
Transcription/Replication Conflicts in Tumorigenesis and Their Potential Role as Novel Therapeutic Targets in Multiple Myeloma

 ,
,  ,
, 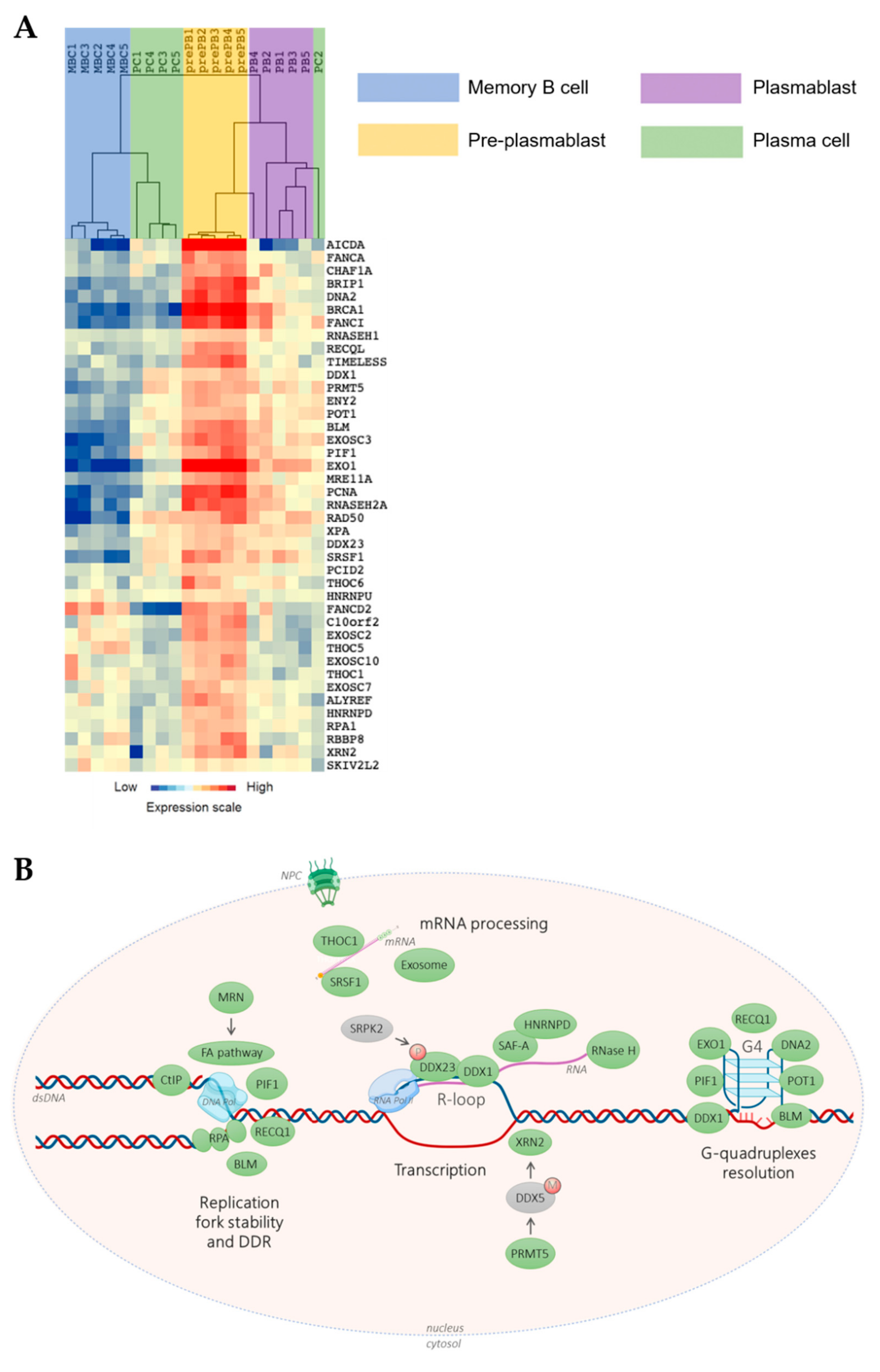{kind=link}
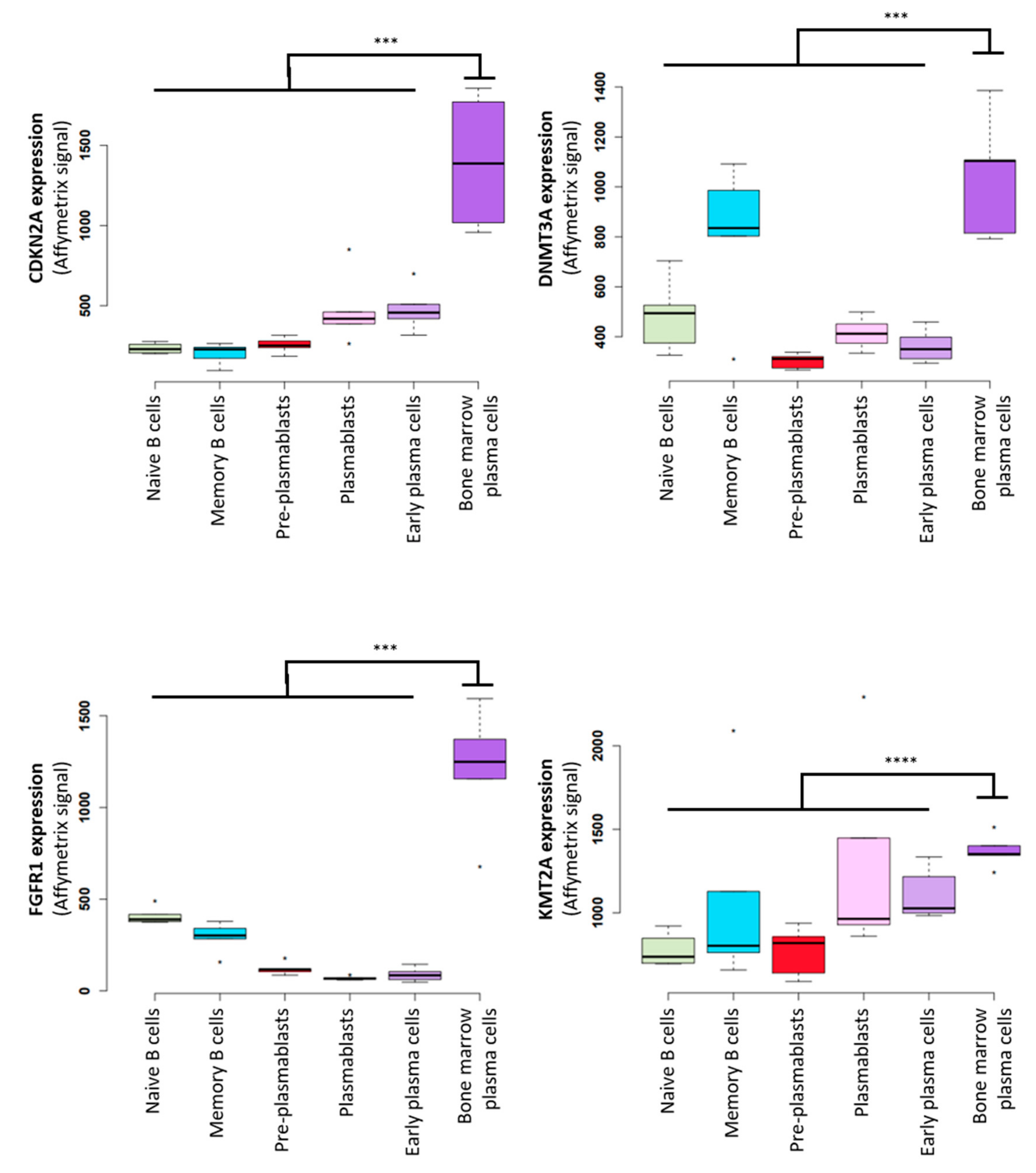{kind=link}
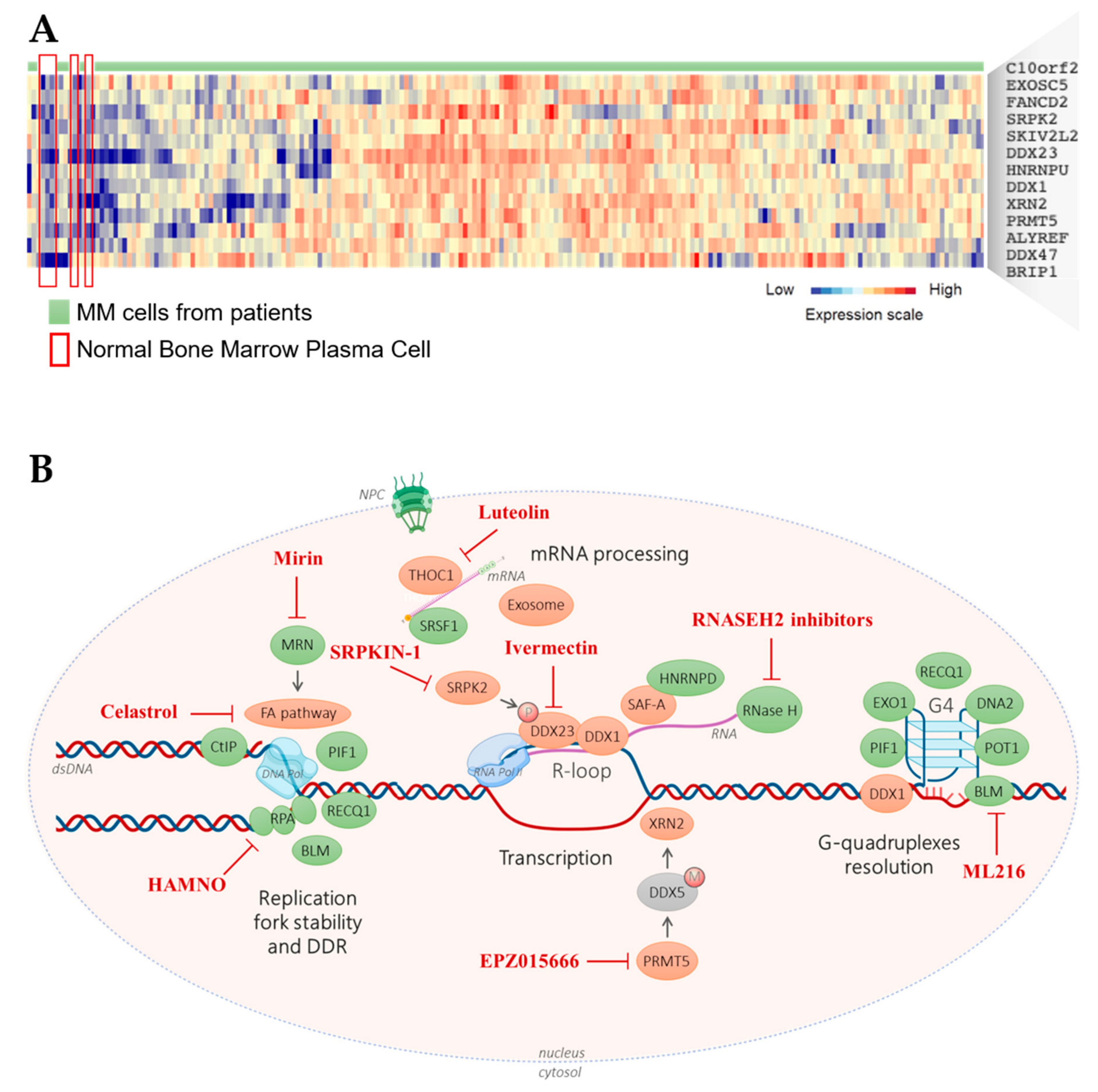{kind=link}
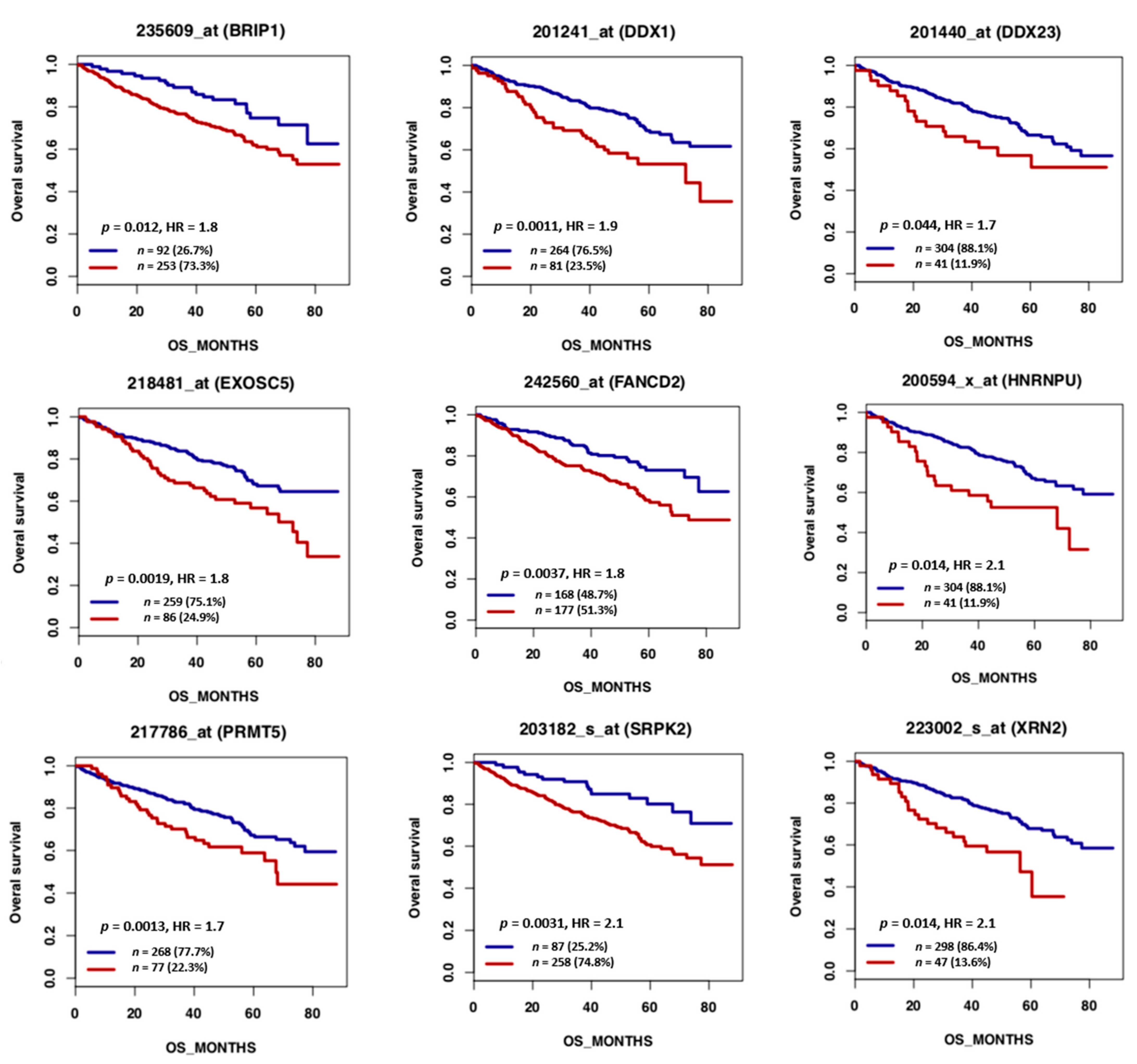{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Management of Transcription/Replication Conflicts (TRCs) in Normal Cells Is Critical to Prevent Genomic Instability
2.1. R-Loop Resolution Genes
2.1.1. RNase H1/2, Replication Protein A (RPA)
2.1.2. The DEAD-Box Protein Family of Helicases
2.2. mRNA Maturation
2.2.1. Serine/Arginine Splicing Factor 1 (SRSF1)
2.2.2. Heterogeneous Nuclear Ribonucleoprotein U (HNRNPU) and D0 (HNRNPD)
2.2.3. The THO/TREX Complex and TREX2
2.3. RNA Processing and Degradation
2.3.1. The RNA Exosome
2.3.2. 5′-3′ Exoribonuclease 2 (XRN2)
2.4. Fork Protection and Stability
2.4.1. The Fanconi Anemia Pathway and the MRN Complex
2.4.2. Breast Cancer Susceptibility Gene 1 and 2 (BRCA1 and BRCA2)
2.4.3. Proliferating Cell Nuclear Antigen (PCNA) SUMOylation
2.4.4. CtBP-Interacting Protein (CtIP)
2.4.5. Exonuclease 1 (EXO1)
2.4.6. Transcription Coupled Nucleotide Excision Repair (TC-NER) Exonucleases
2.5. G-Quadruplexes Resolution
2.5.1. The RecQ family of Helicases
2.5.2. PIF1
2.5.3. DNA2 and POT1 Roles at Telomeres
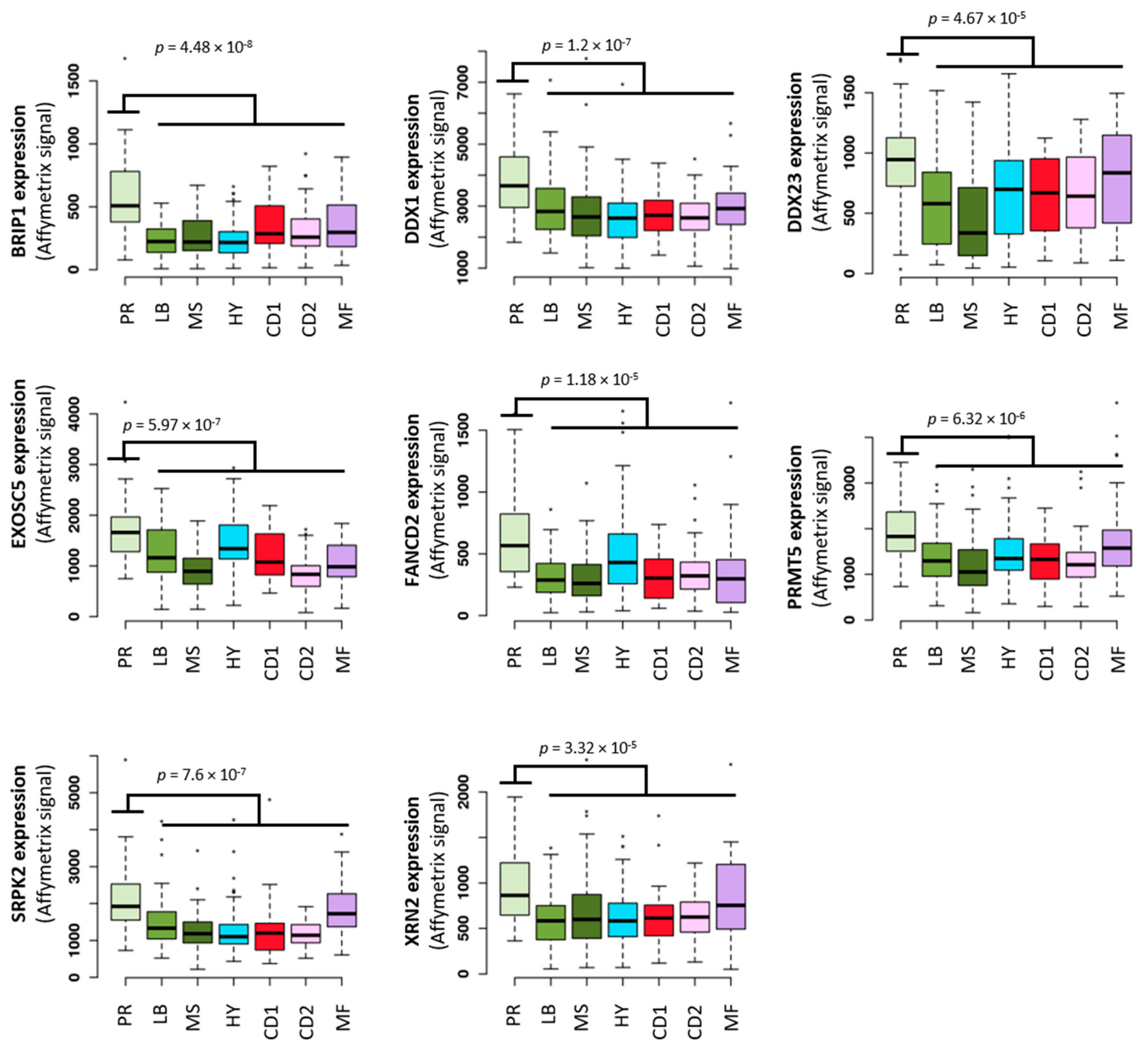
3. The Potential Role of TRC in Multiple Myeloma Pathophysiology
4. TRC Targeting Approaches
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Klein, B.; Tarte, K.; Jourdan, M.; Mathouk, K.; Moreaux, J.; Jourdan, E.; Legouffe, E.; De Vos, J.; Rossi, J.F. Survival and Proliferation Factors of Normal and Malignant Plasma Cells. Int. J. Hematol. 2003, 78, 106–113. [Google Scholar] [CrossRef]
- Nutt, S.L.; Hodgkin, P.D.; Tarlinton, D.M.; Corcoran, L.M. The Generation of Antibody-Secreting Plasma Cells. Nat. Rev. Immunol. 2015, 15, 160–171. [Google Scholar] [CrossRef]
- Cyster, J.G. Homing of Antibody Secreting Cells. Immunol. Rev. 2003, 194, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Hamperl, S.; Cimprich, K.A. The Contribution of Co-Transcriptional RNA:DNA Hybrid Structures to DNA Damage and Genome Instability. DNA Repair 2014, 19, 84–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sollier, J.; Cimprich, K.A. R-Loops Breaking Bad. Trends Cell Biol. 2015, 25, 514–522. [Google Scholar] [CrossRef] [Green Version]
- Gan, W.; Guan, Z.; Liu, J.; Gui, T.; Shen, K.; Manley, J.L.; Li, X. R-Loop-Mediated Genomic Instability Is Caused by Impairment of Replication Fork Progression. Genes Dev. 2011, 25, 2041–2056. [Google Scholar] [CrossRef] [Green Version]
- Hamperl, S.; Cimprich, K.A. Conflict Resolution in the Genome: How Transcription and Replication Make It Work. Cell 2016, 167, 1455–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niehrs, C.; Luke, B. Regulatory R-Loops as Facilitators of Gene Expression and Genome Stability. Nat. Rev. Mol. Cell Biol. 2020, 21, 167–178. [Google Scholar] [CrossRef]
- Alagpulinsa, D.A.; Szalat, R.E.; Poznansky, M.C.; Reis, R.J.S. Genomic Instability in Multiple Myeloma. Trends Cancer 2020, 6, 858–873. [Google Scholar] [CrossRef] [PubMed]
- Hoang, P.H.; Cornish, A.J.; Chubb, D.; Jackson, G.; Kaiser, M.; Houlston, R.S. Impact of Mitochondrial DNA Mutations in Multiple Myeloma. Blood Cancer J. 2020, 10. [Google Scholar] [CrossRef]
- Jourdan, M.; Caraux, A.; De Vos, J.; Fiol, G.; Larroque, M.; Cognot, C.; Bret, C.; Duperray, C.; Hose, D.; Klein, B. An in vitro Model of Differentiation of Memory B Cells into Plasmablasts and Plasma Cells Including Detailed Phenotypic and Molecular Characterization. Blood 2009, 114, 5173–5181. [Google Scholar] [CrossRef] [Green Version]
- Jourdan, M.; Caraux, A.; Caron, G.; Robert, N.; Fiol, G.; Rème, T.; Bolloré, K.; Vendrell, J.-P.; Gallou, S.L.; Mourcin, F.; et al. Characterization of a Transitional Preplasmablast Population in the Process of Human B Cell to Plasma Cell Differentiation. J. Immunol. 2011, 187, 3931–3941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jourdan, M.; Cren, M.; Robert, N.; Bolloré, K.; Fest, T.; Duperray, C.; Guilloton, F.; Hose, D.; Tarte, K.; Klein, B. IL-6 Supports the Generation of Human Long-Lived Plasma Cells in Combination with Either APRIL or Stromal Cell-Soluble Factors. Leukemia 2014, 28, 1647–1656. [Google Scholar] [CrossRef] [PubMed]
- Leung-Hagesteijn, C.; Erdmann, N.; Cheung, G.; Keats, J.J.; Stewart, A.K.; Reece, D.E.; Chung, K.C.; Tiedemann, R.E. Xbp1s-Negative Tumor B Cells and Pre-Plasmablasts Mediate Therapeutic Proteasome Inhibitor Resistance in Multiple Myeloma. Cancer Cell 2013, 24, 289–304. [Google Scholar] [CrossRef] [Green Version]
- Herviou, L.; Jourdan, M.; Martinez, A.-M.; Cavalli, G.; Moreaux, J. EZH2 Is Overexpressed in Transitional Preplasmablasts and Is Involved in Human Plasma Cell Differentiation. Leukemia 2019, 33, 2047–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristini, A.; Groh, M.; Kristiansen, M.S.; Gromak, N. RNA/DNA Hybrid Interactome Identifies DXH9 as a Molecular Player in Transcriptional Termination and R-Loop-Associated DNA Damage. Cell Rep. 2018, 23, 1891–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergsagel, P.L.; Kuehl, W.M.; Zhan, F.; Sawyer, J.; Barlogie, B.; Shaughnessy, J. Cyclin D Dysregulation: An Early and Unifying Pathogenic Event in Multiple Myeloma. Blood 2005, 106, 296–303. [Google Scholar] [CrossRef] [Green Version]
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic Complexity of Multiple Myeloma and Its Clinical Implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113. [Google Scholar] [CrossRef] [PubMed]
- García-Muse, T.; Aguilera, A. Transcription–Replication Conflicts: How They Occur and How They Are Resolved. Nat. Rev. Mol. Cell Biol. 2016, 17, 553–563. [Google Scholar] [CrossRef]
- Gómez-González, B.; Aguilera, A. Transcription-Mediated Replication Hindrance: A Major Driver of Genome Instability. Genes Dev. 2019, 33, 1008–1026. [Google Scholar] [CrossRef] [Green Version]
- Kassambara, A.; Rème, T.; Jourdan, M.; Fest, T.; Hose, D.; Tarte, K.; Klein, B. GenomicScape: An Easy-to-Use Web Tool for Gene Expression Data Analysis. Application to Investigate the Molecular Events in the Differentiation of B Cells into Plasma Cells. PLoS Comput. Biol. 2015, 11. [Google Scholar] [CrossRef]
- Cerritelli, S.M.; Crouch, R.J. Ribonuclease H: The Enzymes in Eukaryotes. FEBS J. 2009, 276, 1494–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyjek, M.; Figiel, M.; Nowotny, M. RNases H: Structure and Mechanism. DNA Repair 2019, 84, 102672. [Google Scholar] [CrossRef]
- Ruhanen, H.; Ushakov, K.; Yasukawa, T. Involvement of DNA Ligase III and Ribonuclease H1 in Mitochondrial DNA Replication in Cultured Human Cells. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 2000. [Google Scholar] [CrossRef] [Green Version]
- Lima, W.F.; Murray, H.M.; Damle, S.S.; Hart, C.E.; Hung, G.; De Hoyos, C.L.; Liang, X.-H.; Crooke, S.T. Viable RNaseH1 Knockout Mice Show RNaseH1 Is Essential for R Loop Processing, Mitochondrial and Liver Function. Nucleic Acids Res. 2016, 44, 5299–5312. [Google Scholar] [CrossRef] [Green Version]
- Cerritelli, S.M.; Frolova, E.G.; Feng, C.; Grinberg, A.; Love, P.E.; Crouch, R.J. Failure to Produce Mitochondrial DNA Results in Embryonic Lethality in Rnaseh1 Null Mice. Mol. Cell 2003, 11, 807–815. [Google Scholar] [CrossRef]
- Parajuli, S.; Teasley, D.C.; Murali, B.; Jackson, J.; Vindigni, A.; Stewart, S.A. Human Ribonuclease H1 Resolves R-Loops and Thereby Enables Progression of the DNA Replication Fork. J. Biol. Chem. 2017, 292, 15216–15224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rydberg, B.; Game, J. Excision of Misincorporated Ribonucleotides in DNA by RNase H (Type 2) and FEN-1 in Cell-Free Extracts. Proc. Natl. Acad. Sci. USA 2002, 99, 16654–16659. [Google Scholar] [CrossRef] [Green Version]
- Nick McElhinny, S.A.; Watts, B.E.; Kumar, D.; Watt, D.L.; Lundström, E.-B.; Burgers, P.M.J.; Johansson, E.; Chabes, A.; Kunkel, T.A. Abundant Ribonucleotide Incorporation into DNA by Yeast Replicative Polymerases. Proc. Natl. Acad. Sci. USA 2010, 107, 4949–4954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.-X.; Williams, J.S.; Lujan, S.A.; Kunkel, T.A. Ribonucleotide Incorporation into DNA during DNA Replication and Its Consequences. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 109–124. [Google Scholar] [CrossRef]
- Crow, Y.J.; Leitch, A.; Hayward, B.E.; Garner, A.; Parmar, R.; Griffith, E.; Ali, M.; Semple, C.; Aicardi, J.; Babul-Hirji, R.; et al. Mutations in Genes Encoding Ribonuclease H2 Subunits Cause Aicardi-Goutières Syndrome and Mimic Congenital Viral Brain Infection. Nat. Genet. 2006, 38, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, K.J.; Carroll, P.; Martin, C.-A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. CGAS Surveillance of Micronuclei Links Genome Instability to Innate Immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wold, M.S. Replication Protein A: A Heterotrimeric, Single-Stranded DNA-Binding Protein Required for Eukaryotic DNA Metabolism. Annu. Rev. Biochem. 1997, 66, 61–92. [Google Scholar] [CrossRef]
- Iftode, C.; Daniely, Y.; Borowiec, J.A. Replication Protein A (RPA): The Eukaryotic SSB. Crit. Rev. Biochem. Mol. Biol. 1999, 34, 141–180. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.D.; Yadav, T.; Giri, S.; Saez, B.; Graubert, T.A.; Zou, L. Functions of Replication Protein A as a Sensor of R Loops and a Regulator of RNaseH1. Mol. Cell 2017, 65, 832–847.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazina, O.M.; Somarowthu, S.; Kadyrova, L.Y.; Baranovskiy, A.G.; Tahirov, T.H.; Kadyrov, F.A.; Mazin, A.V. Replication Protein A Binds RNA and Promotes R-Loop Formation. J. Biol. Chem. 2020, 295, 14203–14213. [Google Scholar] [CrossRef]
- Murakawa, Y.; Sonoda, E.; Barber, L.J.; Zeng, W.; Yokomori, K.; Kimura, H.; Niimi, A.; Lehmann, A.; Zhao, G.Y.; Hochegger, H.; et al. Inhibitors of the Proteasome Suppress Homologous DNA Recombination in Mammalian Cells. Cancer Res. 2007, 67, 8536–8543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquemont, C.; Taniguchi, T. Proteasome Function Is Required for DNA Damage Response and Fanconi Anemia Pathway Activation. Cancer Res. 2007, 67, 7395–7405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panero, J.; Stella, F.; Schutz, N.; Fantl, D.B.; Slavutsky, I. Differential Expression of Non-Shelterin Genes Associated with High Telomerase Levels and Telomere Shortening in Plasma Cell Disorders. PLoS ONE 2015, 10, e0137972. [Google Scholar] [CrossRef]
- Godbout, R.; Li, L.; Liu, R.-Z.; Roy, K. Role of DEAD Box 1 in Retinoblastoma and Neuroblastoma. Future Oncol. 2007, 3, 575–587. [Google Scholar] [CrossRef] [Green Version]
- Germain, D.R.; Graham, K.; Glubrecht, D.D.; Hugh, J.C.; Mackey, J.R.; Godbout, R. DEAD Box 1: A Novel and Independent Prognostic Marker for Early Recurrence in Breast Cancer. Breast Cancer Res. Treat. 2011, 127, 53–63. [Google Scholar] [CrossRef]
- Li, L.; Monckton, E.A.; Godbout, R. A Role for DEAD Box 1 at DNA Double-Strand Breaks. Mol. Cell. Biol. 2008, 28, 6413–6425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Germain, D.R.; Poon, H.-Y.; Hildebrandt, M.R.; Monckton, E.A.; McDonald, D.; Hendzel, M.J.; Godbout, R. DEAD Box 1 Facilitates Removal of RNA and Homologous Recombination at DNA Double-Strand Breaks. Mol. Cell. Biol. 2016, 36, 2794–2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro de Almeida, C.; Dhir, S.; Dhir, A.; Moghaddam, A.E.; Sattentau, Q.; Meinhart, A.; Proudfoot, N.J. RNA Helicase DDX1 Converts RNA G-Quadruplex Structures into R-Loops to Promote IgH Class Switch Recombination. Mol. Cell 2018, 70, 650–662.e8. [Google Scholar] [CrossRef] [Green Version]
- Hänsel-Hertsch, R.; Di Antonio, M.; Balasubramanian, S. DNA G-Quadruplexes in the Human Genome: Detection, Functions and Therapeutic Potential. Nat. Rev. Mol. Cell Biol. 2017, 18, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.; Lipps, H.J. G-Quadruplexes and Their Regulatory Roles in Biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [Green Version]
- Prioleau, M.-N. G-Quadruplexes and DNA Replication Origins. Adv. Exp. Med. Biol. 2017, 1042, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Prorok, P.; Artufel, M.; Aze, A.; Coulombe, P.; Peiffer, I.; Lacroix, L.; Guédin, A.; Mergny, J.-L.; Damaschke, J.; Schepers, A.; et al. Involvement of G-Quadruplex Regions in Mammalian Replication Origin Activity. Nat. Commun. 2019, 10, 3274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsov, V.A.; Bondarenko, V.; Wongsurawat, T.; Yenamandra, S.P.; Jenjaroenpun, P. Toward Predictive R-Loop Computational Biology: Genome-Scale Prediction of R-Loops Reveals Their Association with Complex Promoter Structures, G-Quadruplexes and Transcriptionally Active Enhancers. Nucleic Acids Res. 2018, 46, 7566–7585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duquette, M.L. Intracellular Transcription of G-Rich DNAs Induces Formation of G-Loops, Novel Structures Containing G4 DNA. Genes Dev. 2004, 18, 1618–1629. [Google Scholar] [CrossRef] [Green Version]
- De Magis, A.; Manzo, S.G.; Russo, M.; Marinello, J.; Morigi, R.; Sordet, O.; Capranico, G. DNA Damage and Genome Instability by G-Quadruplex Ligands Are Mediated by R Loops in Human Cancer Cells. Proc. Natl. Acad. Sci. USA 2019, 116, 816–825. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.; Lieber, M.R. Current Insights into the Mechanism of Mammalian Immunoglobulin Class Switch Recombination. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 333–351. [Google Scholar] [CrossRef]
- Stavnezer, J.; Schrader, C.E. Ig Heavy Chain Class Switch Recombination: Mechanism and Regulation. J. Immunol. 2014, 193, 5370–5378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, R.; Hartmuth, K.; Möhlmann, S.; Urlaub, H.; Ficner, R.; Lührmann, R. Phosphorylation of Human PRP28 by SRPK2 Is Required for Integration of the U4/U6-U5 Tri-SnRNP into the Spliceosome. Nat. Struct. Mol. Biol. 2008, 15, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Sridhara, S.C.; Carvalho, S.; Grosso, A.R.; Gallego-Paez, L.M.; Carmo-Fonseca, M.; de Almeida, S.F. Transcription Dynamics Prevent RNA-Mediated Genomic Instability through SRPK2-Dependent DDX23 Phosphorylation. Cell Rep. 2017, 18, 334–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.D. The Superfamily of Arginine/Serine-Rich Splicing Factors. RNA 1995, 1, 663–680. [Google Scholar]
- Mayeda, A.; Krainer, A.R. Regulation of Alternative Pre-MRNA Splicing by HnRNP A1 and Splicing Factor SF2. Cell 1992, 68, 365–375. [Google Scholar] [CrossRef]
- Cáceres, J.F.; Kornblihtt, A.R. Alternative Splicing: Multiple Control Mechanisms and Involvement in Human Disease. Trends Genet. 2002, 18, 186–193. [Google Scholar] [CrossRef]
- Li, X.; Manley, J.L. Inactivation of the SR Protein Splicing Factor ASF/SF2 Results in Genomic Instability. Cell 2005, 122, 365–378. [Google Scholar] [CrossRef] [Green Version]
- Promonet, A.; Padioleau, I.; Liu, Y.; Sanz, L.; Biernacka, A.; Schmitz, A.-L.; Skrzypczak, M.; Sarrazin, A.; Mettling, C.; Rowicka, M.; et al. Topoisomerase 1 Prevents Replication Stress at R-Loop-Enriched Transcription Termination Sites. Nat. Commun. 2020, 11, 3940. [Google Scholar] [CrossRef]
- Li, M.; Pokharel, S.; Wang, J.-T.; Xu, X.; Liu, Y. RECQ5-Dependent SUMOylation of DNA Topoisomerase I Prevents Transcription-Associated Genome Instability. Nat. Commun. 2015, 6, 6720. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.C. Cellular Roles of DNA Topoisomerases: A Molecular Perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.Y.; Shyy, S.H.; Wang, J.C.; Liu, L.F. Transcription Generates Positively and Negatively Supercoiled Domains in the Template. Cell 1988, 53, 433–440. [Google Scholar] [CrossRef]
- Marinello, J.; Bertoncini, S.; Aloisi, I.; Cristini, A.; Malagoli Tagliazucchi, G.; Forcato, M.; Sordet, O.; Capranico, G. Dynamic Effects of Topoisomerase I Inhibition on R-Loops and Short Transcripts at Active Promoters. PLoS ONE 2016, 11, e0147053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obrdlik, A.; Kukalev, A.; Louvet, E.; Farrants, A.-K.O.; Caputo, L.; Percipalle, P. The Histone Acetyltransferase PCAF Associates with Actin and HnRNP U for RNA Polymerase II Transcription. Mol. Cell. Biol. 2008, 28, 6342–6357. [Google Scholar] [CrossRef] [Green Version]
- Yugami, M.; Kabe, Y.; Yamaguchi, Y.; Wada, T.; Handa, H. HnRNP-U Enhances the Expression of Specific Genes by Stabilizing MRNA. FEBS Lett. 2007, 581, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Xiao, R.; Tang, P.; Yang, B.; Huang, J.; Zhou, Y.; Shao, C.; Li, H.; Sun, H.; Zhang, Y.; Fu, X.-D. Nuclear Matrix Factor HnRNP U/SAF-A Exerts a Global Control of Alternative Splicing by Regulating U2 SnRNP Maturation. Mol. Cell 2012, 45, 656–668. [Google Scholar] [CrossRef] [Green Version]
- Britton, S.; Dernoncourt, E.; Delteil, C.; Froment, C.; Schiltz, O.; Salles, B.; Frit, P.; Calsou, P. DNA Damage Triggers SAF-A and RNA Biogenesis Factors Exclusion from Chromatin Coupled to R-Loops Removal. Nucleic Acids Res. 2014, 42, 9047–9062. [Google Scholar] [CrossRef] [Green Version]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [Green Version]
- Alfano, L.; Caporaso, A.; Altieri, A.; Dell’Aquila, M.; Landi, C.; Bini, L.; Pentimalli, F.; Giordano, A. Depletion of the RNA Binding Protein HNRNPD Impairs Homologous Recombination by Inhibiting DNA-End Resection and Inducing R-Loop Accumulation. Nucleic Acids Res. 2019, 47, 4068–4085. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.-H.; De, S.; Srikantan, S.; Abdelmohsen, K.; Grammatikakis, I.; Kim, J.; Kim, K.M.; Noh, J.H.; White, E.J.F.; Martindale, J.L.; et al. PAR-CLIP Analysis Uncovers AUF1 Impact on Target RNA Fate and Genome Integrity. Nat. Commun. 2014, 5, 5248. [Google Scholar] [CrossRef] [Green Version]
- Marchesini, M.; Ogoti, Y.; Fiorini, E.; Aktas Samur, A.; Nezi, L.; D’Anca, M.; Storti, P.; Samur, M.K.; Ganan-Gomez, I.; Fulciniti, M.T.; et al. ILF2 Is a Regulator of RNA Splicing and DNA Damage Response in 1q21-Amplified Multiple Myeloma. Cancer Cell 2017, 32, 88–100.e6. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, Y.; Abe, M.; Itaya, A.; Tomida, J.; Ishiai, M.; Takaori-Kondo, A.; Taoka, M.; Isobe, T.; Takata, M. FANCD2 Protects Genome Stability by Recruiting RNA Processing Enzymes to Resolve R-Loops during Mild Replication Stress. FEBS J. 2019, 286, 139–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimeno, S.; Rondón, A.G.; Luna, R.; Aguilera, A. The Yeast THO Complex and MRNA Export Factors Link RNA Metabolism with Transcription and Genome Instability. EMBO J. 2002, 21, 3526–3535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strässer, K.; Masuda, S.; Mason, P.; Pfannstiel, J.; Oppizzi, M.; Rodriguez-Navarro, S.; Rondón, A.G.; Aguilera, A.; Struhl, K.; Reed, R.; et al. TREX Is a Conserved Complex Coupling Transcription with Messenger RNA Export. Nature 2002, 417, 304–308. [Google Scholar] [CrossRef]
- Luna, R.; Rondón, A.G.; Pérez-Calero, C.; Salas-Armenteros, I.; Aguilera, A. The THO Complex as a Paradigm for the Prevention of Cotranscriptional R-Loops. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2019; Volume 84, pp. 105–114. [Google Scholar] [CrossRef]
- Gómez-González, B.; Felipe-Abrio, I.; Aguilera, A. The S-Phase Checkpoint Is Required To Respond to R-Loops Accumulated in THO Mutants. Mol. Cell. Biol. 2009, 29, 5203–5213. [Google Scholar] [CrossRef] [Green Version]
- Gómez-González, B.; García-Rubio, M.; Bermejo, R.; Gaillard, H.; Shirahige, K.; Marín, A.; Foiani, M.; Aguilera, A. Genome-Wide Function of THO/TREX in Active Genes Prevents R-Loop-Dependent Replication Obstacles. EMBO J. 2011, 30, 3106–3119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domínguez-Sánchez, M.S.; Barroso, S.; Gómez-González, B.; Luna, R.; Aguilera, A. Genome Instability and Transcription Elongation Impairment in Human Cells Depleted of THO/TREX. PLoS Genet. 2011, 7. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Calero, C.; Bayona-Feliu, A.; Xue, X.; Barroso, S.I.; Muñoz, S.; González-Basallote, V.M.; Sung, P.; Aguilera, A. UAP56/DDX39B Is a Major Cotranscriptional RNA–DNA Helicase That Unwinds Harmful R Loops Genome-Wide. Genes Dev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Salas-Armenteros, I.; Pérez-Calero, C.; Bayona-Feliu, A.; Tumini, E.; Luna, R.; Aguilera, A. Human THO–Sin3A Interaction Reveals New Mechanisms to Prevent R-loops That Cause Genome Instability. EMBO J. 2017, 36, 3532–3547. [Google Scholar] [CrossRef]
- Cai, S.; Bai, Y.; Wang, H.; Zhao, Z.; Ding, X.; Zhang, H.; Zhang, X.; Liu, Y.; Jia, Y.; Li, Y.; et al. Knockdown of THOC1 Reduces the Proliferation of Hepatocellular Carcinoma and Increases the Sensitivity to Cisplatin. J. Exp. Clin. Cancer Res. 2020, 39, 135. [Google Scholar] [CrossRef]
- Faza, M.B.; Kemmler, S.; Jimeno, S.; González-Aguilera, C.; Aguilera, A.; Hurt, E.; Panse, V.G. Sem1 Is a Functional Component of the Nuclear Pore Complex-Associated Messenger RNA Export Machinery. J. Cell Biol. 2009, 184, 833–846. [Google Scholar] [CrossRef] [Green Version]
- Wilmes, G.M.; Bergkessel, M.; Bandyopadhyay, S.; Shales, M.; Braberg, H.; Cagney, G.; Collins, S.R.; Whitworth, G.B.; Kress, T.L.; Weissman, J.S.; et al. A Genetic Interaction Map of RNA-Processing Factors Reveals Links between Sem1/Dss1-Containing Complexes and MRNA Export and Splicing. Mol. Cell 2008, 32, 735–746. [Google Scholar] [CrossRef] [Green Version]
- Gallardo, M.; Luna, R.; Erdjument-Bromage, H.; Tempst, P.; Aguilera, A. Nab2p and the Thp1p-Sac3p Complex Functionally Interact at the Interface between Transcription and MRNA Metabolism. J. Biol. Chem. 2003, 278, 24225–24232. [Google Scholar] [CrossRef] [Green Version]
- Santos-Pereira, J.M.; García-Rubio, M.L.; González-Aguilera, C.; Luna, R.; Aguilera, A. A Genome-Wide Function of THSC/TREX-2 at Active Genes Prevents Transcription–Replication Collisions. Nucleic Acids Res. 2014, 42, 12000–12014. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, V.; Barroso, S.I.; García-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 Prevents R-Loop Accumulation and Associates with TREX-2 MRNA Export Factor PCID2. Nature 2014, 511, 362–365. [Google Scholar] [CrossRef]
- Eberle, A.B.; Visa, N. Quality Control of MRNP Biogenesis: Networking at the Transcription Site. In Seminars in Cell and Developmental Biology; Academic Press: New York, NY, USA, 2014; Volume 32, pp. 37–46. [Google Scholar] [CrossRef]
- Schmid, M.; Jensen, T.H. The Exosome: A Multipurpose RNA-Decay Machine. Trends Biochem. Sci. 2008, 33, 501–510. [Google Scholar] [CrossRef]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.-P.; Ahmann, G.J.; Adli, M.; et al. Initial Genome Sequencing and Analysis of Multiple Myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Boyle, E.M.; Ashby, C.; Tytarenko, R.G.; Deshpande, S.; Wang, H.; Wang, Y.; Rosenthal, A.; Sawyer, J.; Tian, E.; Flynt, E.; et al. BRAF and DIS3 Mutations Associate with Adverse Outcome in a Long-Term Follow-up of Patients with Multiple Myeloma. Clin. Cancer Res. 2020, 26, 2422–2432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todoerti, K.; Ronchetti, D.; Favasuli, V.; Maura, F.; Morabito, F.; Bolli, N.; Taiana, E.; Neri, A. DIS3 Mutations in Multiple Myeloma Impact the Transcriptional Signature and Clinical Outcome. Arch. Ist. Ric. 2021. [Google Scholar] [CrossRef]
- Pertesi, M.; Vallée, M.; Wei, X.; Revuelta, M.V.; Galia, P.; Demangel, D.; Oliver, J.; Foll, M.; Chen, S.; Perrial, E.; et al. Exome Sequencing Identifies Germline Variants in DIS3 in Familial Multiple Myeloma. Leukemia 2019, 33, 2324–2330. [Google Scholar] [CrossRef] [Green Version]
- Domingo-Prim, J.; Endara-Coll, M.; Bonath, F.; Jimeno, S.; Prados-Carvajal, R.; Friedländer, M.R.; Huertas, P.; Visa, N. EXOSC10 Is Required for RPA Assembly and Controlled DNA End Resection at DNA Double-Strand Breaks. Nat. Commun. 2019, 10, 2135. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.; Feng, S.; Manley, J.L. A SUMO-Dependent Interaction between Senataxin and the Exosome, Disrupted in the Neurodegenerative Disease AOA2, Targets the Exosome to Sites of Transcription-Induced DNA Damage. Genes Dev. 2013, 27, 2227–2232. [Google Scholar] [CrossRef] [Green Version]
- Ogami, K.; Chen, Y.; Manley, J.L. RNA Surveillance by the Nuclear RNA Exosome: Mechanisms and Significance. Noncoding RNA 2018, 4, 8. [Google Scholar] [CrossRef] [Green Version]
- Kazadi, D.; Lim, J.; Rothschild, G.; Grinstein, V.; Laffleur, B.; Becherel, O.; Lavin, M.J.; Basu, U. Effects of Senataxin and RNA Exosome on B-Cell Chromosomal Integrity. Heliyon 2020, 6. [Google Scholar] [CrossRef]
- Basu, U.; Meng, F.-L.; Keim, C.; Grinstein, V.; Pefanis, E.; Eccleston, J.; Zhang, T.; Myers, D.; Wasserman, C.R.; Wesemann, D.R.; et al. The RNA Exosome Targets the AID Cytidine Deaminase to Both Strands of Transcribed Duplex DNA Substrates. Cell 2011, 144, 353–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laffleur, B.; Lim, J.; Zhang, W.; Chen, Y.; Pefanis, E.; Bizarro, J.; Batista, C.R.; Wu, L.; Economides, A.N.; Wang, J.; et al. Noncoding RNA Processing by DIS3 Regulates Chromosomal Architecture and Somatic Hypermutation in B Cells. Nat. Genet. 2021, 53, 230–242. [Google Scholar] [CrossRef]
- Kim, M.; Vasiljeva, L.; Rando, O.J.; Zhelkovsky, A.; Moore, C.; Buratowski, S. Distinct Pathways for SnoRNA and MRNA Termination. Mol. Cell 2006, 24, 723–734. [Google Scholar] [CrossRef]
- West, S.; Gromak, N.; Proudfoot, N.J. Human 5′→3′ Exonuclease Xrn2 Promotes Transcription Termination at Co-Transcriptional Cleavage Sites. Nature 2004, 432, 522–525. [Google Scholar] [CrossRef]
- Morales, J.C.; Richard, P.; Patidar, P.L.; Motea, E.A.; Dang, T.T.; Manley, J.L.; Boothman, D.A. XRN2 Links Transcription Termination to DNA Damage and Replication Stress. PLoS Genet. 2016, 12. [Google Scholar] [CrossRef] [Green Version]
- Dang, T.T.; Morales, J.C. XRN2 Links RNA:DNA Hybrid Resolution to Double Strand Break Repair Pathway Choice. Cancers 2020, 12, 1821. [Google Scholar] [CrossRef] [PubMed]
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human Senataxin Resolves RNA/DNA Hybrids Formed at Transcriptional Pause Sites to Promote Xrn2-Dependent Termination. Mol. Cell 2011, 42, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Hirling, H.; Scheffner, M.; Restle, T.; Stahl, H. RNA Helicase Activity Associated with the Human P68 Protein. Nature 1989, 339, 562–564. [Google Scholar] [CrossRef] [PubMed]
- Xing, Z.; Wang, S.; Tran, E.J. Characterization of the Mammalian DEAD-Box Protein DDX5 Reveals Functional Conservation with S. Cerevisiae Ortholog Dbp2 in Transcriptional Control and Glucose Metabolism. RNA 2017, 23, 1125–1138. [Google Scholar] [CrossRef] [Green Version]
- Mersaoui, S.Y.; Yu, Z.; Coulombe, Y.; Karam, M.; Busatto, F.F.; Masson, J.; Richard, S. Arginine Methylation of the DDX5 Helicase RGG/RG Motif by PRMT5 Regulates Resolution of RNA:DNA Hybrids. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Villarreal, O.D.; Mersaoui, S.Y.; Yu, Z.; Masson, J.-Y.; Richard, S. Genome-Wide R-Loop Analysis Defines Unique Roles for DDX5, XRN2, and PRMT5 in DNA/RNA Hybrid Resolution. Life Sci. Alliance 2020, 3. [Google Scholar] [CrossRef]
- Gullà, A.; Hideshima, T.; Bianchi, G.; Fulciniti, M.; Kemal Samur, M.; Qi, J.; Tai, Y.-T.; Harada, T.; Morelli, E.; Amodio, N.; et al. Protein Arginine Methyltransferase 5 Has Prognostic Relevance and Is a Druggable Target in Multiple Myeloma. Leukemia 2018, 32, 996–1002. [Google Scholar] [CrossRef]
- Bogliolo, M.; Surrallés, J. Fanconi Anemia: A Model Disease for Studies on Human Genetics and Advanced Therapeutics. Curr. Opin. Genet. Dev. 2015, 33, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Tischkowitz, M.D.; Hodgson, S.V. Fanconi Anaemia. J. Med. Genet. 2003, 40, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P.; Greene, M.H.; Velazquez, I.; Rosenberg, P.S. Cancer in Fanconi Anemia. Blood 2003, 101, 2072. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi Anaemia Pathway: New Players and New Functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef]
- Mamrak, N.E.; Shimamura, A.; Howlett, N.G. Recent Discoveries in the Molecular Pathogenesis of the Inherited Bone Marrow Failure Syndrome Fanconi Anemia. Blood Rev. 2017, 31, 93–99. [Google Scholar] [CrossRef] [Green Version]
- García-Rubio, M.L.; Pérez-Calero, C.; Barroso, S.I.; Tumini, E.; Herrera-Moyano, E.; Rosado, I.V.; Aguilera, A. The Fanconi Anemia Pathway Protects Genome Integrity from R-Loops. PLoS Genet. 2015, 11, e1005674. [Google Scholar] [CrossRef] [Green Version]
- Schwab, R.A.; Nieminuszczy, J.; Shah, F.; Langton, J.; Lopez Martinez, D.; Liang, C.-C.; Cohn, M.A.; Gibbons, R.J.; Deans, A.J.; Niedzwiedz, W. The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol. Cell 2015, 60, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Han, S.-S.; Tompkins, V.S.; Son, D.-J.; Han, S.; Yun, H.; Kamberos, N.L.; Dehoedt, C.L.; Gu, C.; Holman, C.; Tricot, G.; et al. CDKN1A and FANCD2 Are Potential Oncotargets in Burkitt Lymphoma and Multiple Myeloma. Exp. Hematol. Oncol. 2015, 4, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madireddy, A.; Kosiyatrakul, S.T.; Gerhardt, J.; Boisvert, R.A.; Vuono, E.A.; Moyano, E.H.; Garcia Rubio, M.L.; Owen, N.; Yan, Z.; Olson, S.; et al. FANCD2 Facilitates Replication through Common Fragile Sites. Mol. Cell 2016, 64, 388–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Z.; Liang, F.; Teng, Y.; Chen, X.; Liu, J.; Longerich, S.; Rao, T.; Green, A.M.; Collins, N.B.; Xiong, Y.; et al. Binding of FANCI-FANCD2 Complex to RNA and R-Loops Stimulates Robust FANCD2 Monoubiquitination. Cell Rep. 2019, 26, 564–572.e5. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.-P.; Yin, X.-H.; Meng, X.-Y.; Yan, X.-H.; Wang, F.; He, L. Development and Validation of a 9-Gene Prognostic Signature in Patients with Multiple Myeloma. Front. Oncol. 2019, 8, 615. [Google Scholar] [CrossRef] [PubMed]
- Bohl, S.R.; Schmalbrock, L.K.; Bauhuf, I.; Meyer, T.; Dolnik, A.; Szyska, M.; Blätte, T.J.; Knödler, S.; Röhner, L.; Miller, D.; et al. Comprehensive CRISPR-Cas9 Screens Identify Genetic Determinants of Drug Responsiveness in Multiple Myeloma. Blood Adv. 2021, 5, 2391–2402. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.Y.-C.; Tsai, S.; Aristizabal, M.J.; Wells, J.P.; Coulombe, Y.; Busatto, F.F.; Chan, Y.A.; Kumar, A.; Dan Zhu, Y.; Wang, A.Y.-H.; et al. MRE11-RAD50-NBS1 Promotes Fanconi Anemia R-Loop Suppression at Transcription–Replication Conflicts. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Wu, Y.; Shin-ya, K.; Brosh, R.M. FANCJ Helicase Defective in Fanconia Anemia and Breast Cancer Unwinds G-Quadruplex DNA To Defend Genomic Stability. Mol. Cell. Biol. 2008, 28, 4116–4128. [Google Scholar] [CrossRef] [Green Version]
- London, T.B.C.; Barber, L.J.; Mosedale, G.; Kelly, G.P.; Balasubramanian, S.; Hickson, I.D.; Boulton, S.J.; Hiom, K. FANCJ Is a Structure-Specific DNA Helicase Associated with the Maintenance of Genomic G/C Tracts. J. Biol. Chem. 2008, 283, 36132–36139. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.G.; Spies, M. G-Quadruplex Recognition and Remodeling by the FANCJ Helicase. Nucleic Acids Res. 2016, 44, 8742–8753. [Google Scholar] [CrossRef] [Green Version]
- Castillo Bosch, P.; Segura-Bayona, S.; Koole, W.; van Heteren, J.T.; Dewar, J.M.; Tijsterman, M.; Knipscheer, P. FANCJ Promotes DNA Synthesis through G-Quadruplex Structures. EMBO J. 2014, 33, 2521–2533. [Google Scholar] [CrossRef] [Green Version]
- Cantor, S.; Drapkin, R.; Zhang, F.; Lin, Y.; Han, J.; Pamidi, S.; Livingston, D.M. The BRCA1-Associated Protein BACH1 Is a DNA Helicase Targeted by Clinically Relevant Inactivating Mutations. Proc. Natl. Acad. Sci. USA 2004, 101, 2357–2362. [Google Scholar] [CrossRef] [Green Version]
- Odermatt, D.C.; Lee, W.T.C.; Wild, S.; Jozwiakowski, S.K.; Rothenberg, E.; Gari, K. Cancer-Associated Mutations in the Iron-Sulfur Domain of FANCJ Affect G-Quadruplex Metabolism. PLoS Genet. 2020, 16. [Google Scholar] [CrossRef]
- Lowran, K.; Campbell, L.; Popp, P.; Wu, C.G. Assembly of a G-Quadruplex Repair Complex by the FANCJ DNA Helicase and the REV1 Polymerase. Genes 2019, 11, 5. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.; Ketkar, A.; Zafar, M.K.; Maddukuri, L.; Choi, J.-Y.; Eoff, R.L. Human Rev1 Polymerase Disrupts G-Quadruplex DNA. Nucleic Acids Res. 2014, 42, 3272–3285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ketkar, A.; Smith, L.; Johnson, C.; Richey, A.; Berry, M.; Hartman, J.H.; Maddukuri, L.; Reed, M.R.; Gunderson, J.E.C.; Leung, J.W.C.; et al. Human Rev1 Relies on Insert-2 to Promote Selective Binding and Accurate Replication of Stabilized G-Quadruplex Motifs. Nucleic Acids Res. 2021, 49, 2065–2084. [Google Scholar] [CrossRef] [PubMed]
- Nalepa, G.; Clapp, D.W. Fanconi Anaemia and Cancer: An Intricate Relationship. Nat. Rev. Cancer 2018, 18, 168–185. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.I.; Matchett, K.B.; Barros, E.M.; Cooper, K.M.; Irwin, G.W.; Gorski, J.J.; Orr, K.S.; Vohhodina, J.; Kavanagh, J.N.; Madden, A.F.; et al. BRCA1 Deficiency Exacerbates Estrogen-Induced DNA Damage and Genomic Instability. Cancer Res. 2014, 74, 2773–2784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarde, D.N.; Oliveira, V.; Mathews, L.; Wang, X.; Villagra, A.; Boulware, D.; Shain, K.H.; Hazlehurst, L.A.; Alsina, M.; Chen, D.-T.; et al. Targeting the Fanconi Anemia/BRCA Pathway Circumvents Drug Resistance in Multiple Myeloma. Cancer Res. 2009, 69, 9367–9375. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Van der Sluis, P.C.; Boulware, D.; Hazlehurst, L.A.; Dalton, W.S. The FA/BRCA Pathway Is Involved in Melphalan-Induced DNA Interstrand Cross-Link Repair and Accounts for Melphalan Resistance in Multiple Myeloma Cells. Blood 2005, 106, 698–705. [Google Scholar] [CrossRef]
- Volcic, M.; Karl, S.; Baumann, B.; Salles, D.; Daniel, P.; Fulda, S.; Wiesmüller, L. NF-ΚB Regulates DNA Double-Strand Break Repair in Conjunction with BRCA1–CtIP Complexes. Nucleic Acids Res. 2012, 40, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, Y.N.; Kuehl, W.M. A Critical Role for the NFkB Pathway in Multiple Myeloma. Oncotarget 2010, 1, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 Recruitment to Transcriptional Pause Sites Is Required for R-Loop-Driven DNA Damage Repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, S.J.; Rolland, T.; Adelmant, G.; Xia, X.; Owen, M.S.; Dricot, A.; Zack, T.I.; Sahni, N.; Jacob, Y.; Hao, T.; et al. Systematic Screening Reveals a Role for BRCA1 in the Response to Transcription-Associated DNA Damage. Genes Dev. 2014, 28, 1957–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Chiang, H.-C.; Wang, Y.; Zhang, C.; Smith, S.; Zhao, X.; Nair, S.J.; Michalek, J.; Jatoi, I.; Lautner, M.; et al. Attenuation of RNA Polymerase II Pausing Mitigates BRCA1-Associated R-Loop Accumulation and Tumorigenesis. Nat. Commun. 2017, 8, 15908. [Google Scholar] [CrossRef] [PubMed]
- Sessa, G.; Gómez-González, B.; Silva, S.; Pérez-Calero, C.; Beaurepere, R.; Barroso, S.; Martineau, S.; Martin, C.; Ehlén, Å.; Martínez, J.S.; et al. BRCA2 Promotes R-Loop Resolution by DDX5 Helicase at DNA Breaks to Facilitate Their Repair by Homologous Recombination. EMBO J. 2021, 40, e106018. [Google Scholar] [CrossRef]
- Yu, Z.; Mersaoui, S.Y.; Guitton-Sert, L.; Coulombe, Y.; Song, J.; Masson, J.-Y.; Richard, S. DDX5 Resolves R-Loops at DNA Double-Strand Breaks to Promote DNA Repair and Avoid Chromosomal Deletions. NAR Cancer 2020, 2. [Google Scholar] [CrossRef]
- Mailand, N.; Gibbs-Seymour, I.; Bekker-Jensen, S. Regulation of PCNA-Protein Interactions for Genome Stability. Nat. Rev. Mol. Cell Biol. 2013, 14, 269–282. [Google Scholar] [CrossRef]
- Alexandrakis, M.G.; Passam, F.H.; Pappa, C.A.; Dambaki, C.; Sfakiotaki, G.; Alegakis, A.K.; Kyriakou, D.S.; Stathopoulos, E. Expression of Proliferating Cell Nuclear Antigen (PCNA) in Multiple Myeloma: Its Relationship to Bone Marrow Microvessel Density and Other Factors of Disease Activity. Int. J. Immunopathol. Pharmacol. 2004, 17, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Tsirakis, G.; Pappa, C.A.; Kaparou, M.; Katsomitrou, V.; Hatzivasili, A.; Alegakis, T.; Xekalou, A.; Stathopoulos, E.N.; Alexandrakis, M.G. Assessment of Proliferating Cell Nuclear Antigen and Its Relationship with Proinflammatory Cytokines and Parameters of Disease Activity in Multiple Myeloma Patients. Eur. J. Histochem. 2011, 55, e21. [Google Scholar] [CrossRef]
- Müller, R.; Misund, K.; Holien, T.; Bachke, S.; Gilljam, K.M.; Våtsveen, T.K.; Rø, T.B.; Bellacchio, E.; Sundan, A.; Otterlei, M. Targeting Proliferating Cell Nuclear Antigen and Its Protein Interactions Induces Apoptosis in Multiple Myeloma Cells. PLoS ONE 2013, 8, e70430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moldovan, G.-L.; Dejsuphong, D.; Petalcorin, M.I.R.; Hofmann, K.; Takeda, S.; Boulton, S.J.; D’Andrea, A.D. Inhibition of Homologous Recombination by the PCNA-Interacting Protein PARI. Mol. Cell 2012, 45, 75–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gali, H.; Juhasz, S.; Morocz, M.; Hajdu, I.; Fatyol, K.; Szukacsov, V.; Burkovics, P.; Haracska, L. Role of SUMO Modification of Human PCNA at Stalled Replication Fork. Nucleic Acids Res. 2012, 40, 6049–6059. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Xu, X.; Chang, C.-W.; Zheng, L.; Shen, B.; Liu, Y. SUMO2 Conjugation of PCNA Facilitates Chromatin Remodeling to Resolve Transcription-Replication Conflicts. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Gu, Y.; Wang, W.; Wang, X.; Ye, X.; Xin, C.; Lu, M.; Reddy, B.A.; Shu, P. Silencing of SENP2 in Multiple Myeloma Induces Bortezomib Resistance by Activating NF-ΚB Through the Modulation of IκBα Sumoylation. Sci. Rep. 2020, 10, 766. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP Promotes DNA End Resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Symington, L.S. Mechanism and Regulation of DNA End Resection in Eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 195–212. [Google Scholar] [CrossRef] [Green Version]
- Makharashvili, N.; Tubbs, A.T.; Yang, S.-H.; Wang, H.; Barton, O.; Zhou, Y.; Deshpande, R.A.; Lee, J.-H.; Lobrich, M.; Sleckman, B.P.; et al. Catalytic and Noncatalytic Roles of the CtIP Endonuclease in Double-Strand Break End Resection. Mol. Cell 2014, 54, 1022–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Song, Y.; He, X.; Liu, X.; Zhang, Y.; Yang, Z.; Yang, P.; Wang, J.; Hu, K.; Liu, W.; et al. Prognosis Value of RBBP8 Expression in Plasma Cell Myeloma. Cancer Gene Ther. 2020, 27, 22–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makharashvili, N.; Arora, S.; Yin, Y.; Fu, Q.; Wen, X.; Lee, J.-H.; Kao, C.-H.; Leung, J.W.; Miller, K.M.; Paull, T.T. Sae2/CtIP Prevents R-Loop Accumulation in Eukaryotic Cells. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Keijzers, G.; Liu, D.; Rasmussen, L.J. Exonuclease 1 and Its Versatile Roles in DNA Repair. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Stroik, S.; Kurtz, K.; Lin, K.; Karachenets, S.; Myers, C.L.; Bielinsky, A.-K.; Hendrickson, E.A. EXO1 Resection at G-Quadruplex Structures Facilitates Resolution and Replication. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef] [Green Version]
- Dehé, P.-M.; Gaillard, P.-H.L. Control of Structure-Specific Endonucleases to Maintain Genome Stability. Nat. Rev. Mol. Cell Biol. 2017, 18, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Alt, F.W. Transcription-Induced Cleavage of Immunoglobulin Switch Regions by Nucleotide Excision Repair Nucleases in vitro. J. Biol. Chem. 2000, 275, 24163–24172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sollier, J.; Stork, C.T.; García-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-Coupled Nucleotide Excision Repair Factors Promote R-Loop-Induced Genome Instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef] [Green Version]
- Cristini, A.; Ricci, G.; Britton, S.; Salimbeni, S.; Huang, S.N.; Marinello, J.; Calsou, P.; Pommier, Y.; Favre, G.; Capranico, G.; et al. Dual Processing of R-Loops and Topoisomerase I Induces Transcription-Dependent DNA Double-Strand Breaks. Cell Rep. 2019, 28, 3167–3181. [Google Scholar] [CrossRef] [Green Version]
- Yasuhara, T.; Kato, R.; Hagiwara, Y.; Shiotani, B.; Yamauchi, M.; Nakada, S.; Shibata, A.; Miyagawa, K. Human Rad52 Promotes XPG-Mediated R-Loop Processing to Initiate Transcription-Associated Homologous Recombination Repair. Cell 2018, 175, 558–570.e11. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-L.; Pasero, P. Caught in the Act: R-Loops Are Cleaved by Structure-Specific Endonucleases to Generate DSBs. Mol. Cell 2014, 56, 721–722. [Google Scholar] [CrossRef] [Green Version]
- de Larrea, C.F.; Navarro, A.; Tovar, N.; Pedrosa, F.; Díaz, T.; Cibeira, M.T.; Magnano, L.; Rosiñol, L.; Rovira, M.; Rozman, M.; et al. Impact of Single Nucleotide Polymorphisms in Genes Involved in DNA Repair and Drug Metabolism On Survival After Autologous Stem Cell Transplantation in Patients with Multiple Myeloma. Blood 2012, 120, 2934. [Google Scholar] [CrossRef]
- He, Y.; Pasupala, N.; Zhi, H.; Dorjbal, B.; Hussain, I.; Shih, H.-M.; Bhattacharyya, S.; Biswas, R.; Miljkovic, M.; Semmes, O.J.; et al. NF-ΚB–Induced R-Loop Accumulation and DNA Damage Select for Nucleotide Excision Repair Deficiencies in Adult T Cell Leukemia. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Ellis, N.A.; Groden, J.; Ye, T.Z.; Straughen, J.; Lennon, D.J.; Ciocci, S.; Proytcheva, M.; German, J. The Bloom’s Syndrome Gene Product Is Homologous to RecQ Helicases. Cell 1995, 83, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Karow, J.K.; Hickson, I.D.; Maizels, N. The Bloom’s Syndrome Helicase Unwinds G4 DNA. J. Biol. Chem. 1998, 273, 27587–27592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popuri, V.; Bachrati, C.Z.; Muzzolini, L.; Mosedale, G.; Costantini, S.; Giacomini, E.; Hickson, I.D.; Vindigni, A. The Human RecQ Helicases, BLM and RECQ1, Display Distinct DNA Substrate Specificities. J. Biol. Chem. 2008, 283, 17766–17776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.-Q.; Hou, X.-M.; Li, M.; Dou, S.-X.; Xi, X.-G. BLM Unfolds G-Quadruplexes in Different Structural Environments through Different Mechanisms. Nucleic Acids Res. 2015, 43, 4614–4626. [Google Scholar] [CrossRef] [Green Version]
- Chang, E.Y.-C.; Novoa, C.A.; Aristizabal, M.J.; Coulombe, Y.; Segovia, R.; Chaturvedi, R.; Shen, Y.; Keong, C.; Tam, A.S.; Jones, S.J.M.; et al. RECQ-like Helicases Sgs1 and BLM Regulate R-Loop–Associated Genome Instability. J. Cell Biol. 2017, 216, 3991–4005. [Google Scholar] [CrossRef] [Green Version]
- van Wietmarschen, N.; Merzouk, S.; Halsema, N.; Spierings, D.C.J.; Guryev, V.; Lansdorp, P.M. BLM Helicase Suppresses Recombination at G-Quadruplex Motifs in Transcribed Genes. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, G.H.; Tang, W.; Robles, A.I.; Beyer, R.P.; Gray, L.T.; Welsh, J.A.; Schetter, A.J.; Kumamoto, K.; Wang, X.W.; Hickson, I.D.; et al. Regulation of Gene Expression by the BLM Helicase Correlates with the Presence of G-Quadruplex DNA Motifs. Proc. Natl. Acad. Sci. USA 2014, 111, 9905–9910. [Google Scholar] [CrossRef] [Green Version]
- Huppert, J.L.; Balasubramanian, S. Prevalence of Quadruplexes in the Human Genome. Nucleic Acids Res. 2005, 33, 2908–2916. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Rokutanda, N.; Takeuchi, J.; Lai, Y.; Maruyama, R.; Togashi, Y.; Nishikawa, H.; Arai, N.; Miyoshi, Y.; Suzuki, N.; et al. HERC2 Facilitates BLM and WRN Helicase Complex Interaction with RPA to Suppress G-Quadruplex DNA. Cancer Res. 2018, 78, 6371–6385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, R.; Müller, S.; Yeoman, J.A.; Trentesaux, C.; Riou, J.-F.; Balasubramanian, S. A Novel Small Molecule That Alters Shelterin Integrity and Triggers a DNA-Damage Response at Telomeres. J. Am. Chem. Soc. 2008, 130, 15758–15759. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.; Zhu, M.; Wu, W.; Rokutanda, N.; Togashi, Y.; Liang, W.; Ohta, T. HERC2 Regulates RPA2 by Mediating ATR-Induced Ser33 Phosphorylation and Ubiquitin-Dependent Degradation. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Grierson, P.M.; Acharya, S.; Groden, J. Collaborating Functions of BLM and DNA Topoisomerase I in Regulating Human RDNA Transcription. Mutat. Res. 2013, 743, 89–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassambara, A.; Herviou, L.; Ovejero, S.; Jourdan, M.; Thibaut, C.; Vikova, V.; Pasero, P.; Elemento, O.; Moreaux, J. RNA-Sequencing Data-Driven Dissection of Human Plasma Cell Differentiation Reveals New Potential Transcription Regulators. Leukemia 2021. [Google Scholar] [CrossRef]
- Popuri, V.; Croteau, D.L.; Brosh, R.M.; Bohr, V.A. RECQ1 Is Required for Cellular Resistance to Replication Stress and Catalyzes Strand Exchange on Stalled Replication Fork Structures. Cell Cycle 2012, 11, 4252–4265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Parvathaneni, S.; Hara, T.; Lal, A.; Sharma, S. Replication Stress Induces Specific Enrichment of RECQ1 at Common Fragile Sites FRA3B and FRA16D. Mol. Cancer 2013, 12, 29. [Google Scholar] [CrossRef] [Green Version]
- Cui, S.; Arosio, D.; Doherty, K.M.; Brosh, R.M.; Falaschi, A.; Vindigni, A. Analysis of the Unwinding Activity of the Dimeric RECQ1 Helicase in the Presence of Human Replication Protein A. Nucleic Acids Res. 2004, 32, 2158–2170. [Google Scholar] [CrossRef]
- Viziteu, E.; Klein, B.; Basbous, J.; Lin, Y.-L.; Hirtz, C.; Gourzones, C.; Tiers, L.; Bruyer, A.; Vincent, L.; Grandmougin, C.; et al. RECQ1 Helicase Is Involved in Replication Stress Survival and Drug Resistance in Multiple Myeloma. Leukemia 2017, 31, 2104–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parvathaneni, S.; Lu, X.; Chaudhary, R.; Lal, A.; Madhusudan, S.; Sharma, S. RECQ1 Expression Is Upregulated in Response to DNA Damage and in a P53-Dependent Manner. Oncotarget 2017, 8, 75924–75942. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Lu, X.; Parvathaneni, S.; Bilke, S.; Zhang, H.; Thangavel, S.; Vindigni, A.; Hara, T.; Zhu, Y.; Meltzer, P.S.; et al. Identification of RECQ1-Regulated Transcriptome Uncovers a Role of RECQ1 in Regulation of Cancer Cell Migration and Invasion. Cell Cycle 2014, 13, 2431–2445. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Parvathaneni, S.; Li, X.L.; Lal, A.; Sharma, S. Transcriptome Guided Identification of Novel Functions of RECQ1 Helicase. Methods 2016, 108, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Debnath, S.; Sharma, S. RECQ1 Helicase in Genomic Stability and Cancer. Genes 2020, 11, 622. [Google Scholar] [CrossRef]
- Edwards, A.D.; Marecki, J.C.; Byrd, A.K.; Gao, J.; Raney, K.D. G-Quadruplex Loops Regulate PARP-1 Enzymatic Activation. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Bochman, M.L.; Sabouri, N.; Zakian, V.A. Unwinding the Functions of the Pif1 Family Helicases. DNA Repair 2010, 9, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Byrd, A.K.; Raney, K.D. A Parallel Quadruplex DNA Is Bound Tightly but Unfolded Slowly by Pif1 Helicase. J. Biol. Chem. 2015, 290, 6482–6494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, X.-M.; Wu, W.-Q.; Duan, X.-L.; Liu, N.-N.; Li, H.-H.; Fu, J.; Dou, S.-X.; Li, M.; Xi, X.-G. Molecular Mechanism of G-Quadruplex Unwinding Helicase: Sequential and Repetitive Unfolding of G-Quadruplex by Pif1 Helicase. Biochem. J. 2015, 466, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Zhang, J.; Bochman, M.L.; Zakian, V.A.; Ha, T. Periodic DNA Patrolling Underlies Diverse Functions of Pif1 on R-Loops and G-Rich DNA. eLife 2014, 3, e02190. [Google Scholar] [CrossRef]
- Dahan, D.; Tsirkas, I.; Dovrat, D.; Sparks, M.A.; Singh, S.P.; Galletto, R.; Aharoni, A. Pif1 Is Essential for Efficient Replisome Progression through Lagging Strand G-Quadruplex DNA Secondary Structures. Nucleic Acids Res. 2018, 46, 11847–11857. [Google Scholar] [CrossRef] [Green Version]
- Paeschke, K.; Capra, J.A.; Zakian, V.A. DNA Replication through G-Quadruplex Motifs Is Promoted by the Saccharomyces Cerevisiae Pif1 DNA Helicase. Cell 2011, 145, 678–691. [Google Scholar] [CrossRef] [Green Version]
- Ribeyre, C.; Lopes, J.; Boulé, J.-B.; Piazza, A.; Guédin, A.; Zakian, V.A.; Mergny, J.-L.; Nicolas, A. The Yeast Pif1 Helicase Prevents Genomic Instability Caused by G-Quadruplex-Forming CEB1 Sequences In Vivo. PLoS Genet. 2009, 5. [Google Scholar] [CrossRef] [Green Version]
- Sparks, M.A.; Singh, S.P.; Burgers, P.M.; Galletto, R. Complementary Roles of Pif1 Helicase and Single Stranded DNA Binding Proteins in Stimulating DNA Replication through G-Quadruplexes. Nucleic Acids Res. 2019, 47, 8595–8605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maestroni, L.; Audry, J.; Luciano, P.; Coulon, S.; Géli, V.; Corda, Y. RPA and Pif1 Cooperate to Remove G-Rich Structures at Both Leading and Lagging Strand. Cell Stress 2020, 4, 48–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safa, L.; Gueddouda, N.M.; Thiébaut, F.; Delagoutte, E.; Petruseva, I.; Lavrik, O.; Mendoza, O.; Bourdoncle, A.; Alberti, P.; Riou, J.-F.; et al. 5′ to 3′ Unfolding Directionality of DNA Secondary Structures by Replication Protein A. J. Biol. Chem. 2016, 291, 21246–21256. [Google Scholar] [CrossRef] [Green Version]
- Salas, T.R.; Petruseva, I.; Lavrik, O.; Bourdoncle, A.; Mergny, J.-L.; Favre, A.; Saintomé, C. Human Replication Protein A Unfolds Telomeric G-Quadruplexes. Nucleic Acids Res. 2006, 34, 4857–4865. [Google Scholar] [CrossRef]
- Paeschke, K.; Bochman, M.L.; Garcia, P.D.; Cejka, P.; Friedman, K.L.; Kowalczykowski, S.C.; Zakian, V.A. Pif1 Family Helicases Suppress Genome Instability at G-Quadruplex Motifs. Nature 2013, 497, 458–462. [Google Scholar] [CrossRef] [Green Version]
- Audry, J.; Maestroni, L.; Delagoutte, E.; Gauthier, T.; Nakamura, T.M.; Gachet, Y.; Saintomé, C.; Géli, V.; Coulon, S. RPA Prevents G-Rich Structure Formation at Lagging-Strand Telomeres to Allow Maintenance of Chromosome Ends. EMBO J. 2015, 34, 1942–1958. [Google Scholar] [CrossRef] [Green Version]
- Chib, S.; Byrd, A.K.; Raney, K.D. Yeast Helicase Pif1 Unwinds RNA:DNA Hybrids with Higher Processivity than DNA:DNA Duplexes. J. Biol. Chem. 2016, 291, 5889–5901. [Google Scholar] [CrossRef] [Green Version]
- Tran, P.L.T.; Pohl, T.J.; Chen, C.-F.; Chan, A.; Pott, S.; Zakian, V.A. PIF1 Family DNA Helicases Suppress R-Loop Mediated Genome Instability at TRNA Genes. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Budd, M.E.; Choe, W.C.; Campbell, J.L. DNA2 Encodes a DNA Helicase Essential for Replication of Eukaryotic Chromosomes. J. Biol. Chem. 1995, 270, 26766–26769. [Google Scholar] [CrossRef] [Green Version]
- Budd, M.E.; Reis, C.C.; Smith, S.; Myung, K.; Campbell, J.L. Evidence Suggesting That Pif1 Helicase Functions in DNA Replication with the Dna2 Helicase/Nuclease and DNA Polymerase Delta. Mol. Cell. Biol. 2006, 26, 2490–2500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda-Sasa, T.; Polaczek, P.; Peng, X.P.; Chen, L.; Campbell, J.L. Processing of G4 DNA by Dna2 Helicase/Nuclease and Replication Protein A (RPA) Provides Insights into the Mechanism of Dna2/RPA Substrate Recognition. J. Biol. Chem. 2008, 283, 24359–24373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.; Sampathi, S.; Dai, H.; Liu, C.; Zhou, M.; Hu, J.; Huang, Q.; Campbell, J.; Shin-Ya, K.; Zheng, L.; et al. Mammalian DNA2 Helicase/Nuclease Cleaves G-Quadruplex DNA and Is Required for Telomere Integrity. EMBO J. 2013, 32, 1425–1439. [Google Scholar] [CrossRef] [Green Version]
- Choe, W.; Budd, M.; Imamura, O.; Hoopes, L.; Campbell, J.L. Dynamic Localization of an Okazaki Fragment Processing Protein Suggests a Novel Role in Telomere Replication. Mol. Cell. Biol. 2002, 22, 4202–4217. [Google Scholar] [CrossRef] [Green Version]
- Karanja, K.K.; Cox, S.W.; Duxin, J.P.; Stewart, S.A.; Campbell, J.L. DNA2 and EXO1 in Replication-Coupled, Homology-Directed Repair and in the Interplay between HDR and the FA/BRCA Network. Cell Cycle 2012, 11, 3983–3996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, G.; Dai, H.; Zhang, W.; Hsieh, H.-J.; Pan, M.-R.; Park, Y.-Y.; Tsai, Y.-L.; Bedrosian, I.; Lee, J.-S.; Ira, G.; et al. Human Nuclease/Helicase DNA2 Alleviates Replication Stress by Promoting DNA End Resection. Cancer Res. 2012, 72, 2802–2813. [Google Scholar] [CrossRef] [Green Version]
- Daley, J.M.; Chiba, T.; Xue, X.; Niu, H.; Sung, P. Multifaceted Role of the Topo IIIα-RMI1-RMI2 Complex and DNA2 in the BLM-Dependent Pathway of DNA Break End Resection. Nucleic Acids Res. 2014, 42, 11083–11091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palm, W.; de Lange, T. How Shelterin Protects Mammalian Telomeres. Annu. Rev. Genet. 2008, 42, 301–334. [Google Scholar] [CrossRef] [Green Version]
- Zaug, A.J.; Podell, E.R.; Cech, T.R. Human POT1 Disrupts Telomeric G-Quadruplexes Allowing Telomerase Extension in vitro. Proc. Natl. Acad. Sci. USA 2005, 102, 10864–10869. [Google Scholar] [CrossRef] [Green Version]
- Gomez, D.; O’Donohue, M.-F.; Wenner, T.; Douarre, C.; Macadré, J.; Koebel, P.; Giraud-Panis, M.-J.; Kaplan, H.; Kolkes, A.; Shin-ya, K.; et al. The G-Quadruplex Ligand Telomestatin Inhibits POT1 Binding to Telomeric Sequences in vitro and Induces GFP-POT1 Dissociation from Telomeres in Human Cells. Cancer Res. 2006, 66, 6908–6912. [Google Scholar] [CrossRef] [Green Version]
- Di Antonio, M.; Biffi, G.; Mariani, A.; Raiber, E.-A.; Rodriguez, R.; Balasubramanian, S. Selective RNA versus DNA G-Quadruplex Targeting by in Situ Click Chemistry. Angew. Chem. 2012, 51, 11073–11078. [Google Scholar] [CrossRef] [Green Version]
- Ray, S.; Bandaria, J.N.; Qureshi, M.H.; Yildiz, A.; Balci, H. G-Quadruplex Formation in Telomeres Enhances POT1/TPP1 Protection against RPA Binding. Proc. Natl. Acad. Sci. USA 2014, 111, 2990–2995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaires, J.B.; Gray, R.D.; Dean, W.L.; Monsen, R.; DeLeeuw, L.W.; Stribinskis, V.; Trent, J.O. Human POT1 Unfolds G-Quadruplexes by Conformational Selection. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paeschke, K.; Simonsson, T.; Postberg, J.; Rhodes, D.; Lipps, H.J. Telomere End-Binding Proteins Control the Formation of G-Quadruplex DNA Structures in Vivo. Nat. Struct. Mol. Biol. 2005, 12, 847–854. [Google Scholar] [CrossRef]
- Panero, J.; Stanganelli, C.; Arbelbide, J.; Fantl, D.B.; Kohan, D.; García Rivello, H.; Rabinovich, G.A.; Slavutsky, I. Expression Profile of Shelterin Components in Plasma Cell Disorders. Clinical Significance of POT1 Overexpression. Blood Cells Mol. Dis. 2014, 52, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Venturutti, L.; Melnick, A.M. The Dangers of Déjà vu: Memory B Cells as the Cells of Origin of ABC-DLBCLs. Blood 2020, 136, 2263–2274. [Google Scholar] [CrossRef]
- Rustad, E.H.; Yellapantula, V.; Leongamornlert, D.; Bolli, N.; Ledergor, G.; Nadeu, F.; Angelopoulos, N.; Dawson, K.J.; Mitchell, T.J.; Osborne, R.J.; et al. Timing the Initiation of Multiple Myeloma. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Braggio, E.; Kortüm, K.M.; Stewart, A.K. SnapShot: Multiple Myeloma. Cancer Cell 2015, 28, 678–678.e1. [Google Scholar] [CrossRef] [Green Version]
- González, D.; van der Burg, M.; García-Sanz, R.; Fenton, J.A.; Langerak, A.W.; González, M.; van Dongen, J.J.M.; San Miguel, J.F.; Morgan, G.J. Immunoglobulin Gene Rearrangements and the Pathogenesis of Multiple Myeloma. Blood 2007, 110, 3112–3121. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and Consequences of Replication Stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, P.H.; Cornish, A.J.; Dobbins, S.E.; Kaiser, M.; Houlston, R.S. Mutational Processes Contributing to the Development of Multiple Myeloma. Blood Cancer J. 2019, 9, 60. [Google Scholar] [CrossRef]
- Vikova, V.; Jourdan, M.; Robert, N.; Requirand, G.; Boireau, S.; Bruyer, A.; Vincent, L.; Cartron, G.; Klein, B.; Elemento, O.; et al. Comprehensive Characterization of the Mutational Landscape in Multiple Myeloma Cell Lines Reveals Potential Drivers and Pathways Associated with Tumor Progression and Drug Resistance. Theranostics 2019, 9, 540–553. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread Genetic Heterogeneity in Multiple Myeloma: Implications for Targeted Therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Moreaux, J.; Rème, T.; Leonard, W.; Veyrune, J.-L.; Requirand, G.; Goldschmidt, H.; Hose, D.; Klein, B. Development of Gene Expression–Based Score to Predict Sensitivity of Multiple Myeloma Cells to DNA Methylation Inhibitors. Mol. Cancer Ther. 2012, 11, 2685–2692. [Google Scholar] [CrossRef] [Green Version]
- Zhan, F.; Huang, Y.; Colla, S.; Stewart, J.P.; Hanamura, I.; Gupta, S.; Epstein, J.; Yaccoby, S.; Sawyer, J.; Burington, B.; et al. The Molecular Classification of Multiple Myeloma. Blood 2006, 108, 2020–2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, J.P.; White, J.; Stirling, P.C. R Loops and Their Composite Cancer Connections. Trends Cancer 2019, 5, 619–631. [Google Scholar] [CrossRef]
- Miglietta, G.; Russo, M.; Capranico, G. G-Quadruplex–R-Loop Interactions and the Mechanism of Anticancer G-Quadruplex Binders. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Kim, N. The Interplay between G-Quadruplex and Transcription. Curr. Med. Chem. 2019, 26, 2898–2917. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Hurley, L.H. Structures, Folding Patterns, and Functions of Intramolecular DNA G-Quadruplexes Found in Eukaryotic Promoter Regions. Biochimie 2008, 90, 1149–1171. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct Evidence for a G-Quadruplex in a Promoter Region and Its Targeting with a Small Molecule to Repress c-MYC Transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, R.; Bhattacharya, S.; Dash, J.; Bhattacharya, S. Recent Update on Targeting c-MYC G-Quadruplexes by Small Molecules for Anticancer Therapeutics. J. Med. Chem. 2021, 64, 42–70. [Google Scholar] [CrossRef]
- Yang, D.; Hurley, L.H. Structure of the Biologically Relevant G-Quadruplex in the c-MYC Promoter. Nucleosides Nucleotides Nucleic Acids 2006, 25, 951–968. [Google Scholar] [CrossRef]
- Dexheimer, T.S.; Sun, D.; Hurley, L.H. Deconvoluting the Structural and Drug-Recognition Complexity of the G-Quadruplex-Forming Region Upstream of the Bcl-2 P1 Promoter. J. Am. Chem. Soc. 2006, 128, 5404–5415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Dexheimer, T.S.; Chen, D.; Carver, M.; Ambrus, A.; Jones, R.A.; Yang, D. An Intramolecular G-Quadruplex Structure with Mixed Parallel/Antiparallel G-Strands Formed in the Human BCL-2 Promoter Region in Solution. J. Am. Chem. Soc. 2006, 128, 1096–1098. [Google Scholar] [CrossRef] [Green Version]
- Eddy, J.; Maizels, N. Gene Function Correlates with Potential for G4 DNA Formation in the Human Genome. Nucleic Acids Res. 2006, 34, 3887–3896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaikwad, S.M.; Phyo, Z.; Arteaga, A.Q.; Gorjifard, S.; Calabrese, D.R.; Connors, D.; Huang, J.; Michalowski, A.M.; Zhang, S.; Liu, Z.-G.; et al. A Small Molecule Stabilizer of the MYC G4-Quadruplex Induces Endoplasmic Reticulum Stress, Senescence and Pyroptosis in Multiple Myeloma. Cancers 2020, 12, 2952. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.; Miller, K.M.; Forment, J.V.; Bradshaw, C.R.; Nikan, M.; Britton, S.; Oelschlaegel, T.; Xhemalce, B.; Balasubramanian, S.; Jackson, S.P. Small Molecule-Induced DNA Damage Identifies Alternative DNA Structures in Human Genes. Nat. Chem. Biol. 2012, 8, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Müller, S.; Kumari, S.; Rodriguez, R.; Balasubramanian, S. Small-Molecule-Mediated G-Quadruplex Isolation from Human Cells. Nat. Chem. 2010, 2, 1095–1098. [Google Scholar] [CrossRef]
- Koirala, D.; Dhakal, S.; Ashbridge, B.; Sannohe, Y.; Rodriguez, R.; Sugiyama, H.; Balasubramanian, S.; Mao, H. A Single-Molecule Platform for Investigation of Interactions between G-Quadruplexes and Small-Molecule Ligands. Nat. Chem. 2011, 3, 782–787. [Google Scholar] [CrossRef] [Green Version]
- Olivieri, M.; Cho, T.; Álvarez-Quilón, A.; Li, K.; Schellenberg, M.J.; Zimmermann, M.; Hustedt, N.; Rossi, S.E.; Adam, S.; Melo, H.; et al. A Genetic Map of the Response to DNA Damage in Human Cells. Cell 2020, 182, 481–496.e21. [Google Scholar] [CrossRef]
- Zimmer, J.; Tacconi, E.M.C.; Folio, C.; Badie, S.; Porru, M.; Klare, K.; Tumiati, M.; Markkanen, E.; Halder, S.; Ryan, A.; et al. Targeting BRCA1 and BRCA2 Deficiencies with G-Quadruplex-Interacting Compounds. Mol. Cell 2016, 61, 449–460. [Google Scholar] [CrossRef] [Green Version]
- McLuckie, K.I.E.; Di Antonio, M.; Zecchini, H.; Xian, J.; Caldas, C.; Krippendorff, B.-F.; Tannahill, D.; Lowe, C.; Balasubramanian, S. G-Quadruplex DNA as a Molecular Target for Induced Synthetic Lethality in Cancer Cells. J. Am. Chem. Soc. 2013, 135, 9640–9643. [Google Scholar] [CrossRef] [PubMed]
- Neckles, C.; Boer, R.E.; Aboreden, N.; Cross, A.M.; Walker, R.L.; Kim, B.-H.; Kim, S.; Schneekloth, J.S.; Caplen, N.J. HNRNPH1-Dependent Splicing of a Fusion Oncogene Reveals a Targetable RNA G-Quadruplex Interaction. RNA 2019, 25, 1731–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maleki, P.; Mustafa, G.; Gyawali, P.; Budhathoki, J.B.; Ma, Y.; Nagasawa, K.; Balci, H. Quantifying the Impact of Small Molecule Ligands on G-Quadruplex Stability against Bloom Helicase. Nucleic Acids Res. 2019, 47, 10744–10753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendoza, O.; Gueddouda, N.M.; Boulé, J.-B.; Bourdoncle, A.; Mergny, J.-L. A Fluorescence-Based Helicase Assay: Application to the Screening of G-Quadruplex Ligands. Nucleic Acids Res. 2015, 43, e71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, S.P.; Das, P. Novel Splicing in IGFN1 Intron 15 and Role of Stable G-Quadruplex in the Regulation of Splicing in Renal Cell Carcinoma. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Moruno-Manchon, J.F.; Koellhoffer, E.C.; Gopakumar, J.; Hambarde, S.; Kim, N.; McCullough, L.D.; Tsvetkov, A.S. The G-Quadruplex DNA Stabilizing Drug Pyridostatin Promotes DNA Damage and Downregulates Transcription of Brca1 in Neurons. Aging 2017, 9, 1957–1970. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, D.C.; Fones, L.; Vivinetto, A.L.; Caufield, J.T.; Ratan, R.R.; Cave, J.W. Manipulating Adult Neural Stem and Progenitor Cells with G-Quadruplex Ligands. ACS Chem. Neurosci. 2020, 11, 1504–1518. [Google Scholar] [CrossRef]
- Bywater, M.J.; Poortinga, G.; Sanij, E.; Hein, N.; Peck, A.; Cullinane, C.; Wall, M.; Cluse, L.; Drygin, D.; Anderes, K.; et al. Inhibition of RNA Polymerase I as a Therapeutic Strategy to Promote Cancer-Specific Activation of P53. Cancer Cell 2012, 22, 51–65. [Google Scholar] [CrossRef] [Green Version]
- Drygin, D.; Lin, A.; Bliesath, J.; Ho, C.B.; O’Brien, S.E.; Proffitt, C.; Omori, M.; Haddach, M.; Schwaebe, M.K.; Siddiqui-Jain, A.; et al. Targeting RNA Polymerase I with an Oral Small Molecule CX-5461 Inhibits Ribosomal RNA Synthesis and Solid Tumor Growth. Cancer Res. 2011, 71, 1418–1430. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Santos, N.D.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 Is a DNA G-Quadruplex Stabilizer with Selective Lethality in BRCA1/2 Deficient Tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef]
- Boros-Oláh, B.; Dobos, N.; Hornyák, L.; Szabó, Z.; Karányi, Z.; Halmos, G.; Roszik, J.; Székvölgyi, L. Drugging the R-Loop Interactome: RNA-DNA Hybrid Binding Proteins as Targets for Cancer Therapy. DNA Repair 2019, 84, 102642. [Google Scholar] [CrossRef]
- Shaw, N.N.; Arya, D.P. Recognition of the Unique Structure of DNA:RNA Hybrids. Biochimie 2008, 90, 1026–1039. [Google Scholar] [CrossRef] [PubMed]
- Takusagawa, F.; Takusagawa, K.T.; Carlson, R.G.; Weaver, R.F. Selectivity of F8-Actinomycin D for RNA:DNA Hybrids and Its Anti-Leukemia Activity. Bioorganic Med. Chem. 1997, 5, 1197–1207. [Google Scholar] [CrossRef]
- Wang, I.X.; Grunseich, C.; Fox, J.; Burdick, J.; Zhu, Z.; Ravazian, N.; Hafner, M.; Cheung, V.G. Human Proteins That Interact with RNA/DNA Hybrids. Genome Res. 2018, 28, 1405–1414. [Google Scholar] [CrossRef] [Green Version]
- Shuck, S.C.; Turchi, J.J. Targeted Inhibition of Replication Protein A Reveals Cytotoxic Activity, Synergy with Chemotherapeutic DNA-Damaging Agents, and Insight into Cellular Function. Cancer Res. 2010, 70, 3189–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minas, T.Z.; Han, J.; Javaheri, T.; Hong, S.-H.; Schlederer, M.; Saygideğer-Kont, Y.; Çelik, H.; Mueller, K.M.; Temel, I.; Özdemirli, M.; et al. YK-4-279 Effectively Antagonizes EWS-FLI1 Induced Leukemia in a Transgenic Mouse Model. Oncotarget 2015, 6, 37678–37694. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, P.; Huang, J.T.J.; Hiom, K. DHX9 Helicase Promotes R-Loop Formation in Cells with Impaired RNA Splicing. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, J.-Y.; Huang, Y.-J.; Gu, Y.; Qiu, J.; Qian, H.; Shao, C.; Zhang, X.; Hu, J.; Li, H.; et al. The Augmented R-Loop Is a Unifying Mechanism for Myelodysplastic Syndromes Induced by High-Risk Splicing Factor Mutations. Mol. Cell 2018, 69, 412–425.e6. [Google Scholar] [CrossRef] [Green Version]
- Sorrells, S.; Nik, S.; Casey, M.J.; Cameron, R.C.; Truong, H.; Toruno, C.; Gulfo, M.; Lowe, A.; Jette, C.; Stewart, R.A.; et al. Spliceosomal Components Protect Embryonic Neurons from R-Loop-Mediated DNA Damage and Apoptosis. Dis. Models Mech. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Tumini, E.; Herrera-Moyano, E.; San Martín-Alonso, M.; Barroso, S.; Galmarini, C.M.; Aguilera, A. The Antitumor Drugs Trabectedin and Lurbinectedin Induce Transcription-Dependent Replication Stress and Genome Instability. Mol. Cancer Res. 2019, 17, 773–782. [Google Scholar] [CrossRef] [Green Version]
- White, R.; Saxty, B.; Large, J.; Kettleborough, C.A.; Jackson, A.P. Identification of Small-Molecule Inhibitors of the Ribonuclease H2 Enzyme. J. Biomol. Screen. 2013, 18, 610–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flanagan, J.M.; Funes, J.M.; Henderson, S.; Wild, L.; Carey, N.; Boshoff, C. Genomics Screen in Transformed Stem Cells Reveals RNASEH2A, PPAP2C, and ADARB1 as Putative Anticancer Drug Targets. Mol. Cancer Ther. 2009, 8, 249–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, G.H.; Dexheimer, T.S.; Rosenthal, A.S.; Chu, W.K.; Singh, D.K.; Mosedale, G.; Bachrati, C.Z.; Schultz, L.; Sakurai, M.; Savitsky, P.; et al. A Small Molecule Inhibitor of the BLM Helicase Modulates Chromosome Stability in Human Cells. Chem. Biol. 2013, 20, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Peng, X.; Daley, J.; Yang, L.; Shen, J.; Nguyen, N.; Bae, G.; Niu, H.; Peng, Y.; Hsieh, H.-J.; et al. Inhibition of DNA2 Nuclease as a Therapeutic Strategy Targeting Replication Stress in Cancer Cells. Oncogenesis 2017, 6, e319. [Google Scholar] [CrossRef] [Green Version]
- Brosh, R.M., Jr.; Karow, J.K.; White, E.J.; Shaw, N.D.; Hickson, I.D.; Bohr, V.A. Potent Inhibition of Werner and Bloom Helicases by DNA Minor Groove Binding Drugs. Nucleic Acids Res. 2000, 28, 2420–2430. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Park, G.; Lee, J.E.; Choi, E.Y.; Park, J.Y.; Kim, T.-H.; Park, N.; Jin, X.; Jung, J.-E.; Shin, D.; et al. DEAD-Box RNA Helicase DDX23 Modulates Glioma Malignancy via Elevating MiR-21 Biogenesis. Brain 2015, 138, 2553–2570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupré, A.; Boyer-Chatenet, L.; Sattler, R.M.; Modi, A.P.; Lee, J.-H.; Nicolette, M.L.; Kopelovich, L.; Jasin, M.; Baer, R.; Paull, T.T.; et al. A Forward Chemical Genetic Screen Reveals an Inhibitor of the Mre11-Rad50-Nbs1 Complex. Nat. Chem. Biol. 2008, 4, 119–125. [Google Scholar] [CrossRef]
- Shibata, A.; Moiani, D.; Arvai, A.S.; Perry, J.; Harding, S.M.; Genois, M.-M.; Maity, R.; van Rossum-Fikkert, S.; Kertokalio, A.; Romoli, F.; et al. DNA Double-Strand Break Repair Pathway Choice Is Directed by Distinct MRE11 Nuclease Activities. Mol. Cell 2014, 53, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.-Z.; Liu, Y.-Q.; Cheng, X.; Zhou, G.-B. Celastrol Induces Proteasomal Degradation of FANCD2 to Sensitize Lung Cancer Cells to DNA Crosslinking Agents. Cancer Sci. 2015, 106, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Gallmeier, E.; Hucl, T.; Brody, J.R.; Dezentje, D.A.; Tahir, K.; Kasparkova, J.; Brabec, V.; Bachman, K.E.; Kern, S.E. High-Throughput Screening Identifies Novel Agents Eliciting Hypersensitivity in Fanconi Pathway-Deficient Cancer Cells. Cancer Res. 2007, 67, 2169–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landais, I.; Sobeck, A.; Stone, S.; LaChapelle, A.; Hoatlin, M.E. A Novel Cell-Free Screen Identifies a Potent Inhibitor of the Fanconi Anemia Pathway. Int. J. Cancer 2009, 124, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Shi, R.; Wang, X.; Shen, H.-M. Luteolin, a Flavonoid with Potentials for Cancer Prevention and Therapy. Curr. Cancer Drug Targets 2008, 8, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Glanzer, J.G.; Liu, S.; Wang, L.; Mosel, A.; Peng, A.; Oakley, G.G. RPA Inhibition Increases Replication Stress and Suppresses Tumor Growth. Cancer Res. 2014, 74, 5165–5172. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dutrieux, L.; Lin, Y.-L.; Lutzmann, M.; Rodriguez, R.; Cogné, M.; Pasero, P.; Moreaux, J. Transcription/Replication Conflicts in Tumorigenesis and Their Potential Role as Novel Therapeutic Targets in Multiple Myeloma. Cancers 2021, 13, 3755. https://doi.org/10.3390/cancers13153755
Dutrieux L, Lin Y-L, Lutzmann M, Rodriguez R, Cogné M, Pasero P, Moreaux J. Transcription/Replication Conflicts in Tumorigenesis and Their Potential Role as Novel Therapeutic Targets in Multiple Myeloma. Cancers. 2021; 13(15):3755. https://doi.org/10.3390/cancers13153755
Chicago/Turabian StyleDutrieux, Laure, Yea-Lih Lin, Malik Lutzmann, Raphaël Rodriguez, Michel Cogné, Philippe Pasero, and Jérôme Moreaux. 2021. "Transcription/Replication Conflicts in Tumorigenesis and Their Potential Role as Novel Therapeutic Targets in Multiple Myeloma" Cancers 13, no. 15: 3755. https://doi.org/10.3390/cancers13153755
APA StyleDutrieux, L., Lin, Y. -L., Lutzmann, M., Rodriguez, R., Cogné, M., Pasero, P., & Moreaux, J. (2021). Transcription/Replication Conflicts in Tumorigenesis and Their Potential Role as Novel Therapeutic Targets in Multiple Myeloma. Cancers, 13(15), 3755. https://doi.org/10.3390/cancers13153755