosis of breast dancer subtypes. Clin. Cancer Res. 2020, 26, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Hammerl, D.; Smid, M.; Timmermans, A.; Sleijfer, S.; Martens, J.; Debets, R. Breast cancer genomics and immuno-oncological markers to guide immune therapies. Semin. Cancer Biol. 2018, 52, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Josefowicz, S.Z.; Lu, L.-F.; Rudensky, A.Y. Regulatory T Cells: Mechanisms of Differentiation and Function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef]
- Liu, S.; Foulkes, W.D.; Leung, S.; Gao, D.; Lau, S.; Kos, Z.; Nielsen, O.T. Prognostic significance of FOXP3+ tumor-infiltrating lymphocytes in breast cancer depends on estrogen receptor and human epidermal growth factor receptor-2 expression status and concurrent cytotoxic T-cell infiltration. Breast Cancer Res. 2014, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Qian, F.; Qingping, Y.; Linquan, W.; Xiaojin, H.; Rongshou, W.; Shanshan, R.; Wenjun, L.; Yong, H.; Enliang, L. High tumor-infiltrating FoxP3 + T cells predict poor survival in estrogen receptor-positive breast cancer: A meta-analysis. Eur. J. Surg. Oncol. 2017, 43, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Gao, Z.; Cai, Z.; Wang, M.; He, J. Clinicopathological and prognostic significance of FOXP3+ tumor infiltrating lymphocytes in patients with breast cancer: A meta-analysis. BMC Cancer 2015, 15, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Silva-Santos, B.; Serre, K.; Norell, H. Gammadelta T cells in cancer. Nat. Rev. Immunol. 2015, 15, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Morrow, E.S.; Roseweir, A.; Edwards, J. The role of gamma delta T lymphocytes in breast cancer: A review. Transl. Res. 2019, 203, 88–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chabab, G.; Barjon, C.; Bonnefoy, N.; Lafont, V. Pro-tumor gammadelta T Cells in human cancer: Polarization, mechanisms of action, and implications for therapy. Front Immunol. 2020, 11, 2186. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Ma, C.; Wang, F.; Hsueh, E.C.; Toth, K.; Huang, Y.; Mo, W.; Liu, S.; Han, B.; Varvares, M.A.; et al. Specific recruitment of gammadelta regulatory T cells in human breast cancer. Cancer Res. 2013, 73, 6137–6148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebestyen, Z.; Prinz, I.; Déchanet-Merville, J.; Silva-Santos, B.; Kuball, J. Translating gammadelta (γδ) T cells and their receptors into cancer cell therapies. Nat. Rev. Drug Discov. 2019, 19, 169–184. [Google Scholar] [CrossRef] [Green Version]
- Garaud, S.; Buisseret, L.; Solinas, C.; Gu-Trantien, C.; De Wind, A.; Eynden, G.V.D.; Naveaux, C.; Lodewyckx, J.-N.; Boisson, A.; Duvillier, H.; et al. Tumor-infiltrating B cells signal functional humoral immune responses in breast cancer. JCI Insight 2019, 4, e129641. [Google Scholar] [CrossRef] [Green Version]
- O’Meara, T.; Marczyk, M.; Qing, T.; Yaghoobi, V.; Blenman, K.; Cole, K.; Pelekanou, V.; Rimm, D.L.; Pusztai, L. Immunological Differences Between Immune-Rich Estrogen Receptor–Positive and Immune-Rich Triple-Negative Breast Cancers. JCO Precis. Oncol. 2020, 767–779. [Google Scholar] [CrossRef]
- Ishigami, E.; Sakakibara, M.; Sakakibara, J.; Masuda, T.; Fujimoto, H.; Hayama, S.; Nagashima, T.; Sangai, T.; Nakagawa, A.; Nakatani, Y.; et al. Coexistence of regulatory B cells and regulatory T cells in tumor-infiltrating lymphocyte aggregates is a prognostic factor in patients with breast cancer. Breast Cancer 2018, 26, 180–189. [Google Scholar] [CrossRef]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308.e36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anurag, M.; Zhu, M.; Huang, C.; Vasaikar, S.; Wang, J.; Hoog, J.; Burugu, S.; Gao, D.; Suman, V.; Zhang, X.H.; et al. Immune Checkpoint Profiles in Luminal B Breast Cancer (Alliance). J. Natl. Cancer Inst. 2019, 112, 737–746. [Google Scholar] [CrossRef]
- Svensson, S.; Abrahamsson, A.; Rodriguez, G.V.; Olsson, A.-K.; Jensen, L.; Cao, Y.; Dabrosin, C. CCL2 and CCL5 Are Novel Therapeutic Targets for Estrogen-Dependent Breast Cancer. Clin. Cancer Res. 2015, 21, 3794–3805. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Plüddemann, A.; Estrada, F.O.M. Macrophage heterogeneity in tissues: Phenotypic diversity and functions. Immunol. Rev. 2014, 262, 36–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Sanchez, L.R.; Borriello, L.; Entenberg, D.; Condeelis, J.S.; Oktay, M.H.; Karagiannis, G.S. The emerging roles of macrophages in cancer metastasis and response to chemotherapy. J. Leukoc. Biol. 2019, 106, 259–274. [Google Scholar] [CrossRef]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 123–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swierczak, A.; Pollard, J.W. Myeloid cells in metastasis. Cold Spring Harb. Perspect. Med. 2019, 10, a038026. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.J.; Ponik, S.M. Biomechanical Contributions to Macrophage Activation in the Tumor Microenvironment. Front. Oncol. 2020, 10, 787. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000 Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [Green Version]
- Chung, W.; Eum, H.H.; Lee, H.-O.; Lee, K.-M.; Lee, H.-B.; Kim, K.-T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef] [Green Version]
- Clark, N.M.; Martinez, L.M.; Murdock, S.; de Ligio, J.T.; Olex, A.L.; Effi, C.; Dozmorov, M.G.; Bos, P.D. Regulatory T cells support breast cancer progression by opposing IFN-gamma-dependent functional reprogramming of myeloid cells. Cell Rep. 2020, 33, 108482. [Google Scholar] [CrossRef]
- Pelekanou, V.; Villarroel-Espindola, F.; Schalper, K.A.; Pusztai, L.; Rimm, D.L. CD68, CD163, and matrix metalloproteinase 9 (MMP-9) co-localization in breast tumor microenvironment predicts survival differently in ER-positive and -negative cancers. Breast Cancer Res. 2018, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hollmén, M.; Roudnicky, F.; Karaman, S.; Detmar, M. Characterization of macrophage—cancer cell crosstalk in estrogen receptor positive and triple-negative breast cancer. Sci. Rep. 2015, 5, srep09188. [Google Scholar] [CrossRef]
- Hollmén, M.; Karaman, S.; Schwager, S.; Lisibach, A.; Christiansen, A.J.; Maksimow, M.; Varga, Z.; Jalkanen, S.; Detmar, M. G-CSF regulates macrophage phenotype and associates with poor overall survival in human triple-negative breast cancer. Onco. Immunol. 2015, 5, e1115177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrand-Rosenberg, S.; Fenselau, C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J. Immunol. 2018, 200, 422–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozga, A.J.; Chow, M.T.; Luster, A.D. Chemokines and the immune response to cancer. Immunity 2021, 54, 859–874. [Google Scholar] [CrossRef]
- Varn, F.S.; Andrews, E.H.; Mullins, D.W.; Cheng, C. Integrative analysis of breast cancer reveals prognostic haematopoietic activity and patient-specific immune response profiles. Nat. Commun. 2016, 7, 10248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, A.M.; Lim, E.; Ormandy, C.J.; Gallego-Ortega, D. The innate and adaptive infiltrating immune systems as targets for breast cancer immunotherapy. Endocr. Relat. Cancer 2017, 24, R123–R144. [Google Scholar] [CrossRef] [Green Version]
- Ali, H.R.; Chlon, L.; Pharoah, P.D.P.; Markowetz, F.; Caldas, C. Patterns of Immune Infiltration in Breast Cancer and Their Clinical Implications: A Gene-Expression-Based Retrospective Study. PLoS Med. 2016, 13, e1002194. [Google Scholar] [CrossRef]
- Bergenfelz, C.; Roxå, A.; Mehmeti, M.; Leandersson, K.; Larsson, A.-M. Clinical relevance of systemic monocytic-MDSCs in patients with metastatic breast cancer. Cancer Immunol. Immunother. 2020, 69, 435–448. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Shen, Y.; Huang, H.; Pan, S.; Jiang, J.; Chen, W.; Zhang, T.; Zhang, C.; Ni, C. A Rosetta Stone for Breast Cancer: Prognostic Value and Dynamic Regulation of Neutrophil in Tumor Microenvironment. Front. Immunol. 2020, 11, 1779. [Google Scholar] [CrossRef] [PubMed]
- Michea, P.; Noël, F.; Zakine, E.; Czerwinska, U.; Sirven, P.; Abouzid, O.; Goudot, C.; Scholer-Dahirel, A.; Vincent-Salomon, A.; Reyal, F.; et al. Adjustment of dendritic cells to the breast-cancer microenvironment is subset specific. Nat. Immunol. 2018, 19, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Marcus, A.; Gowen, B.; Thompson, T.W.; Iannello, A.; Ardolino, M.; Deng, W.; Wang, L.; Shifrin, N.; Raulet, D.H. Recognition of Tumors by the Innate Immune System and Natural Killer Cells. Adv. Immunol. 2014, 122, 91–128. [Google Scholar] [CrossRef] [Green Version]
- Franks, S.E.; Wolfson, B.; Hodge, J.W. Natural Born Killers: NK Cells in Cancer Therapy. Cancers 2020, 12, 2131. [Google Scholar] [CrossRef] [PubMed]
- Wellenstein, M.D.; de Visser, K.E. Cancer-cell-intrinsic mechanisms shaping the tumor immune landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef] [Green Version]
- Sobral-Leite, M.; Salomon, I.; Opdam, M.; Kruger, D.T.; Beelen, K.J.; Van Der Noort, V.; Van Vlierberghe, R.L.P.; Blok, E.J.; Giardiello, D.; Sanders, J.; et al. Cancer-immune interactions in ER-positive breast cancers: PI3K pathway alterations and tumor-infiltrating lymphocytes. Breast Cancer Res. 2019, 21, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Wright, K.L.; Adams, J.R.; Liu, J.C.; Loch, A.J.; Wong, R.G.; Jo, C.E.; Beck, L.A.; Santhanam, D.R.; Weiss, L.; Mei, X.; et al. Ras Signaling Is a Key Determinant for Metastatic Dissemination and Poor Survival of Luminal Breast Cancer Patients. Cancer Res. 2015, 75, 4960–4972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, S.N.; Wronski, A.; Castaño, Z.; Dake, B.; Malone, C.; De Raedt, T.; Enos, M.; Derose, Y.S.; Zhou, W.; Guerra, S.; et al. Loss of RasGAP Tumor Suppressors Underlies the Aggressive Nature of Luminal B Breast Cancers. Cancer Discov. 2016, 7, 202–217. [Google Scholar] [CrossRef] [Green Version]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertucci, F.; Ng, C.K.Y.; Patsouris, A.; Droin, N.; Piscuoglio, S.; Carbuccia, N.; Soria, J.C.; Dien, A.T.; Adnani, Y.; Kamal, M.; et al. Genomic characterization of metastatic breast cancers. Nat. Cell Biol. 2019, 569, 560–564. [Google Scholar] [CrossRef]
- Griffith, O.L.; Spies, N.C.; Anurag, M.; Griffith, M.; Luo, J.; Tu, D.; Yeo, B.; Kunisaki, J.; Miller, C.A.; Krysiak, K.; et al. The prognostic effects of somatic mutations in ER-positive breast cancer. Nat. Commun. 2018, 9, 3476. [Google Scholar] [CrossRef]
- Cullis, J.; Das, S.; Bar-Sagi, D. Kras and Tumor Immunity: Friend or Foe? Cold Spring Harb. Perspect. Med. 2017, 8, a031849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, P.D.; Guimarães, C.; Cardoso, A.; Mendonça, S.; Costa, A.; Oliveira, M.J.; Velho, S. KRAS Oncogenic Signaling Extends beyond Cancer Cells to Orchestrate the Microenvironment. Cancer Res. 2017, 78, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Hamarsheh, S.; Groß, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Szekely, B.; Bossuyt, V.; Li, X.; Wali, V.; Patwardhan, G.; Frederick, C.; Silber, A.; Park, T.; Harigopal, M.; Pelekanou, V.; et al. Immunological differences between primary and metastatic breast cancer. Ann. Oncol. 2018, 29, 2232–2239. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Narloch, J.L.; Onkar, S.; Joy, M.; Broadwater, G.; Luedke, C.; Hall, A.; Kim, R.; Pogue-Geile, K.; Sammons, S.; et al. Metastatic breast cancers have reduced immune cell recruitment but harbor increased macrophages relative to their matched primary tumors. J. Immunother. Cancer 2019, 7, 265. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.M.; Spoelstra, N.S.; Arnesen, S.; O’Neill, K.I.; Christenson, J.L.; Reese, J.; Torkko, K.C.; Goodspeed, A.; Rosas, E.; Hanamura, T.; et al. Steroid Hormone Receptor and Infiltrating Immune Cell Status Reveals Therapeutic Vulnerabilities of ESR1-Mutant Breast Cancer. Cancer Res. 2020, 81, 732–746. [Google Scholar] [CrossRef]
- Smid, M.; Wang, Y.; Zhang, Y.; Sieuwerts, A.M.; Yu, J.; Klijn, J.G.M.; Foekens, J.A.; Martens, J.W.M. Subtypes of Breast Cancer Show Preferential Site of Relapse. Cancer Res. 2008, 68, 3108–3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.U.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic Behavior of Breast Cancer Subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Molnár, I.A.; Molnár, B.; Vízkeleti, L.; Fekete, K.; Tamás, J.; Deák, P.; Szundi, C.; Székely, B.; Moldvay, J.; Vári-Kakas, S.; et al. Breast carcinoma subtypes show different patterns of metastatic behavior. Virchows Archiv. 2017, 470, 275–283. [Google Scholar] [CrossRef]
- Lee, H.; Na, K.J.; Choi, H. Differences in Tumor Immune Microenvironment in Metastatic Sites of Breast Cancer. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- de Lara, P.T.; Castañón, H.; Vermeer, M.; Núñez, N.; Silina, K.; Sobottka, B.; Urdinez, J.; Cecconi, V.; Yagita, H.; Attar, F.M.; et al. CD39+PD-1+CD8+ T cells mediate metastatic dormancy in breast cancer. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Farach-Carson, M.C.; Lin, S.-H.; Nalty, T.; Satcher, R.L. Sex Differences and Bone Metastases of Breast, Lung, and Prostate Cancers: Do Bone Homing Cancers Favor Feminized Bone Marrow? Front. Oncol. 2017, 7, 163. [Google Scholar] [CrossRef] [Green Version]
- Salvador, F.; Llorente, A.; Gomis, R.R. From latency to overt bone metastasis in breast cancer: Potential for treatment and prevention. J. Pathol. 2019, 249, 6–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonnell, D.P.; Wardell, S.E. The molecular mechanisms underlying the pharmacological actions of ER modulators: Implications for new drug discovery in breast cancer. Curr. Opin. Pharmacol. 2010, 10, 620–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traboulsi, T.; El Ezzy, M.; Gleason, J.; Mader, S. Antiestrogens: Structure-Activity relationships and use in breast cancer treatment. J. Mol. Endocrinol. 2017, 58, R15–R31. [Google Scholar] [CrossRef] [PubMed]
- Binder, N.B.; Niederreiter, B.; Hoffmann, O.; Stange, R.; Pap, T.; Stulnig, T.; Mack, M.; Erben, R.G.; Smolen, J.S.; Redlich, K. Estrogen-dependent and C-C chemokine receptor-2–dependent pathways determine osteoclast behavior in osteoporosis. Nat. Med. 2009, 15, 417–424. [Google Scholar] [CrossRef]
- Liu, C.; Zhao, Q.; Yu, X. Bone Marrow Adipocytes, Adipocytokines, and Breast Cancer Cells: Novel Implications in Bone Metastasis of Breast Cancer. Front. Oncol. 2020, 10, 561595. [Google Scholar] [CrossRef]
- Bado, I.L.; Zhang, W.; Hu, J.; Xu, Z.; Wang, H.; Sarkar, P.; Li, L.; Wan, Y.-W.; Liu, J.; Wu, W.; et al. The bone microenvironment increases phenotypic plasticity of ER+ breast cancer cells. Dev. Cell 2021, 56, 1100–1117.e9. [Google Scholar] [CrossRef]
- Harvey, J.M.; Clark, G.M.; Osborne, C.K.; Allred, D.C. Estrogen Receptor Status by Immunohistochemistry Is Superior to the Ligand-Binding Assay for Predicting Response to Adjuvant Endocrine Therapy in Breast Cancer. J. Clin. Oncol. 1999, 17, 1474. [Google Scholar] [CrossRef] [PubMed]
- Heindl, A.; Sestak, I.; Naidoo, K.; Cuzick, J.; Dowsett, M.; Yuan, Y. Relevance of Spatial Heterogeneity of Immune Infiltration for Predicting Risk of Recurrence After Endocrine Therapy of ER+ Breast Cancer. J. Natl. Cancer Inst. 2017, 110, 166–175. [Google Scholar] [CrossRef]
- Keely, P.J. Mechanisms by Which the Extracellular Matrix and Integrin Signaling Act to Regulate the Switch Between Tumor Suppression and Tumor Promotion. J. Mammary Gland. Biol. Neoplasia 2011, 16, 205–219. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Boulter, L.; Bullock, E.; Mabruk, Z.; Brunton, V.G. The fibrotic and immune microenvironments as targetable drivers of metastasis. Br. J. Cancer 2020, 124, 27–36. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Houthuijzen, J.M.; Jonkers, J. Cancer-associated fibroblasts as key regulators of the breast cancer tumor microenvironment. Cancer Metastasis Rev. 2018, 37, 577–597. [Google Scholar] [CrossRef] [PubMed]
- Pires, A.; Greenshields-Watson, A.; Jones, E.; Smart, K.; Lauder, S.N.; Somerville, M.; Milutinovic, S.; Kendrick, H.; Hindley, J.P.; French, R.; et al. Immune Remodeling of the Extracellular Matrix Drives Loss of Cancer Stem Cells and Tumor Rejection. Cancer Immunol. Res. 2020, 8, 1520–1531. [Google Scholar] [CrossRef]
- Maller, O.; Drain, A.P.; Barrett, A.S.; Borgquist, S.; Ruffell, B.; Zakharevich, I.; Pham, T.T.; Gruosso, T.; Kuasne, H.; Lakins, J.N.; et al. Tumour-associated macrophages drive stromal cell-dependent collagen crosslinking and stiffening to promote breast cancer aggression. Nat. Mater. 2020, 20, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Tomko, L.A.; Hill, R.C.; Barrett, A.; Szulczewski, J.M.; Conklin, M.; Eliceiri, K.; Keely, P.J.; Hansen, K.C.; Ponik, S.M. Targeted matrisome analysis identifies thrombospondin-2 and tenascin-C in aligned collagen stroma from invasive breast carcinoma. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Pickup, M.W.; Weaver, V.M. From transformation to metastasis: Deconstructing the extracellular matrix in breast cancer. Cancer Metastasis Rev. 2016, 35, 655–667. [Google Scholar] [CrossRef]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef]
- Lowy, C.M.; Oskarsson, T. Tenascin C in metastasis: A view from the invasive front. Cell Adhes. Migr. 2015, 9, 112–124. [Google Scholar] [CrossRef] [Green Version]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell 2019, 49, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Panciera, T.; Citron, A.; Di Biagio, D.; Battilana, G.; Gandin, A.; Giulitti, S.; Forcato, M.; Bicciato, S.; Panzetta, V.; Fusco, S.; et al. Reprogramming normal cells into tumour precursors requires ECM stiffness and oncogene-mediated changes of cell mechanical properties. Nat. Mater. 2020, 19, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Levental, K.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [Green Version]
- Barcus, C.E.; Keely, P.J.; Eliceiri, K.W.; Schuler, L.A. Prolactin signaling through focal adhesion complexes is amplified by stiff extracellular matrices in breast cancer cells. Oncotarget 2016, 7, 48093–48106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcus, C.E.; O’Leary, K.A.; Brockman, J.L.; Rugowski, D.E.; Liu, Y.; Garcia, N.; Yu, M.; Keely, P.J.; Eliceiri, K.W.; Schuler, L.A. Elevated collagen-I augments tumor progressive signals, intravasation and metastasis of prolactin-induced estrogen receptor alpha positive mammary tumor cells. Breast Cancer Res. 2017, 19, 9. [Google Scholar] [CrossRef] [Green Version]
- Mushtaq, M.U.; Papadas, A.; Pagenkopf, A.; Flietner, E.; Morrow, Z.; Chaudhary, S.G.; Asimakopoulos, F. Tumor matrix remodeling and novel immunotherapies: The promise of matrix-derived immune biomarkers. J. Immunother. Cancer 2018, 6, 65. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [Green Version]
- Shea, M.P.; O’Leary, K.A.; Wegner, K.A.; Vezina, C.M.; Schuler, L.A. High collagen density augments mTOR-dependent cancer stem cells in ERα+ mammary carcinomas and increases mTOR-independent lung metastases. Cancer Lett. 2018, 433, 1–9. [Google Scholar] [CrossRef]
- Pan, Z.; Tian, Y.; Cao, C.; Niu, G. The Emerging Role of YAP/TAZ in Tumor Immunity. Mol. Cancer Res. 2019, 17, 1777–1786. [Google Scholar] [CrossRef] [Green Version]
- Morciano, G.; Vezzani, B.; Missiroli, S.; Boncompagni, C.; Pinton, P.; Giorgi, C. An Updated Understanding of the Role of YAP in Driving Oncogenic Responses. Cancers 2021, 13, 3100. [Google Scholar] [CrossRef]
- Martinez, V.G.; Park, D.; Acton, S.E. Immunotherapy: Breaching the barriers for cancer treatment. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180214. [Google Scholar] [CrossRef]
- Conklin, M.; Eickhoff, J.C.; Riching, K.M.; Pehlke, C.A.; Eliceiri, K.; Provenzano, P.; Friedl, A.; Keely, P.J. Aligned Collagen Is a Prognostic Signature for Survival in Human Breast Carcinoma. Am. J. Pathol. 2011, 178, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Eliceiri, K.W.; Campbell, J.M.; Inman, D.R.; White, J.G.; Keely, P.J. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006, 4, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esbona, K.; Yi, Y.; Saha, S.; Yu, M.; Van Doorn, R.R.; Conklin, M.W.; Graham, D.S.; Wisinski, K.B.; Ponik, S.M.; Eliceiri, K.W.; et al. The Presence of Cyclooxygenase 2, Tumor-Associated Macrophages, and Collagen Alignment as Prognostic Markers for Invasive Breast Carcinoma Patients. Am. J. Pathol. 2018, 188, 559–573. [Google Scholar] [CrossRef] [Green Version]
- Brechbuhl, H.M.; Finlay-Schultz, J.; Yamamoto, T.M.; Gillen, A.; Cittelly, D.; Tan, A.C.; Sams, S.B.; Pillai, M.M.; Elias, A.D.; Robinson, W.; et al. Fibroblast Subtypes Regulate Responsiveness of Luminal Breast Cancer to Estrogen. Clin. Cancer Res. 2016, 23, 1710–1721. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2018, 18, 99–115. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejarano, L.; Jordāo, M.J.; Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef]
- Brechbuhl, H.M.; Barrett, A.S.; Kopin, E.; Hagen, J.C.; Han, A.L.; Gillen, A.E.; Finlay-Schultz, J.; Cittelly, D.M.; Owens, P.; Horwitz, K.B.; et al. Fibroblast subtypes define a metastatic matrisome in breast cancer. JCI Insight 2020, 5, e130751. [Google Scholar] [CrossRef] [Green Version]
- Pelon, F.; Bourachot, B.; Kieffer, Y.; Magagna, I.; Mermet-Meillon, F.; Bonnet, I.; Costa, A.; Givel, A.-M.; Attieh, Y.; Barbazan, J.; et al. Cancer-associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat. Commun. 2020, 11, 404. [Google Scholar] [CrossRef] [Green Version]
- Barcus, C.E.; Holt, E.C.; Keely, P.J.; Eliceiri, K.W.; Schuler, L.A. Dense Collagen-I Matrices Enhance Pro-Tumorigenic Estrogen-Prolactin Crosstalk in MCF-7 and T47D Breast Cancer Cells. PLoS ONE 2015, 10, e0116891. [Google Scholar] [CrossRef] [PubMed]
- Jallow, F.; O’Leary, K.A.; Rugowski, D.E.; Guerrero, J.F.; Ponik, S.; Schuler, L.A. Dynamic interactions between the extracellular matrix and estrogen activity in progression of ER+ breast cancer. Oncogene 2019, 38, 6913–6925. [Google Scholar] [CrossRef]
- Cortes, E.; Lachowski, D.; Robinson, B.; Sarper, M.; Teppo, J.S.; Thorpe, S.; Lieberthal, T.; Iwamoto, K.; Lee, A.D.; Okada-Hatakeyama, M.; et al. Tamoxifen mechanically reprograms the tumor microenvironment via HIF–1A and reduces cancer cell survival. EMBO Rep. 2018, 20, e46557. [Google Scholar] [CrossRef]
- Carpenter, R.; Miller, W.R. Role of aromatase inhibitors in breast cancer. Br. J. Cancer 2005, 93, S1–S5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, I.E.; Dowsett, M. Aromatase Inhibitors in Breast Cancer. N. Engl. J. Med. 2003, 348, 2431–2442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, J.; Come, S.; Jones, S.; Beex, L.; Kaufmann, M.; Makris, A.; Nortier, J.; Possinger, K.; Rutqvist, L.-E. Endocrine treatment options for advanced breast cancer—The role of fulvestrant. Eur. J. Cancer 2005, 41, 346–356. [Google Scholar] [CrossRef]
- Nardone, A.; Weir, H.; Delpuech, O.; Brown, H.; De Angelis, C.; Cataldo, M.L.; Fu, X.; Shea, M.J.; Mitchell, T.; Veeraraghavan, J.; et al. The oral selective oestrogen receptor degrader (SERD) AZD9496 is comparable to fulvestrant in antagonising ER and circumventing endocrine resistance. Br. J. Cancer 2018, 120, 331–339. [Google Scholar] [CrossRef]
- Márquez-Garbán, D.C.; Deng, G.; Comin-Anduix, B.; Garcia, A.J.; Xing, Y.; Chen, H.-W.; Cheung-Lau, G.; Hamilton, N.; Jung, M.E.; Pietras, R.J. Antiestrogens in combination with immune checkpoint inhibitors in breast cancer immunotherapy. J. Steroid Biochem. Mol. Biol. 2019, 193, 105415. [Google Scholar] [CrossRef]
- Lupien, M.; Meyer, C.A.; Bailey, S.T.; Eeckhoute, J.; Cook, J.; Westerling, T.; Zhang, X.; Carroll, J.S.; Rhodes, D.R.; Liu, X.S.; et al. Growth factor stimulation induces a distinct ER cistrome underlying breast cancer endocrine resistance. Genes Dev. 2010, 24, 2219–2227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammed, H.; Russell, I.A.; Stark, R.; Rueda, O.M.; Hickey, T.E.; Tarulli, G.A.; Serandour, A.A.; Birrell, S.N.; Bruna, A.; Saadi, A.; et al. Progesterone receptor modulates ER alpha action in breast cancer. Nature 2015, 523, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Hickey, T.E.; Selth, L.A.; Chia, K.M.; Laven-Law, G.; Milioli, H.H.; Roden, D.; Jindal, S.; Hui, M.; Finlay-Schultz, J.; Ebrahimie, E.; et al. The androgen receptor is a tumor suppressor in estrogen receptor–positive breast cancer. Nat. Med. 2021, 27, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Siersbæk, R.D.; Kumar, S.; Carroll, J. Signaling pathways and steroid receptors modulating estrogen receptor α function in breast cancer. Genes Dev. 2018, 32, 1141–1154. [Google Scholar] [CrossRef] [Green Version]
- Farcas, A.M.; Nagarajan, S.; Cosulich, S.; Carroll, J.S. Genome-Wide Estrogen Receptor Activity in Breast Cancer. Endocrinol. 2020, 162, bqaa224. [Google Scholar] [CrossRef] [PubMed]
- Franco, H.L.; Nagari, A.; Kraus, W.L.; Falpha, T.N. Signaling exposes latent estrogen receptor binding sites to alter the breast cancer cell transcriptome. Mol. Cell 2015, 58, 21–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frasor, J.; El-Shennawy, L.; Stender, J.D.; Kastrati, I. NFkB affects estrogen receptor expression and activity in breast cancer through multiple mechanisms. Mol. Cell Endocrinol. 2015, 418, 235–239. [Google Scholar] [CrossRef] [Green Version]
- Siersbæk, R.D.; Scabia, V.; Nagarajan, S.; Chernukhin, I.; Papachristou, E.K.; Broome, R.; Johnston, S.J.; Joosten, S.E.; Green, A.R.; Kumar, S.; et al. IL6/STAT3 Signaling Hijacks Estrogen Receptor α Enhancers to Drive Breast Cancer Metastasis. Cancer Cell 2020, 38, 412–423.e9. [Google Scholar] [CrossRef]
- Stender, J.D.; Nwachukwu, J.; Kastrati, I.; Kim, Y.; Strid, T.; Yakir, M.; Srinivasan, S.; Nowak, J.; Izard, T.; Rangarajan, E.S.; et al. Structural and Molecular Mechanisms of Cytokine-Mediated Endocrine Resistance in Human Breast Cancer Cells. Mol. Cell 2017, 65, 1122–1135.e5. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, R.; Picon-Ruiz, M.; Aurrekoetxea-Rodriguez, I.; de Paiva, V.N.; D’Amico, M.; Yoon, H.; Radhakrishnan, R.; Morata-Tarifa, C.; Ince, T.; Lippman, M.E.; et al. The Major Pre- and Postmenopausal Estrogens Play Opposing Roles in Obesity-Driven Mammary Inflammation and Breast Cancer Development. Cell Metab. 2020, 31, 1154–1172.e9. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-kB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Kastrati, I.; Joosten, S.E.P.; Semina, S.E.; Alejo, L.H.; Brovkovych, S.D.; Stender, J.D.; Horlings, H.M.; Kok, M.; Alarid, E.T.; Greene, G.L.; et al. The NF-kB pathway promotes tamoxifen tolerance and disease recurrence in estrogen receptor-positive breast cancers. Mol. Cancer Res. 2020, 18, 1018–1027. [Google Scholar] [CrossRef] [Green Version]
- Steele, C.W.; Jamieson, N.; Evans, T.R.J.; McKay, C.J.; Sansom, O.J.; Morton, J.; Carter, C.R. Exploiting inflammation for therapeutic gain in pancreatic cancer. Br. J. Cancer 2013, 108, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeselsohn, R.; De Angelis, C.; Brown, M.; Schiff, R. The evolving role of the estrogen receptor mutations in endocrine therapy-resistant creast cancer. Curr. Oncol. Rep. 2017, 19, 35. [Google Scholar] [CrossRef]
- Bahreini, A.; Li, Z.; Wang, P.; Levine, K.M.; Tasdemir, N.; Cao, L.; Weir, H.M.; Puhalla, S.L.; Davidson, N.E.; Stern, A.M.; et al. Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models. Breast Cancer Res. 2017, 19, 60. [Google Scholar] [CrossRef] [Green Version]
- Jeselsohn, R.; Bergholz, J.S.; Pun, M.; Cornwell, M.; Liu, W.; Nardone, A.; Xiao, T.; Li, W.; Qiu, X.; Buchwalter, G.; et al. Allele-Specific Chromatin Recruitment and Therapeutic Vulnerabilities of ESR1 Activating Mutations. Cancer Cell 2018, 33, 173–186.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laws, M.J.; Ziegler, Y.; Shahoei, S.H.; Dey, P.; Kim, S.H.; Yasuda, M.; Park, B.H.; Nettles, K.W.; Katzenellenbogen, J.A.; Nelson, E.R.; et al. Suppression of breast cancer metastasis and extension of survival by a new antiestrogen in a preclinical model driven by mutant estrogen receptors. Breast Cancer Res. Treat. 2020, 181, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Groenendijk, F.H.; Treece, T.; Yoder, E.; Baron, P.; Beitsch, P.; Audeh, W.; Dinjens, W.N.M.; Bernards, R.; Whitworth, P. Estrogen receptor variants in ER-positive basal-type breast cancers responding to therapy like ER-negative breast cancers. NPJ Breast Cancer 2019, 5, 15. [Google Scholar] [CrossRef]
- Bertucci, F.; Finetti, P.; Goncalves, A.; Birnbaum, D. The therapeutic response of ER+/HER2− breast cancers differs according to the molecular Basal or Luminal subtype. NPJ Breast Cancer 2020, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Ström, A.; Treuter, E.; Warner, M.; et al. Estrogen Receptors: How Do They Signal and What Are Their Targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Omoto, Y.; Iwase, H.; Yamashita, H.; Toyama, T.; Coombes, R.C.; Filipovic, A.; Warner, M.; Gustafsson, J.A. Differential expression of estrogen receptor alpha, beta1, and beta2 in lobular and ductal breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 1933–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawse, J.R.; Carter, J.M.; Aspros, K.; Bruinsma, E.S.; Koepplin, J.W.; Negron, V.; Subramaniam, M.; Ingle, J.N.; Rech, K.L.; Goetz, M.P. Optimized immunohistochemical detection of estrogen receptor beta using two validated monoclonal antibodies confirms its expression in normal and malignant breast tissues. Breast Cancer Res. Treat. 2019, 179, 241–249. [Google Scholar] [CrossRef]
- Chang, E.C.; Frasor, J.; Komm, B.; Katzenellenbogen, B.S. Impact of estrogen receptor beta on gene networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology 2006, 147, 4831–4842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haldosén, L.-A.; Zhao, C.; Dahlman-Wright, K. Estrogen receptor beta in breast cancer. Mol. Cell. Endocrinol. 2014, 382, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Mal, R.; Magner, A.; David, J.; Datta, J.; Vallabhaneni, M.; Kassem, M.; Manouchehri, J.; Willingham, N.; Stover, D.; Vandeusen, J.; et al. Estrogen Receptor Beta (ERbeta): A Ligand Activated Tumor Suppressor. Front Oncol. 2020, 10, 587386. [Google Scholar] [CrossRef]
- Barkhem, T.; Carlsson, B.; Nilsson, Y.; Enmark, E.; Gustafsson, J.; Nilsson, S. Differential response of estrogen receptor alpha and estrogen receptor beta to partial estrogen agonists/antagonists. Mol. Pharmacol. 1998, 54, 105–112. [Google Scholar] [CrossRef]
- Paech, K.; Webb, P.; Kuiper, G.G.; Nilsson, S.; Gustafsson, J.; Kushner, P.J.; Scanlan, T.S. Differential ligand activation of estrogen receptors ER alpha and ER beta at AP1 sites. Science 1997, 277, 1508–1510. [Google Scholar] [CrossRef]
- Peekhaus, N.T.; Chang, T.; Hayes, E.C.; Wilkinson, A.H.; Mitra, S.W.; Schaeffer, J.M.; Rohrer, S.P. Distinct effects of the antiestrogen Faslodex on the stability of estrogen receptors-alpha and -beta in the breast cancer cell line MCF-7. J. Mol. Endocrinol. 2004, 32, 987–995. [Google Scholar] [CrossRef] [Green Version]
- Sjöström, M.; Hartman, L.; Grabau, R.; Fornander, T.; Malmström, P.; Nordenskjöld, B.; Sgroi, D.C.; Skoog, L.; Stål, O.; Leeb-Lundberg, L.M.F.; et al. Lack of G protein-coupled estrogen receptor (GPER) in the plasma membrane is associated with excellent long-term prognosis in breast cancer. Breast Cancer Res. Treat. 2014, 145, 61–71. [Google Scholar] [CrossRef]
- Ignatov, T.; Claus, M.; Nass, N.; Haybaeck, J.; Seifert, B.; Kalinski, T.; Ortmann, O.; Ignatov, A. G-protein-coupled estrogen receptor GPER-1 expression in hormone receptor-positive breast cancer is associated with poor benefit of tamoxifen. Breast Cancer Res. Treat. 2018, 174, 121–127. [Google Scholar] [CrossRef]
- Hsu, L.-H.; Chu, N.-M.; Lin, Y.-F.; Kao, S.-H. G-Protein Coupled Estrogen Receptor in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 306. [Google Scholar] [CrossRef] [Green Version]
- De Marco, P.; Lappano, R.; De Francesco, E.M.; Cirillo, F.; Pupo, M.; Avino, S.; Vivacqua, A.; Abonante, S.; Picard, D.; Maggiolini, M. GPER signalling in both cancer-associated fibroblasts and breast cancer cells mediates a feedforward IL1beta/IL1R1 response. Sci. Rep. 2016, 6, 24354. [Google Scholar] [CrossRef] [Green Version]
- Cortes, E.; Sarper, M.; Robinson, B.; Lachowski, D.; Chronopoulos, A.; Thorpe, S.D.; Lee, D.A.; Hernández, A.E.D.R. GPER is a mechanoregulator of pancreatic stellate cells and the tumor microenvironment. EMBO Rep. 2018, 20, e46556. [Google Scholar] [CrossRef]
- Brunsing, R.L.; Owens, K.S.; Prossnitz, E.R. The G Protein-coupled Estrogen Receptor (GPER) Agonist G-1 Expands the Regulatory T-cell Population Under TH17-polarizing Conditions. J. Immunother. 2013, 36, 190–196. [Google Scholar] [CrossRef] [Green Version]
- Harding, A.T.; Goff, M.A.; Froggatt, H.M.; Lim, J.K.; Heaton, N.S. GPER1 is required to protect fetal health from maternal inflammation. Science 2021, 371, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Gallicchio, L.; Macdonald, R.; Wood, B.; Rushovich, E.; Helzlsouer, K.J. Androgens and musculoskeletal symptoms among breast cancer patients on aromatase inhibitor therapy. Breast Cancer Res. Treat. 2011, 130, 569–577. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets–update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, J.M.; Korach, K. Stromal Cell-Derived Factor 1, a Novel Target of Estrogen Receptor Action, Mediates the Mitogenic Effects of Estradiol in Ovarian and Breast Cancer Cells. Mol. Endocrinol. 2003, 17, 792–803. [Google Scholar] [CrossRef]
- Frasor, J.; Stossi, F.; Danes, J.M.; Komm, B.; Lyttle, C.R.; Katzenellenbogen, B.S. Selective estrogen receptor modulators: Discrimination of agonistic versus antagonistic activities by gene expression profiling in breast cancer cells. Cancer Res. 2004, 64, 1522–1533. [Google Scholar] [CrossRef] [Green Version]
- Pattarozzi, A.; Gatti, M.; Barbieri, F.; Wurth, R.; Porcile, C.; Lunardi, G.; Ratto, A.; Favoni, R.; Bajetto, A.; Ferrari, A.; et al. 17beta-estradiol promotes breast cancer cell proliferation-inducing stromal cell-derived factor-1-mediated epidermal growth factor receptor transactivation: Reversal by gefitinib pretreatment. Mol. Pharmacol. 2008, 73, 191–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Divekar, S.D.; Li, H.-H.; Parodi, D.A.; Ghafouri, T.B.; Chen, R.; Cyrus, K.; Foxworth, A.E.; Fornace, A.J.; Byrne, C.; Martin, M.B. Arsenite and cadmium promote the development of mammary tumors. Carcinogenesis 2019, 41, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Manjegowda, M.; Deb, G.; Kumar, N.; Limaye, A.M. Expression profiling of genes modulated by estrogen, EGCG or both in MCF-7 breast cancer cells. Genom. Data 2015, 5, 210–212. [Google Scholar] [CrossRef] [Green Version]
- Daniel, A.R.; Gaviglio, A.L.; Knutson, T.P.; Ostrander, J.H.; D’Assoro, A.B.; Ravindranathan, P.; Peng, Y.; Raj, G.V.; Yee, D.; Lange, C.A. Progesterone receptor-B enhances estrogen responsiveness of breast cancer cells via scaffolding PELP1- and estrogen receptor-containing transcription complexes. Oncogene 2014, 34, 506–515. [Google Scholar] [CrossRef] [Green Version]
- LeComte, S.; DeMay, F.; Pham, T.H.; Moulis, S.; Efstathiou, T.; Chalmel, F.; Pakdel, F. Deciphering the Molecular Mechanisms Sustaining the Estrogenic Activity of the Two Major Dietary Compounds Zearalenone and Apigenin in ER-Positive Breast Cancer Cell Lines. Nutr. 2019, 11, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, M.; Andreassen, T.; Jensen, L.; Bathen, T.F.; Sinha, I.; Gao, H.; Zhao, C.; Haldosen, L.-A.; Cao, Y.; Girnita, L.; et al. Estrogen Receptor α Promotes Breast Cancer by Reprogramming Choline Metabolism. Cancer Res. 2016, 76, 5634–5646. [Google Scholar] [CrossRef] [Green Version]
- Putnik, M.; Zhao, C.; Gustafsson, J.; Dahlman-Wright, K. Global identification of genes regulated by estrogen signaling and demethylation in MCF-7 breast cancer cells. Biochem. Biophys. Res. Commun. 2012, 426, 26–32. [Google Scholar] [CrossRef]
- Caffa, I.; Spagnolo, V.; Vernieri, C.; Valdemarin, F.; Becherini, P.; Wei, M.; Brandhorst, S.; Zucal, C.; Driehuis, E.; Ferrando, L.; et al. Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nat. Cell Biol. 2020, 583, 620–624. [Google Scholar] [CrossRef]
- Lin, Z.; Reierstad, S.; Huang, C.-C.; Bulun, S.E. Novel Estrogen Receptor-α Binding Sites and Estradiol Target Genes Identified by Chromatin Immunoprecipitation Cloning in Breast Cancer. Cancer Res. 2007, 67, 5017–5024. [Google Scholar] [CrossRef] [Green Version]
- Zielińska, K.A.; Katanaev, V.L. The Signaling Duo CXCL12 and CXCR4: Chemokine Fuel for Breast Cancer Tumorigenesis. Cancers 2020, 12, 3071. [Google Scholar] [CrossRef] [PubMed]
- Kohli, K.; Pillarisetty, V.G.; Kim, T.S. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. 2021, 1–12. [Google Scholar] [CrossRef]
- Mortezaee, K. CXCL12/CXCR4 axis in the microenvironment of solid tumors: A critical mediator of metastasis. Life Sci. 2020, 249, 117534. [Google Scholar] [CrossRef]
- Wu, W.; Qian, L.; Chen, X.; Ding, B. Prognostic significance of CXCL12, CXCR4, and CXCR7 in patients with breast cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 13217–13224. [Google Scholar] [PubMed]
- Ahirwar, D.K.; Nasser, M.W.; Ouseph, M.; Elbaz, M.; Cuitiño, M.C.; Kladney, R.D.; Varikuti, S.; Kaul, K.; Satoskar, A.R.; Ramaswamy, B.; et al. Fibroblast-derived CXCL12 promotes breast cancer metastasis by facilitating tumor cell intravasation. Oncogene 2018, 37, 4428–4442. [Google Scholar] [CrossRef]
- Soria, G.; Ben-Baruch, A. The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett. 2008, 267, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Miyazono, K. TGFbeta signalling: A complex web in cancer progression. Nat. Rev. Cancer 2010, 10, 415–424. [Google Scholar] [CrossRef]
- Bellomo, C.; Caja, L.; Moustakas, A. Transforming growth factor beta as regulator of cancer stemness and metastasis. Br. J. Cancer 2016, 115, 761–769. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Massague, J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Moses, H.; Barcellos-Hoff, M.H. TGF-beta biology in mammary development and breast cancer. Cold Spring Harbor Perspect Bio. 2011, 3, a003277. [Google Scholar]
- Joffroy, C.M.; Buck, M.B.; Stope, M.B.; Popp, S.L.; Pfizenmaier, K.; Knabbe, C. Antiestrogens induce transforming growth factor beta-mediated immunosuppression in breast cancer. Cancer Res. 2010, 70, 1314–1322. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wong, K.; Ouyang, W.; Rutz, S. Targeting IL-10 Family Cytokines for the Treatment of Human Diseases. Cold Spring Harb. Perspect. Biol. 2017, 11, a028548. [Google Scholar] [CrossRef]
- Niess, J.H.; Francés, R. Editorial: The IL-20 Cytokines and Related Family Members in Immunity and Diseases. Front. Immunol. 2019, 10, 1976. [Google Scholar] [CrossRef]
- Lu, S.-W.; Pan, H.-C.; Hsu, Y.-H.; Chang, K.-C.; Wu, L.-W.; Chen, W.-Y.; Chang, M.-S. IL-20 antagonist suppresses PD-L1 expression and prolongs survival in pancreatic cancer models. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Knower, K.C.; Chand, A.; Eriksson, N.; Takagi, K.; Miki, Y.; Sasano, H.; Visvader, J.E.; Lindeman, G.; Funder, J.W.; Fuller, P.; et al. Distinct nuclear receptor expression in stroma adjacent to breast tumors. Breast Cancer Res. Treat. 2013, 142, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsson, A.; Rodriguez, G.V.; Dabrosin, C. Fulvestrant-Mediated Attenuation of the Innate Immune Response Decreases ER+ Breast Cancer Growth In Vivo More Effectively than Tamoxifen. Cancer Res. 2020, 80, 4487–4499. [Google Scholar] [CrossRef] [PubMed]
- Hillers-Ziemer, L.E.; Arendt, L.M. Weighing the Risk: Effects of Obesity on the Mammary Gland and Breast Cancer Risk. J. Mammary Gland. Biol. Neoplasia 2020, 25, 115–131. [Google Scholar] [CrossRef]
- Zhang, Y.; Mikhaylova, L.; Kobzik, L.; Fedulov, A.V. Estrogen-mediated impairment of macrophageal uptake of environmental TiO2particles to explain inflammatory effect of TiO2on airways during pregnancy. J. Immunotoxicol. 2014, 12, 81–91. [Google Scholar] [CrossRef]
- Carreras, E.; Turner, S.; Frank, M.B.; Knowlton, N.; Osban, J.; Centola, M.; Park, C.G.; Simmons, A.; Alberola-Ila, J.; Kovats, S. Estrogen receptor signaling promotes dendritic cell differentiation by increasing expression of the transcription factor IRF4. Blood 2010, 115, 238–246. [Google Scholar] [CrossRef] [Green Version]
- Lemieux, C.; Cloutier, I.; Tanguay, J.-F. Estrogen-Induced Gene Expression in Bone Marrow c-kit+ Stem Cells and Stromal Cells: Identification of Specific Biological Processes Involved in the Functional Organization of the Stem Cell Niche. Stem Cells Dev. 2008, 17, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.; Greaves, E.; Critchley, H.; Saunders, P. Estrogen-dependent regulation of human uterine natural killer cells promotes vascular remodelling via secretion of CCL2. Hum. Reprod. 2015, 30, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Tai, P.; Wang, J.; Jin, H.; Song, X.; Yan, J.; Kang, Y.; Zhao, L.; An, X.; Du, X.; Chen, X.; et al. Induction of regulatory T cells by physiological level estrogen. J. Cell. Physiol. 2007, 214, 456–464. [Google Scholar] [CrossRef]
- Bai, J.; Qi, Q.-R.; Li, Y.; Day, R.; Makhoul, J.; Magness, R.R.; Chen, D.-B. Estrogen Receptors and Estrogen-Induced Uterine Vasodilation in Pregnancy. Int. J. Mol. Sci. 2020, 21, 4349. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Morton, J.S.; Davidge, S.T. Mechanisms of Estrogen Effects on the Endothelium: An Overview. Can. J. Cardiol. 2014, 30, 705–712. [Google Scholar] [CrossRef]
- Bowman, P.D.; Zhao, B.; Bynum, J.A. NCBI GEO Database GSE1486, GSE59558, GSE59558, GSE59558, GSE52649. 2004. Available online: https://www.ncbi.nlm.nih.gov/geo/ (accessed on 23 May 2021).
- Sobrino, A.; Mata, M.; Laguna-Fernández, A.; Novella, S.; Oviedo, P.J.; García-Pérez, M.A.; Tarín, J.J.; Caño, A.; Hermenegildo, C. Estradiol Stimulates Vasodilatory and Metabolic Pathways in Cultured Human Endothelial Cells. PLoS ONE 2009, 4, e8242. [Google Scholar] [CrossRef] [Green Version]
- Krüger-Genge, A.; Blocki, A.; Franke, R.-P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armingol, E.; Officer, A.; Harismendy, O.; Lewis, N.E. Deciphering cell–cell interactions and communication from gene expression. Nat. Rev. Genet. 2020, 22, 71–88. [Google Scholar] [CrossRef]
- Smart, E.; Semina, S.E.; Frasor, J. Update on the role of NFkappaB in promoting aggressive phenotypes of estrogen receptor-positive breast cancer. Endocrinology 2020, 161, bqaa152. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, K.A.; Shea, M.P.; Schuler, L.A. Modeling Prolactin Actions in Breast Cancer In Vivo: Insights from the NRL-PRL Mouse. In Recent Advances in Prolactin Research; Springer: Cham, Switzerland, 2014; Volume 846, pp. 201–220. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, K.A.; Shea, M.P.; Salituro, S.; Blohm, C.E.; Schuler, L.A. Prolactin Alters the Mammary Epithelial Hierarchy, Increasing Progenitors and Facilitating Ovarian Steroid Action. Stem Cell Rep. 2017, 9, 1167–1179. [Google Scholar] [CrossRef] [Green Version]
- Campbell, K.; O’Leary, K.A.; Rugowski, D.E.; Mulligan, W.A.; Barnell, E.; Skidmore, Z.; Krysiak, K.; Griffith, M.; Schuler, L.A.; Griffith, O. A Spontaneous Aggressive ERα+ Mammary Tumor Model Is Driven by Kras Activation. Cell Rep. 2019, 28, 1526–1537.e4. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nat. Cell Biol. 2005, 438, 820–827. [Google Scholar] [CrossRef]
- Oskarsson, T.; Acharyya, S.; Zhang, X.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massague, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874. [Google Scholar] [CrossRef] [Green Version]
- Hanker, A.B.; Sudhan, D.R.; Arteaga, C.L. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 2020, 37, 496–513. [Google Scholar] [CrossRef]
- Liang, X.; Briaux, A.; Becette, V.; Benoist, C.; Boulai, A.; Chemlali, W.; Schnitzler, A.; Baulande, S.; Rivera, S.; Mouret-Reynier, M.-A.; et al. Molecular profiling of hormone receptor-positive, HER2-negative breast cancers from patients treated with neoadjuvant endocrine therapy in the CARMINA 02 trial (UCBG-0609). J. Hematol. Oncol. 2018, 11, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skriver, S.K.; Jensen, M.-B.; Knoop, A.S.; Ejlertsen, B.; Laenkholm, A.-V. Tumour-infiltrating lymphocytes and response to neoadjuvant letrozole in patients with early oestrogen receptor-positive breast cancer: Analysis from a nationwide phase II DBCG trial. Breast Cancer Res. 2020, 22, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dunbier, A.K.; Ghazoui, Z.; Anderson, H.; Salter, J.; Nerurkar, A.; Osin, P.; A’Hern, R.; Miller, W.R.; Smith, I.E.; Dowsett, M. Molecular Profiling of Aromatase Inhibitor–Treated Postmenopausal Breast Tumors Identifies Immune-Related Correlates of Resistance. Clin. Cancer Res. 2013, 19, 2775–2786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patani, N.; Dunbier, A.; Anderson, H.; Ghazoui, Z.; Ribas, R.; Anderson, E.; Gao, Q.; A’Hern, R.; Mackay, A.; Lindemann, J.; et al. Differences in the Transcriptional Response to Fulvestrant and Estrogen Deprivation in ER-Positive Breast Cancer. Clin. Cancer Res. 2014, 20, 3962–3973. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.; Patani, N.; Dunbier, A.; Ghazoui, Z.; Zvelebil, M.; Martin, L.-A.; Dowsett, M. Effect of Aromatase Inhibition on Functional Gene Modules in Estrogen Receptor–Positive Breast Cancer and Their Relationship with Antiproliferative Response. Clin. Cancer Res. 2014, 20, 2485–2494. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.; on behalf of the POETIC Trial Management Group and Trialists; López-Knowles, E.; Cheang, M.C.U.; Morden, J.; Ribas, R.; Sidhu, K.; Evans, D.; Martins, V.; Dodson, A.; et al. Impact of aromatase inhibitor treatment on global gene expression and its association with antiproliferative response in ER+ breast cancer in postmenopausal patients. Breast Cancer Res. 2019, 22, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Generali, D.; Bates, G.; Berruti, A.; Brizzi, M.P.; Campo, L.; Bonardi, S.; Bersiga, A.; Allevi, G.; Milani, M.; Aguggini, S.; et al. Immunomodulation of FOXP3+ Regulatory T Cells by the Aromatase Inhibitor Letrozole in Breast Cancer Patients. Clin. Cancer Res. 2009, 15, 1046–1051. [Google Scholar] [CrossRef] [Green Version]
- Chan, M.S.M.; Wang, L.; Felizola, S.J.A.; Ueno, T.; Toi, M.; Loo, W.; Chow, L.W.C.; Suzuki, T.; Sasano, H. Changes of tumor infiltrating lymphocyte subtypes before and after neoadjuvant endocrine therapy in estrogen receptor-positive breast cancer patients—an immunohistochemical study of cd8+ and foxp3+ using double immunostaining with correlation to the pathobiological response of the patients. Int. J. Biol. Markers 2012, 27, 295–304. [Google Scholar] [CrossRef]
- Eliassen, A.H.; Missmer, S.A.; Tworoger, S.S.; Hankinson, S.E. Endogenous steroid hormone concentrations and risk of breast cancer: Does the association vary by a woman’s predicted breast cancer risk? J. Clin. Oncol. 2006, 24, 1823–1830. [Google Scholar] [CrossRef]
- Ogba, N.; Manning, N.G.; Bliesner, B.S.; Ambler, S.K.; Haughian, J.M.; Pinto, M.P.; Jedlicka, P.; Joensuu, K.; Heikkilä, P.; Horwitz, K.B. Luminal breast cancer metastases and tumor arousal from dormancy are promoted by direct actions of estradiol and progesterone on the malignant cells. Breast Cancer Res. 2014, 16, 489. [Google Scholar] [CrossRef] [Green Version]
- Holmberg, L.; Iversen, O.-E.; Rudenstam, C.M.; Hammar, M.; Kumpulainen, E.; Jaskiewicz, J.; Jassem, J.; Dobaczewska, D.; Fjosne, H.E.; Peralta, O.; et al. Increased Risk of Recurrence After Hormone Replacement Therapy in Breast Cancer Survivors. J. Natl. Cancer Inst. 2008, 100, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Manson, J.E.; Aragaki, A.K.; Rossouw, J.E.; Anderson, G.L.; Prentice, R.L.; LaCroix, A.Z.; Chlebowski, R.T.; Howard, B.V.; Thomson, C.A.; Margolis, K.L.; et al. Investigators, Menopausal hormone rherapy and long-term all-cause and cause-specific mortality: The women’s health initiative randomized trials. JAMA 2017, 318, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Von Schoultz, E.; Rutqvist, L.E. Stockholm Breast Cancer Study, Menopausal hormone therapy after breast cancer: The Stockholm randomized trial. J. Natl. Cancer Inst. 2005, 97, 533–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batur, P.; Blixen, C.E.; Moore, H.C.; Thacker, H.L.; Xu, M. Menopausal hormone therapy (HT) in patients with breast cancer. Maturitas 2006, 53, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcellos-Hoff, M.H.; Akhurst, R.J. Transforming growth factor-beta in breast cancer: Too much, too late. Breast Cancer Res. 2009, 1, 202. [Google Scholar] [CrossRef]
- Cyrus, K.; Wehenkel, M.; Choi, E.-Y.; Lee, H.; Swanson, H.; Kim, K.-B. Jostling for Position: Optimizing Linker Location in the Design of Estrogen Receptor-Targeting PROTACs. Chem. Med. Chem. 2010, 5, 979–985. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Xiang, H.; Luo, G. Targeting estrogen receptor α for degradation with PROTACs: A promising approach to overcome endocrine resistance. Eur. J. Med. Chem. 2020, 206, 112689. [Google Scholar] [CrossRef] [PubMed]
- Terranova-Barberio, M.; Pawlowska, N.; Dhawan, M.; Moasser, M.; Chien, A.J.; Melisko, M.E.; Rugo, H.; Rahimi, R.; Deal, T.; Daud, A.; et al. Exhausted T cell signature predicts immunotherapy response in ER-positive breast cancer. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Chun, B.M.; Page, D.B.; McArthur, H.L. Combination Immunotherapy Strategies in Breast Cancer. Curr. Breast Cancer Rep. 2019, 11, 228–240. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Zhang, X.H.F.; Rosen, J.M. TIME Is a Great Healer—Targeting Myeloid Cells in the Tumor Immune Microenvironment to Improve Triple-Negative Breast Cancer Outcomes. Cells 2020, 10, 11. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Yrigoyen, M.; Cassetta, L.; Pollard, J.W. Macrophage targeting in cancer. Ann. N. Y. Acad. Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kabelitz, D.; Serrano, R.; Kouakanou, L.; Peters, C.; Kalyan, S. Cancer immunotherapy with gammadelta T cells: Many paths ahead of us. Cell Mol. Immunol. 2020, 17, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Jagodinsky, J.C.; Morris, Z.S. Priming and Propagating Anti-tumor Immunity: Focal Hypofractionated Radiation for in Situ Vaccination and Systemic Targeted Radionuclide Theranostics for Immunomodulation of Tumor Microenvironments. Semin. Radiat. Oncol. 2020, 30, 181–186. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Lelliott, E.J.; Kong, I.Y.; Zethoven, M.; Ramsbottom, K.M.; Martelotto, L.G.; Meyran, D.; Jiang Zhu, J.; Costacurta, M.; Kirby, L.; Sandow, J.J.; et al. CDK4/6 inhibition promotes anti-tumor immunity through the induction of T cell memory. Cancer Discov. 2021. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Estrogen Activity Modulator | Activity at ERα | Activity at Mutant ERα (mESR1) | Activity at ERβ | Activity at GPER |
|---|---|---|---|---|
| 17β-estradiol | agonist | no effect; constitutively active | agonist | agonist |
| SERMs, e.g., tamoxifen | competitive antagonist to partial agonist, depending on cell context | potential new antagonists under development | partial agonist | agonist |
| SERDS, e.g., fulvestrant | competitive antagonist and degrades | can degrade; lower affinity | reduced ability to degrade | agonist |
| Aromatase inhibitors | reduces endogenous E2 | no effect; constitutively active | reduces endogenous E2 | reduces endogenous E2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schuler, L.A.; Murdoch, F.E. Endogenous and Therapeutic Estrogens: Maestro Conductors of the Microenvironment of ER+ Breast Cancers. Cancers 2021, 13, 3725. https://doi.org/10.3390/cancers13153725
Schuler LA, Murdoch FE. Endogenous and Therapeutic Estrogens: Maestro Conductors of the Microenvironment of ER+ Breast Cancers. Cancers. 2021; 13(15):3725. https://doi.org/10.3390/cancers13153725
Chicago/Turabian StyleSchuler, Linda A., and Fern E. Murdoch. 2021. "Endogenous and Therapeutic Estrogens: Maestro Conductors of the Microenvironment of ER+ Breast Cancers" Cancers 13, no. 15: 3725. https://doi.org/10.3390/cancers13153725
APA StyleSchuler, L. A., & Murdoch, F. E. (2021). Endogenous and Therapeutic Estrogens: Maestro Conductors of the Microenvironment of ER+ Breast Cancers. Cancers, 13(15), 3725. https://doi.org/10.3390/cancers13153725