
Targeting RB1 Loss in Cancers

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Targeting Gene Products Upregulated Following RB1 Inactivation
3. Partners Other Than E2Fs Liberated upon RB1 Loss
4. Synthetic Lethality with RB1 Inactivation
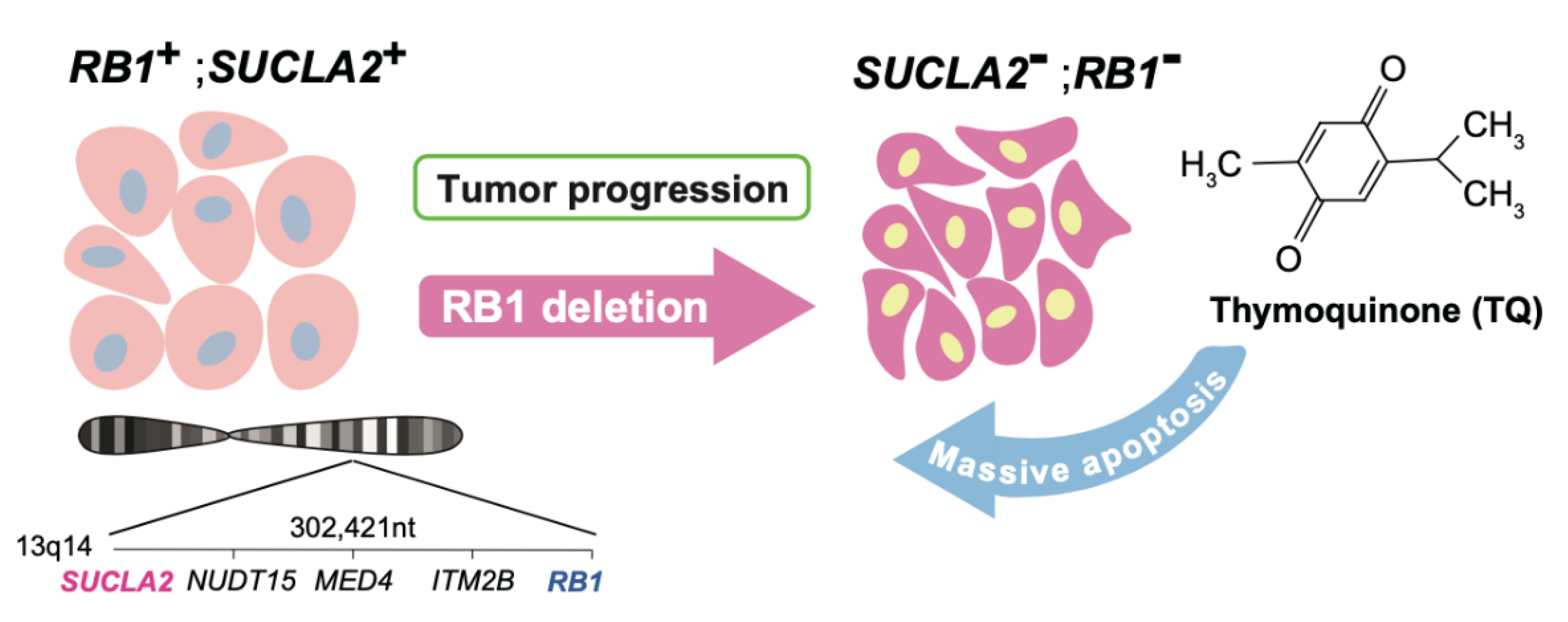
5. Targeting Loss of a Gene Neighboring RB1
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Burkhart, D.L.; Sage, J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 2008, 8, 671–682. [Google Scholar] [CrossRef]
- Harbour, J.W.; Dean, D.C. The Rb/E2F pathway: Expanding roles and emerging paradigms. Genes Dev. 2000, 14, 2393–2409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, J.L.; Polski, A.; Cavenee, W.K.; Dryja, T.P.; Murphree, A.L.; Gallie, B.L. The RB1 Story: Characterization and Cloning of the First Tumor Suppressor Gene. Genes 2019, 10, 879. [Google Scholar] [CrossRef] [Green Version]
- Macpherson, D. Insights from mouse models into human retinoblastoma. Cell Div. 2008, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Wikenheiser-Brokamp, K.A. Retinoblastoma family proteins: Insights gained through genetic manipulation of mice. Cell. Mol. Life Sci. 2006, 63, 767–780. [Google Scholar] [CrossRef]
- Wikenheiser-Brokamp, K.A. Retinoblastoma regulatory pathway in lung cancer. Curr. Mol. Med. 2006, 6, 783–793. [Google Scholar] [CrossRef]
- Viatour, P.; Sage, J. Newly identified aspects of tumor suppression by RB. Dis. Model. Mech. 2011, 4, 581–585. [Google Scholar] [CrossRef] [Green Version]
- Feugeas, O.; Guriec, N.; Babin-Boilletot, A.; Marcellin, L.; Simon, P.; Babin, S.; Thyss, A.; Hofman, P.; Terrier, P.; Kalifa, C.; et al. Loss of heterozygosity of the RB gene is a poor prognostic factor in patients with osteosarcoma. J. Clin. Oncol. 1996, 14, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.J.; Yee, J.K.; Shew, J.Y.; Chen, P.L.; Bookstein, R.; Friedmann, T.; Lee, E.Y.; Lee, W.H. Suppression of the neoplastic phenotype by replacement of the RB gene in human cancer cells. Science 1988, 242, 1563–1566. [Google Scholar] [CrossRef] [PubMed]
- Ookawa, K.; Tsuchida, S.; Adachi, J.; Yokota, J. Differentiation induced by RB expression and apoptosis induced by p53 expression in an osteosarcoma cell line. Oncogene 1997, 14, 1389–1396. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Yeow, W.S.; Ertel, A.; Coleman, I.; Clegg, N.; Thangavel, C.; Morrissey, C.; Zhang, X.; Comstock, C.E.; Witkiewicz, A.K.; et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J. Clin. Investig. 2010, 120, 4478–4492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salah, M.; Nishimoto, Y.; Kohno, S.; Kondoh, A.; Kitajima, S.; Muranaka, H.; Nishiuchi, T.; Ibrahim, A.; Yoshida, A.; Takahashi, C. An in vitro system to characterize prostate cancer progression identified signaling required for self-renewal. Mol. Carcinog. 2016, 55, 1974–1989. [Google Scholar] [CrossRef] [Green Version]
- Kitajima, S.; Yoshida, A.; Kohno, S.; Li, F.; Suzuki, S.; Nagatani, N.; Nishimoto, Y.; Sasaki, N.; Muranaka, H.; Wan, Y.; et al. The RB-IL-6 axis controls self-renewal and endocrine therapy resistance by fine-tuning mitochondrial activity. Oncogene 2017, 36, 5145–5157. [Google Scholar] [CrossRef]
- Yoshida, A.; Kitajima, S.; Li, F.; Chaoyang, C.; Takegami, Y.; Kohno, S.; Wan, Y.; Hayashi, N.; Muranaka, H.; Nishimoto, Y.; et al. MicroRNA-140 mediates RB tumor suppressor function to control stem cell-like activity through interleukin-6. Oncotarget 2017, 8, 13872–13885. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Kitajima, S.; Kohno, S.; Yoshida, A.; Tange, S.; Sasaki, S.; Okada, N.; Nishimoto, Y.; Muranaka, H.; Nagatani, N.; et al. Retinoblastoma Inactivation Induces a Protumoral Microenvironment via Enhanced CCL2 Secretion. Cancer Res. 2019, 79, 3903–3915. [Google Scholar] [CrossRef] [Green Version]
- Kitajima, S.; Kohno, S.; Kondoh, A.; Sasaki, N.; Nishimoto, Y.; Li, F.; Mohammed, M.S.A.; Muranaka, H.; Nagatani, N.; Suzuki, M.; et al. Undifferentiated State Induced by Rb-p53 Double Inactivation in Mouse Thyroid Neuroendocrine Cells and Embryonic Fibroblasts. Stem Cells 2015, 33, 1657–1669. [Google Scholar] [CrossRef]
- Kohno, S.; Kitajima, S.; Sasaki, N.; Takahashi, C. Retinoblastoma tumor suppressor functions shared by stem cell and cancer cell strategies. World J. Stem Cells 2016, 8, 170–184. [Google Scholar] [CrossRef]
- Calo, E.; Quintero-Estades, J.A.; Danielian, P.S.; Nedelcu, S.; Berman, S.D.; Lees, J.A. Rb regulates fate choice and lineage commitment in vivo. Nature 2010, 466, 1110–1114. [Google Scholar] [CrossRef] [Green Version]
- Bracken, A.P.; Ciro, M.; Cocito, A.; Helin, K. E2F target genes: Unraveling the biology. Trends Biochem. Sci. 2004, 29, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, L.; Bronson, R.; Williams, B.O.; Dyson, N.J.; Harlow, E.; Jacks, T. Loss of E2F-1 reduces tumorigenesis and extends the lifespan of Rb1(+/−)mice. Nat. Genet. 1998, 18, 360–364. [Google Scholar] [CrossRef]
- Fujiwara, K.; Yuwanita, I.; Hollern, D.P.; Andrechek, E.R. Prediction and genetic demonstration of a role for activator E2Fs in Myc-induced tumors. Cancer Res. 2011, 71, 1924–1932. [Google Scholar] [CrossRef] [Green Version]
- Rouaud, F.; Hamouda-Tekaya, N.; Cerezo, M.; Abbe, P.; Zangari, J.; Hofman, V.; Ohanna, M.; Mograbi, B.; El-Hachem, N.; Benfodda, Z.; et al. E2F1 inhibition mediates cell death of metastatic melanoma. Cell Death Dis. 2018, 9, 527. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.; Lu, F.; Shao, Y. The E2F family as potential biomarkers and therapeutic targets in colon cancer. PeerJ 2020, 8, e8562. [Google Scholar] [CrossRef]
- Ma, Y.; Kurtyka, C.A.; Boyapalle, S.; Sung, S.S.; Lawrence, H.; Guida, W.; Cress, W.D. A small-molecule E2F inhibitor blocks growth in a melanoma culture model. Cancer Res. 2008, 68, 6292–6299. [Google Scholar] [CrossRef] [Green Version]
- Tadesse, S.; Caldon, E.C.; Tilley, W.; Wang, S. Cyclin-Dependent Kinase 2 Inhibitors in Cancer Therapy: An Update. J. Med. Chem. 2019, 62, 4233–4251. [Google Scholar] [CrossRef]
- Sotillo, R.; Hernando, E.; Diaz-Rodriguez, E.; Teruya-Feldstein, J.; Cordon-Cardo, C.; Lowe, S.W.; Benezra, R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell 2007, 11, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Schvartzman, J.M.; Duijf, P.H.; Sotillo, R.; Coker, C.; Benezra, R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell 2011, 19, 701–714. [Google Scholar] [CrossRef] [Green Version]
- Perez, I.S. MAD2 in the Spotlight as a Cancer Therapy Regulator. Int. J. Cancer Oncol. 2016, 3, 1–8. [Google Scholar] [CrossRef]
- Kastl, J.; Braun, J.; Prestel, A.; Moller, H.M.; Huhn, T.; Mayer, T.U. Mad2 Inhibitor-1 (M2I-1): A Small Molecule Protein-Protein Interaction Inhibitor Targeting the Mitotic Spindle Assembly Checkpoint. ACS Chem. Biol. 2015, 10, 1661–1666. [Google Scholar] [CrossRef]
- Li, J.; Dang, N.; Martinez-Lopez, N.; Jowsey, P.A.; Huang, D.; Lightowlers, R.N.; Gao, F.; Huang, J.Y. M2I-1 disrupts the in vivo interaction between CDC20 and MAD2 and increases the sensitivities of cancer cell lines to anti-mitotic drugs via MCL-1s. Cell Div. 2019, 14, 5. [Google Scholar] [CrossRef]
- Sage, J.; Straight, A.F. RB’s original CIN? Genes Dev. 2010, 24, 1329–1333. [Google Scholar] [CrossRef] [Green Version]
- Amato, A.; Lentini, L.; Schillaci, T.; Iovino, F.; Di Leonardo, A. RNAi mediated acute depletion of retinoblastoma protein (pRb) promotes aneuploidy in human primary cells via micronuclei formation. BMC Cell Biol. 2009, 10, 79. [Google Scholar] [CrossRef] [Green Version]
- Manning, A.L.; Longworth, M.S.; Dyson, N.J. Loss of pRB causes centromere dysfunction and chromosomal instability. Genes Dev. 2010, 24, 1364–1376. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, E.S.; Pruitt, S.C.; Hershberger, P.A.; Witkiewicz, A.K.; Goodrich, D.W. Cell Cycle and Beyond: Exploiting New RB1 Controlled Mechanisms for Cancer Therapy. Trends Cancer 2019, 5, 308–324. [Google Scholar] [CrossRef]
- Shamma, A.; Takegami, Y.; Miki, T.; Kitajima, S.; Noda, M.; Obara, T.; Okamoto, T.; Takahashi, C. Rb Regulates DNA damage response and cellular senescence through E2F-dependent suppression of N-ras isoprenylation. Cancer Cell 2009, 15, 255–269. [Google Scholar] [CrossRef] [Green Version]
- Kitajima, S.; Takahashi, C. Intersection of retinoblastoma tumor suppressor function, stem cells, metabolism, and inflammation. Cancer Sci. 2017, 108, 1726–1731. [Google Scholar] [CrossRef]
- Kitajima, S.; Li, F.; Takahashi, C. Tumor Milieu Controlled by RB Tumor Suppressor. Int. J. Mol. Sci. 2020, 21, 2450. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Ding, D.; Yan, Y.; Li, H.; Wang, B.; Ma, L.; Ye, Z.; Ma, T.; Wu, Q.; Rodrigues, D.N.; et al. Phosphorylated RB Promotes Cancer Immunity by Inhibiting NF-kappaB Activation and PD-L1 Expression. Mol. Cell 2019, 73, 22–35.e26. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Lu, X.; Horvitz, H.R. lin-35 and lin-53, two genes that antagonize a C. elegans Ras pathway, encode proteins similar to Rb and its binding protein RbAp48. Cell 1998, 95, 981–991. [Google Scholar] [CrossRef] [Green Version]
- Ceol, C.J.; Horvitz, H.R. dpl-1 DP and efl-1 E2F act with lin-35 Rb to antagonize Ras signaling in C. elegans vulval development. Mol. Cell 2001, 7, 461–473. [Google Scholar] [CrossRef]
- Lee, K.Y.; Ladha, M.H.; McMahon, C.; Ewen, M.E. The retinoblastoma protein is linked to the activation of Ras. Mol. Cell Biol. 1999, 19, 7724–7732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, C.; Bronson, R.T.; Socolovsky, M.; Contreras, B.; Lee, K.Y.; Jacks, T.; Noda, M.; Kucherlapati, R.; Ewen, M.E. Rb and N-ras function together to control differentiation in the mouse. Mol. Cell Biol. 2003, 23, 5256–5268. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, C.; Contreras, B.; Bronson, R.T.; Loda, M.; Ewen, M.E. Genetic interaction between Rb and K-ras in the control of differentiation and tumor suppression. Mol. Cell Biol. 2004, 24, 10406–10415. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, C.; Contreras, B.; Iwanaga, T.; Takegami, Y.; Bakker, A.; Bronson, R.T.; Noda, M.; Loda, M.; Hunt, J.L.; Ewen, M.E. Nras loss induces metastatic conversion of Rb1-deficient neuroendocrine thyroid tumor. Nat. Genet. 2006, 38, 118–123. [Google Scholar] [CrossRef]
- Takahashi, C.; Ewen, M.E. Genetic interaction between Rb and N-ras: Differentiation control and metastasis. Cancer Res. 2006, 66, 9345–9348. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, C.; Sasaki, N.; Kitajima, S. Twists in views on RB functions in cellular signaling, metabolism and stem cells. Cancer Sci. 2012, 103, 1182–1188. [Google Scholar] [CrossRef]
- Muranaka, H.; Hayashi, A.; Minami, K.; Kitajima, S.; Kohno, S.; Nishimoto, Y.; Nagatani, N.; Suzuki, M.; Kulathunga, L.A.N.; Sasaki, N.; et al. A distinct function of the retinoblastoma protein in the control of lipid composition identified by lipidomic profiling. Oncogenesis 2017, 6, e350. [Google Scholar] [CrossRef]
- Kulathunga, N.; Kohno, S.; Linn, P.; Nishimoto, Y.; Horike, S.I.; Zaraiskii, M.I.; Kumar, S.; Muranaka, H.; Takahashi, C. Peripubertal high-fat diet promotes c-Myc stabilization in mammary gland epithelium. Cancer Sci. 2020, 111, 2336–2348. [Google Scholar] [CrossRef]
- Zhang, J.; Benavente, C.A.; McEvoy, J.; Flores-Otero, J.; Ding, L.; Chen, X.; Ulyanov, A.; Wu, G.; Wilson, M.; Wang, J.; et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature 2012, 481, 329–334. [Google Scholar] [CrossRef]
- Zocchi, L.; Mehta, A.; Wu, S.C.; Wu, J.; Gu, Y.; Wang, J.; Suh, S.; Spitale, R.C.; Benavente, C.A. Chromatin remodeling protein HELLS is critical for retinoblastoma tumor initiation and progression. Oncogenesis 2020, 9, 25. [Google Scholar] [CrossRef] [Green Version]
- Benavente, C.A.; Finkelstein, D.; Johnson, D.A.; Marine, J.C.; Ashery-Padan, R.; Dyer, M.A. Chromatin remodelers HELLS and UHRF1 mediate the epigenetic deregulation of genes that drive retinoblastoma tumor progression. Oncotarget 2014, 5, 9594–9608. [Google Scholar] [CrossRef]
- Sanidas, I.; Morris, R.; Fella, K.A.; Rumde, P.H.; Boukhali, M.; Tai, E.C.; Ting, D.T.; Lawrence, M.S.; Haas, W.; Dyson, N.J. A Code of Mono-phosphorylation Modulates the Function of RB. Mol. Cell 2019, 73, 985–1000.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeCaprio, J.A. How the Rb tumor suppressor structure and function was revealed by the study of Adenovirus and SV40. Virology 2009, 384, 274–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christman, J.K. 5-Azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef] [Green Version]
- Johnstone, R.W.; Licht, J.D. Histone deacetylase inhibitors in cancer therapy: Is transcription the primary target? Cancer Cell 2003, 4, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.S.; Chen, J.Y.; Tsai, H.J.; Chen, T.Y.; Hung, W.C. The SUV39H1 inhibitor chaetocin induces differentiation and shows synergistic cytotoxicity with other epigenetic drugs in acute myeloid leukemia cells. Blood Cancer J. 2015, 5, e313. [Google Scholar] [CrossRef]
- Huang, L.Y.; Lee, Y.S.; Huang, J.J.; Chang, C.C.; Chang, J.M.; Chuang, S.H.; Kao, K.J.; Tsai, Y.J.; Tsai, P.Y.; Liu, C.W.; et al. Characterization of the biological activity of a potent small molecule Hec1 inhibitor TAI-1. J. Exp. Clin. Cancer Res. 2014, 33, 6. [Google Scholar] [CrossRef] [Green Version]
- Ji, P.; Jiang, H.; Rekhtman, K.; Bloom, J.; Ichetovkin, M.; Pagano, M.; Zhu, L. An Rb-Skp2-p27 pathway mediates acute cell cycle inhibition by Rb and is retained in a partial-penetrance Rb mutant. Mol. Cell 2004, 16, 47–58. [Google Scholar] [CrossRef]
- Lu, Z.; Bauzon, F.; Fu, H.; Cui, J.; Zhao, H.; Nakayama, K.; Nakayama, K.I.; Zhu, L. Skp2 suppresses apoptosis in Rb1-deficient tumours by limiting E2F1 activity. Nat. Commun. 2014, 5, 3463. [Google Scholar] [CrossRef] [Green Version]
- Skapek, S.X.; Pan, Y.R.; Lee, E.Y. Regulation of cell lineage specification by the retinoblastoma tumor suppressor. Oncogene 2006, 25, 5268–5276. [Google Scholar] [CrossRef] [Green Version]
- Kareta, M.S.; Gorges, L.L.; Hafeez, S.; Benayoun, B.A.; Marro, S.; Zmoos, A.F.; Cecchini, M.J.; Spacek, D.; Batista, L.F.; O’Brien, M.; et al. Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell 2015, 16, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Du, J.; Parsons, S.H.; Merzoug, F.F.; Webster, Y.; Iversen, P.W.; Chio, L.C.; Van Horn, R.D.; Lin, X.; Blosser, W.; et al. Aurora A Kinase Inhibition Is Synthetic Lethal with Loss of the RB1 Tumor Suppressor Gene. Cancer Discov. 2019, 9, 248–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oser, M.G.; Fonseca, R.; Chakraborty, A.A.; Brough, R.; Spektor, A.; Jennings, R.B.; Flaifel, A.; Novak, J.S.; Gulati, A.; Buss, E.; et al. Cells Lacking the RB1 Tumor Suppressor Gene Are Hyperdependent on Aurora B Kinase for Survival. Cancer Discov. 2019, 9, 230–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, J.; Yang, E.J.; Zhang, B.; Wu, C.; Pardeshi, L.; Shi, C.; Mou, P.K.; Liu, Y.; Tan, K.; Shim, J.S. Synthetic lethality of RB1 and aurora A is driven by stathmin-mediated disruption of microtubule dynamics. Nat. Commun. 2020, 11, 5105. [Google Scholar] [CrossRef] [PubMed]
- Manning, A.L.; Dyson, N.J. RB: Mitotic implications of a tumour suppressor. Nat. Rev. Cancer 2012, 12, 220–226. [Google Scholar] [CrossRef] [Green Version]
- Shamma, A.; Suzuki, M.; Hayashi, N.; Kobayashi, M.; Sasaki, N.; Nishiuchi, T.; Doki, Y.; Okamoto, T.; Kohno, S.; Muranaka, H.; et al. ATM mediates pRB function to control DNMT1 protein stability and DNA methylation. Mol. Cell Biol. 2013, 33, 3113–3124. [Google Scholar] [CrossRef] [Green Version]
- Velez-Cruz, R.; Manickavinayaham, S.; Biswas, A.K.; Clary, R.W.; Premkumar, T.; Cole, F.; Johnson, D.G. RB localizes to DNA double-strand breaks and promotes DNA end resection and homologous recombination through the recruitment of BRG1. Genes Dev. 2016, 30, 2500–2512. [Google Scholar] [CrossRef] [Green Version]
- Marshall, A.E.; Roes, M.V.; Passos, D.T.; DeWeerd, M.C.; Chaikovsky, A.C.; Sage, J.; Howlett, C.J.; Dick, F.A. RB1 Deletion in Retinoblastoma Protein Pathway-Disrupted Cells Results in DNA Damage and Cancer Progression. Mol. Cell Biol. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Witkiewicz, A.K.; Chung, S.; Brough, R.; Vail, P.; Franco, J.; Lord, C.J.; Knudsen, E.S. Targeting the Vulnerability of RB Tumor Suppressor Loss in Triple-Negative Breast Cancer. Cell Rep. 2018, 22, 1185–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farago, A.F.; Yeap, B.Y.; Stanzione, M.; Hung, Y.P.; Heist, R.S.; Marcoux, J.P.; Zhong, J.; Rangachari, D.; Barbie, D.A.; Phat, S.; et al. Combination Olaparib and Temozolomide in Relapsed Small-Cell Lung Cancer. Cancer Discov. 2019, 9, 1372–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, G.; Armenia, J.; Mazzu, Y.Z.; Nandakumar, S.; Stopsack, K.H.; Atiq, M.O.; Komura, K.; Jehane, L.; Hirani, R.; Chadalavada, K.; et al. Significance of BRCA2 and RB1 Co-loss in Aggressive Prostate Cancer Progression. Clin. Cancer Res. 2020, 26, 2047–2064. [Google Scholar] [CrossRef] [PubMed]
- Dick, F.A.; Goodrich, D.W.; Sage, J.; Dyson, N.J. Non-canonical functions of the RB protein in cancer. Nat. Rev. Cancer 2018, 18, 442–451. [Google Scholar] [CrossRef]
- Zagorski, W.A.; Knudsen, E.S.; Reed, M.F. Retinoblastoma deficiency increases chemosensitivity in lung cancer. Cancer Res. 2007, 67, 8264–8273. [Google Scholar] [CrossRef] [Green Version]
- Trere, D.; Brighenti, E.; Donati, G.; Ceccarelli, C.; Santini, D.; Taffurelli, M.; Montanaro, L.; Derenzini, M. High prevalence of retinoblastoma protein loss in triple-negative breast cancers and its association with a good prognosis in patients treated with adjuvant chemotherapy. Ann. Oncol. 2009, 20, 1818–1823. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; Ertel, A.; McFalls, J.; Valsecchi, M.E.; Schwartz, G.; Knudsen, E.S. RB-pathway disruption is associated with improved response to neoadjuvant chemotherapy in breast cancer. Clin. Cancer Res. 2012, 18, 5110–5122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, T.J.; Liu, J.C.; Vizeacoumar, F.; Sun, T.; Maclean, N.; Egan, S.E.; Schimmer, A.D.; Datti, A.; Zacksenhaus, E. RB1 status in triple negative breast cancer cells dictates response to radiation treatment and selective therapeutic drugs. PLoS ONE 2013, 8, e78641. [Google Scholar] [CrossRef] [Green Version]
- Hijioka, S.; Hosoda, W.; Matsuo, K.; Ueno, M.; Furukawa, M.; Yoshitomi, H.; Kobayashi, N.; Ikeda, M.; Ito, T.; Nakamori, S.; et al. Rb Loss and KRAS Mutation Are Predictors of the Response to Platinum-Based Chemotherapy in Pancreatic Neuroendocrine Neoplasm with Grade 3: A Japanese Multicenter Pancreatic NEN-G3 Study. Clin. Cancer Res. 2017, 23, 4625–4632. [Google Scholar] [CrossRef] [Green Version]
- Coussy, F.; El-Botty, R.; Chateau-Joubert, S.; Dahmani, A.; Montaudon, E.; Leboucher, S.; Morisset, L.; Painsec, P.; Sourd, L.; Huguet, L.; et al. BRCAness, SLFN11, and RB1 loss predict response to topoisomerase I inhibitors in triple-negative breast cancers. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.C.; Granieri, L.; Shrestha, M.; Wang, D.Y.; Vorobieva, I.; Rubie, E.A.; Jones, R.; Ju, Y.; Pellecchia, G.; Jiang, Z.; et al. Identification of CDC25 as a Common Therapeutic Target for Triple-Negative Breast Cancer. Cell Rep. 2018, 23, 112–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, L.L.; Piwnica-Worms, H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science 1992, 257, 1955–1957. [Google Scholar] [CrossRef]
- Perry, J.A.; Kornbluth, S. Cdc25 and Wee1: Analogous opposites? Cell Div. 2007, 2, 12. [Google Scholar] [CrossRef] [Green Version]
- Lolli, G.; Johnson, L.N. CAK-Cyclin-dependent Activating Kinase: A key kinase in cell cycle control and a target for drugs? Cell Cycle 2005, 4, 572–577. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Zheng, M.; Lu, R.; Du, J.; Zhao, Q.; Li, Z.; Li, Y.; Zhang, S. The role of CDC25C in cell cycle regulation and clinical cancer therapy: A systematic review. Cancer Cell Int. 2020, 20, 213. [Google Scholar] [CrossRef] [PubMed]
- Lew, D.J.; Kornbluth, S. Regulatory roles of cyclin dependent kinase phosphorylation in cell cycle control. Curr. Opin. Cell Biol. 1996, 8, 795–804. [Google Scholar] [CrossRef]
- Zacksenhaus, E.; Liu, J.C.; Granieri, L.; Vorobieva, I.; Wang, D.Y.; Ghanbari-Azarnier, R.; Li, H.; Ali, A.; Chung, P.E.D.; Ju, Y.; et al. CDC25 as a common therapeutic target for triple-negative breast cancer—The challenges ahead. Mol. Cell Oncol. 2018, 5, e1481814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Gordon, G.M.; Du, C.H.; Xu, J.; Du, W. Specific killing of Rb mutant cancer cells by inactivating TSC2. Cancer Cell 2010, 17, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Gordon, G.M.; Zhang, T.; Zhao, J.; Du, W. Deregulated G1-S control and energy stress contribute to the synthetic-lethal interactions between inactivation of RB and TSC1 or TSC2. J. Cell Sci. 2013, 126, 2004–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceron, J.; Rual, J.F.; Chandra, A.; Dupuy, D.; Vidal, M.; van den Heuvel, S. Large-scale RNAi screens identify novel genes that interact with the C. elegans retinoblastoma pathway as well as splicing-related components with synMuv B activity. BMC Dev. Biol. 2007, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Brough, R.; Gulati, A.; Haider, S.; Kumar, R.; Campbell, J.; Knudsen, E.; Pettitt, S.J.; Ryan, C.J.; Lord, C.J. Identification of highly penetrant Rb-related synthetic lethal interactions in triple negative breast cancer. Oncogene 2018, 37, 5701–5718. [Google Scholar] [CrossRef] [Green Version]
- Shao, Z.; Siegert, J.L.; Ruppert, S.; Robbins, P.D. Rb interacts with TAF(II)250/TFIID through multiple domains. Oncogene 1997, 15, 385–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bignell, G.R.; Greenman, C.D.; Davies, H.; Butler, A.P.; Edkins, S.; Andrews, J.M.; Buck, G.; Chen, L.; Beare, D.; Latimer, C.; et al. Signatures of mutation and selection in the cancer genome. Nature 2010, 463, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Dey, P.; Baddour, J.; Muller, F.; Wu, C.C.; Wang, H.; Liao, W.T.; Lan, Z.; Chen, A.; Gutschner, T.; Kang, Y.; et al. Genomic deletion of malic enzyme 2 confers collateral lethality in pancreatic cancer. Nature 2017, 542, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.L.; Colla, S.; Aquilanti, E.; Manzo, V.E.; Genovese, G.; Lee, J.; Eisenson, D.; Narurkar, R.; Deng, P.; Nezi, L.; et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature 2012, 488, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrozzo, R.; Dionisi-Vici, C.; Steuerwald, U.; Lucioli, S.; Deodato, F.; Di Giandomenico, S.; Bertini, E.; Franke, B.; Kluijtmans, L.A.; Meschini, M.C.; et al. SUCLA2 mutations are associated with mild methylmalonic aciduria, Leigh-like encephalomyopathy, dystonia and deafness. Brain 2007, 130, 862–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matilainen, S.; Isohanni, P.; Euro, L.; Lonnqvist, T.; Pihko, H.; Kivela, T.; Knuutila, S.; Suomalainen, A. Mitochondrial encephalomyopathy and retinoblastoma explained by compound heterozygosity of SUCLA2 point mutation and 13q14 deletion. Eur. J. Hum. Genet. 2015, 23, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Kohno, S.; Linn, P.; Nagatani, N.; Watanabe, Y.; Kumar, S.; Soga, T.; Takahashi, C. Pharmacologically targetable vulnerability in prostate cancer carrying RB1-SUCLA2 deletion. Oncogene 2020, 39, 5690–5707. [Google Scholar] [CrossRef]
- Khader, M.; Eckl, P.M. Thymoquinone: An emerging natural drug with a wide range of medical applications. Iran. J. Basic Med. Sci. 2014, 17, 950–957. [Google Scholar]
- Lee, S.; Jeong, J.; Majewski, T.; Scherer, S.E.; Kim, M.S.; Tuziak, T.; Tang, K.S.; Baggerly, K.; Grossman, H.B.; Zhou, J.H.; et al. Forerunner genes contiguous to RB1 contribute to the development of in situ neoplasia. Proc. Natl. Acad. Sci. USA 2007, 104, 13732–13737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschop, K.; Conery, A.R.; Litovchick, L.; Decaprio, J.A.; Settleman, J.; Harlow, E.; Dyson, N. A kinase shRNA screen links LATS2 and the pRB tumor suppressor. Genes Dev. 2011, 25, 814–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschop, K.; Dyson, N. Identifying players in the functional network around pRB. Cell Cycle 2011, 10, 3814–3815. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Gene | Inhibitor | Phase of Clinical Trial |
|---|---|---|
| Aurora A, B | Alisertib | III |
| Tozasertib | II | |
| Barasertib | I | |
| MNL8054 | I | |
| Danusertib | II | |
| AT9283 | II | |
| KW-2449 | I | |
| SNS-341 | I | |
| ENMD-2076 | II | |
| BI-847325 | I | |
| PLK1 | Volasertib | III |
| Rigosertib | III | |
| BI2536 | II | |
| CHK1 | AZD7762 | I |
| MK-8779 | II | |
| PF-477736 | I | |
| CDC25A, B | K-252a | ND |
| NSC663284 | ND | |
| NSC95397 | ND | |
| WEE1 | Adabosertib | II |
| TSC1, 2 | ND | ND |
| SKP2 | SKPin C1 | ND |
| TAF1 | CeMMEC-1, 13 | ND |
| BAY299 | ND | |
| NUP88 | ND | ND |
| NUP214 | ND | ND |
| PARP | Olaparib | FDA-approved |
| Veliparib | III | |
| Rucapatib | III | |
| Iniparib | III | |
| SYK | Fostamatinib | III |
| R406 | I |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linn, P.; Kohno, S.; Sheng, J.; Kulathunga, N.; Yu, H.; Zhang, Z.; Voon, D.; Watanabe, Y.; Takahashi, C. Targeting RB1 Loss in Cancers. Cancers 2021, 13, 3737. https://doi.org/10.3390/cancers13153737
Linn P, Kohno S, Sheng J, Kulathunga N, Yu H, Zhang Z, Voon D, Watanabe Y, Takahashi C. Targeting RB1 Loss in Cancers. Cancers. 2021; 13(15):3737. https://doi.org/10.3390/cancers13153737
Chicago/Turabian StyleLinn, Paing, Susumu Kohno, Jindan Sheng, Nilakshi Kulathunga, Hai Yu, Zhiheng Zhang, Dominic Voon, Yoshihiro Watanabe, and Chiaki Takahashi. 2021. "Targeting RB1 Loss in Cancers" Cancers 13, no. 15: 3737. https://doi.org/10.3390/cancers13153737
APA StyleLinn, P., Kohno, S., Sheng, J., Kulathunga, N., Yu, H., Zhang, Z., Voon, D., Watanabe, Y., & Takahashi, C. (2021). Targeting RB1 Loss in Cancers. Cancers, 13(15), 3737. https://doi.org/10.3390/cancers13153737