
Reactive Oxygen Species: A Promising Therapeutic Target for SDHx-Mutated Pheochromocytoma and Paraganglioma

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
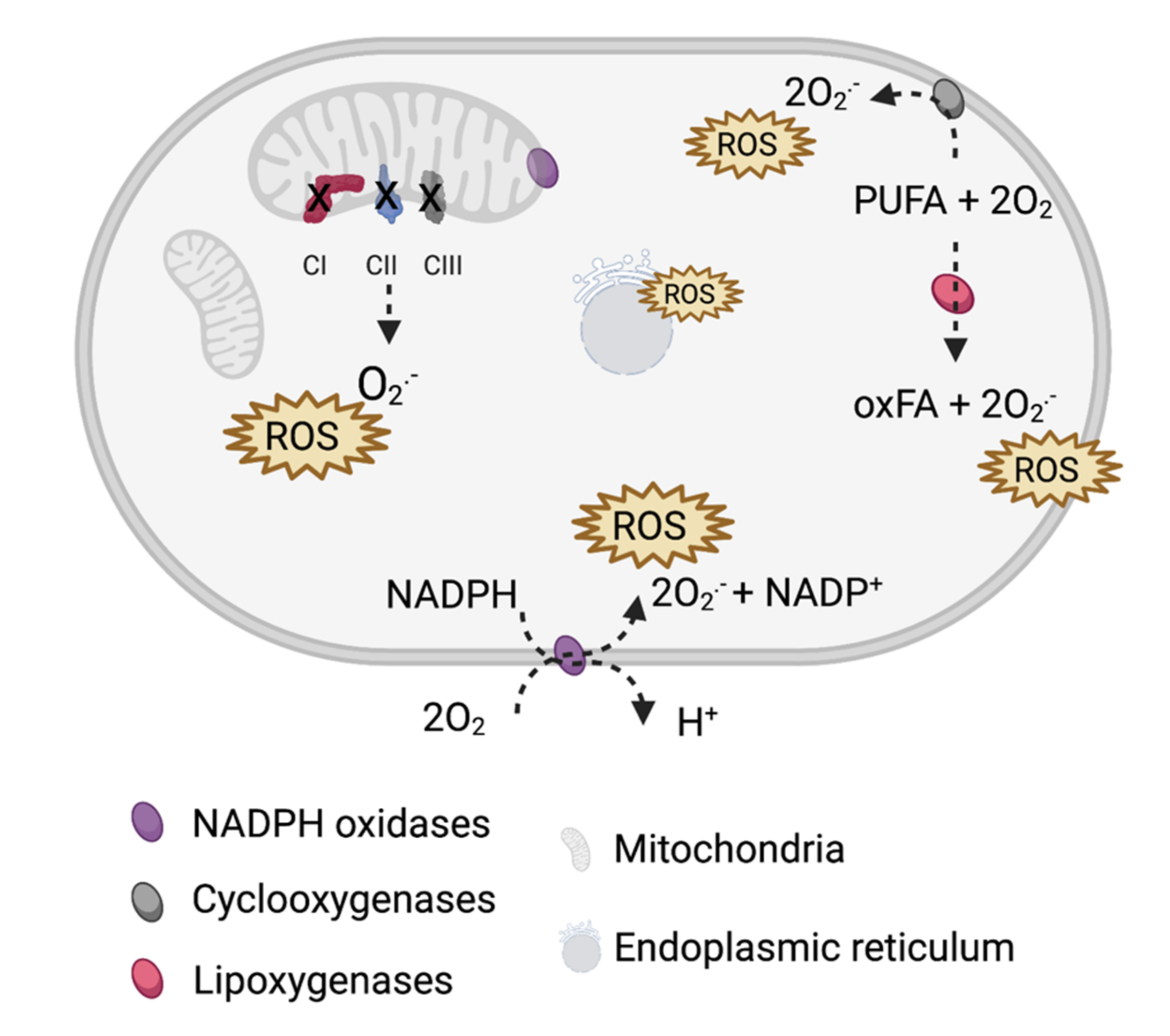
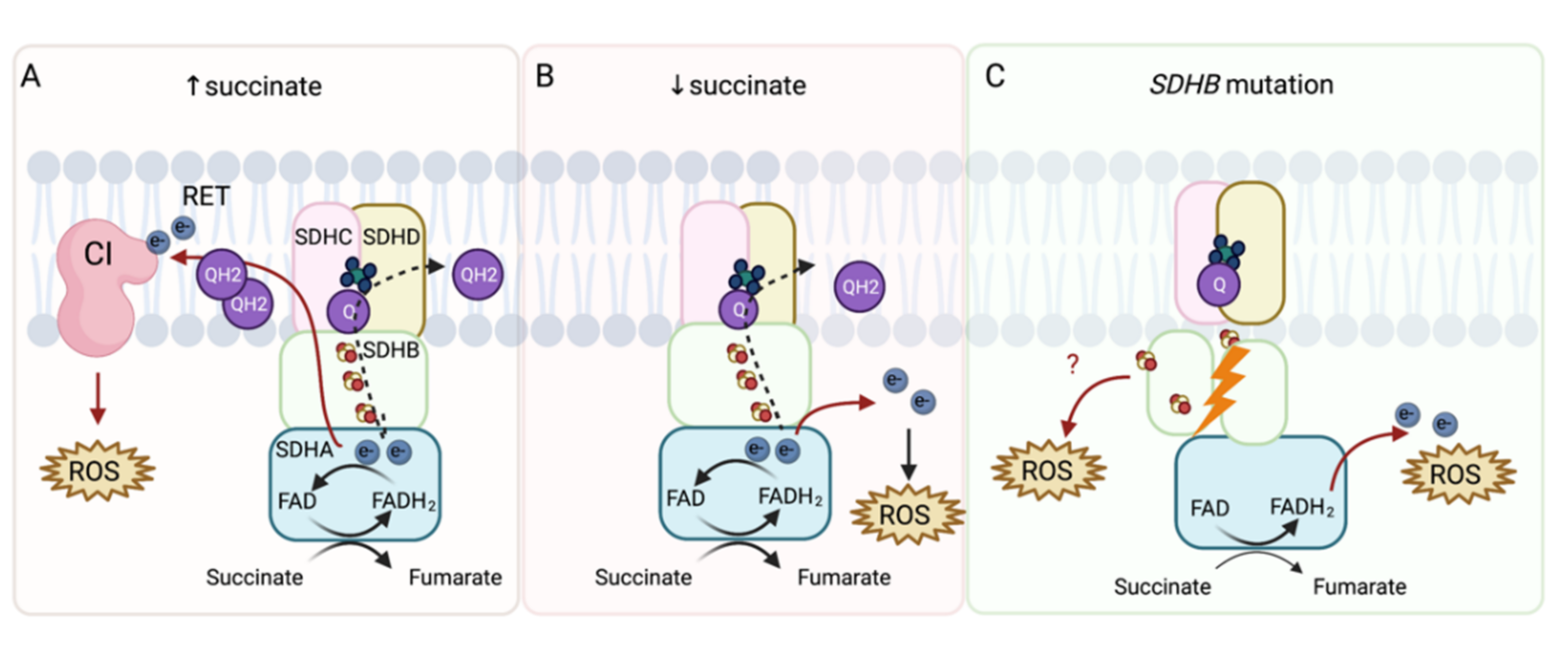
2. ROS in PHEO and PGL
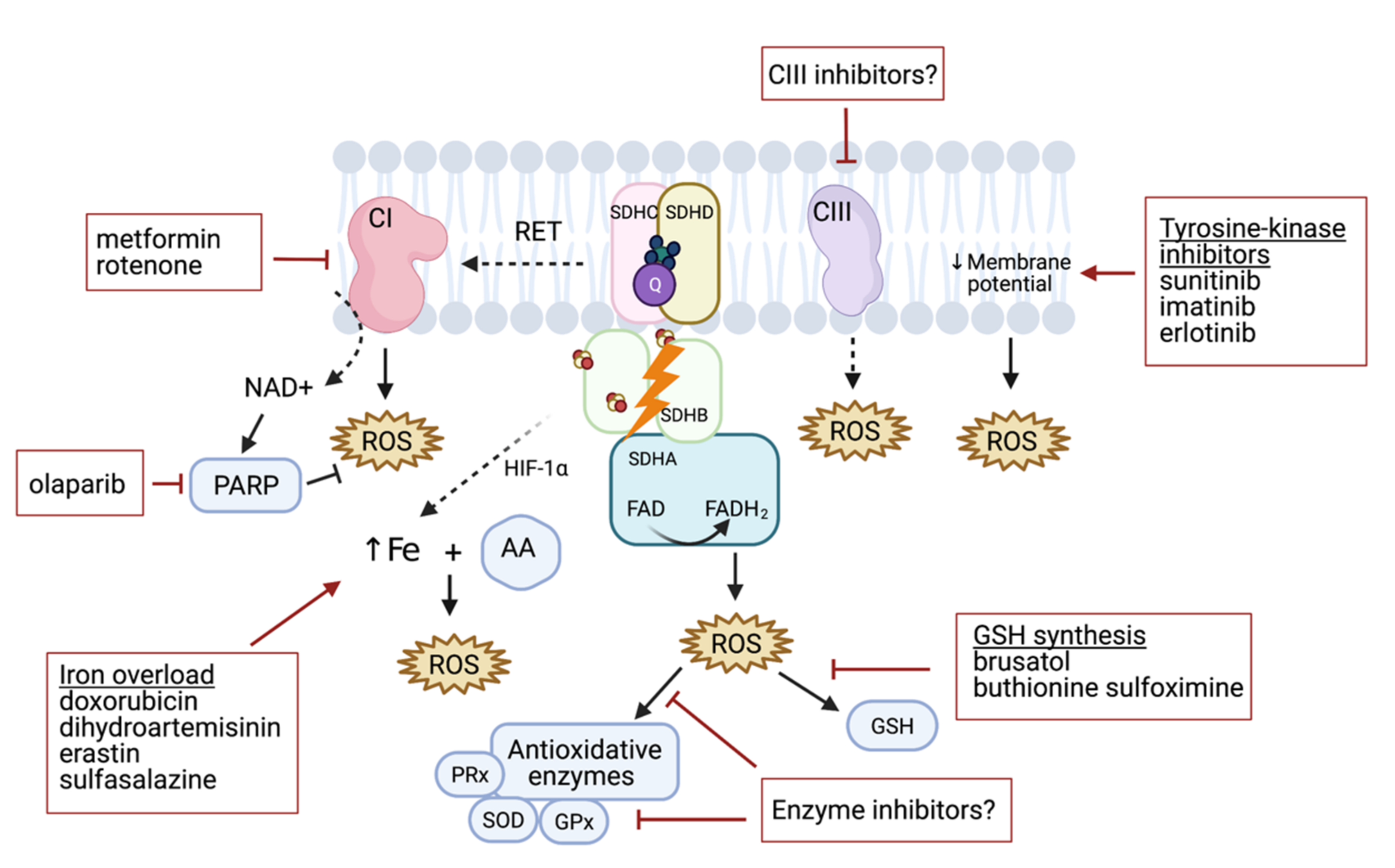
3. Targeting of ROS Production in PHEO and PGL
4. Future Directions
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Ursini, F.; Maiorino, M. An overview of mechanisms of redox signaling. J. Mol. Cell Cardiol. 2014, 73, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, A.; Scholer-Dahirel, A.; Mechta-Grigoriou, F. The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin. Cancer Biol. 2014, 25, 23–32. [Google Scholar] [CrossRef]
- Nishikawa, M. Reactive oxygen species in tumor metastasis. Cancer Lett. 2008, 266, 53–59. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liaison in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, A.G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 2017, 31, 181–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remacha, L.; Comino-Méndez, I.; Richter, S.; Contreras, L.; Currás-Freixes, M.; Pita, G.; Letón, R.; Galarreta, A.; Torres-Pérez, R.; Honrado, E.; et al. Targeted Exome Sequencing of Krebs Cycle Genes Reveals Candidate Cancer-Predisposing Mutations in Pheochromocytomas and Paragangliomas. Clin. Cancer Res. 2017, 23, 6315–6324. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yang, C. Oncometabolites in Cancer: Current Understanding and Challenges. Cancer Res. 2021, 81, 2820–2823. [Google Scholar] [CrossRef]
- Neumann, H.P.; Bausch, B.; McWhinney, S.R.; Bender, B.U.; Gimm, O.; Franke, G.; Schipper, J.; Klisch, J.; Altehoefer, C.; Zerres, K.; et al. Germ-line mutations in nonsyndromic pheochromocytoma. N. Engl. J. Med. 2002, 346, 1459–1466. [Google Scholar] [CrossRef] [Green Version]
- Eijkelenkamp, K.; Osinga, T.E.; Links, T.P.; Van der Horst-Schrivers, A.N.A. Clinical implications of the oncometabolite succinate in SDHx-mutation carriers. Clin. Genet. 2020, 97, 39–53. [Google Scholar] [CrossRef] [Green Version]
- Neumann, H.P.; Pawlu, C.; Peczkowska, M.; Bausch, B.; McWhinney, S.R.; Muresan, M.; Buchta, M.; Franke, G.; Klisch, J.; Bley, T.A.; et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 2004, 292, 943–951. [Google Scholar] [CrossRef] [Green Version]
- Else, T.; Marvin, M.L.; Everett, J.N.; Gruber, S.B.; Arts, H.A.; Stoffel, E.M.; Auchus, R.J.; Raymond, V.M. The clinical phenotype of SDHC-associated hereditary paraganglioma syndrome (PGL3). J. Clin. Endocrinol. Metab. 2014, 99, E1482–E1486. [Google Scholar] [CrossRef] [Green Version]
- Amar, L.; Bertherat, J.; Baudin, E.; Ajzenberg, C.; Bressac-de Paillerets, B.; Chabre, O.; Chamontin, B.; Delemer, B.; Giraud, S.; Murat, A.; et al. Genetic testing in pheochromocytoma or functional paraganglioma. J. Clin. Oncol. 2005, 23, 8812–8818. [Google Scholar] [CrossRef]
- Heesterman, B.L.; Bayley, J.P.; Tops, C.M.; Hes, F.J.; Van Brussel, B.T.; Corssmit, E.P.; Hamming, J.F.; Van der Mey, A.G.; Jansen, J.C. High prevalence of occult paragangliomas in asymptomatic carriers of SDHD and SDHB gene mutations. Eur. J. Hum. Genet. 2013, 21, 469–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Tuin, K.; Mensenkamp, A.R.; Tops, C.M.J.; Corssmit, E.P.M.; Dinjens, W.N.; Van de Horst-Schrivers, A.N.A.; Jansen, J.C.; De Jong, M.M.; Kunst, H.P.M.; Kusters, B.; et al. Clinical Aspects of SDHA-Related Pheochromocytoma and Paraganglioma: A Nationwide Study. J. Clin. Endocrinol. Metab. 2018, 103, 438–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilanchezhian, M.; Jha, A.; Pacak, K.; Del Rivero, J. Emerging Treatments for Advanced/Metastatic Pheochromocytoma and Paraganglioma. Curr. Treat. Options Oncol. 2020, 21, 85. [Google Scholar] [CrossRef] [PubMed]
- Hadrava Vanova, K.; Kraus, M.; Neuzil, J.; Rohlena, J. Mitochondrial complex II and reactive oxygen species in disease and therapy. Redox Rep. 2020, 25, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Moog, S.; Lussey-Lepoutre, C.; Favier, J. Epigenetic and metabolic reprogramming of SDH-deficient paragangliomas. Endocr. Relat. Cancer 2020, 27, R451–R463. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.J.; Brand, M.D. Reactive oxygen species production by mitochondria. Methods Mol. Biol. 2009, 554, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef] [Green Version]
- Robb, E.L.; Hall, A.R.; Prime, T.A.; Eaton, S.; Szibor, M.; Viscomi, C.; James, A.M.; Murphy, M.P. Control of mitochondrial superoxide production by reverse electron transport at complex I. J. Biol. Chem. 2018, 293, 9869–9879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Nakamura, E.; Yang, H.; Wei, W.; Linggi, M.S.; Sajan, M.P.; Farese, R.V.; Freeman, R.S.; Carter, B.D.; Kaelin, W.G.; et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: Developmental culling and cancer. Cancer Cell 2005, 8, 155–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, T.; Yasuda, K.; Akatsuka, A.; Hino, O.; Hartman, P.S.; Ishii, N. A mutation in the SDHC gene of complex II increases oxidative stress, resulting in apoptosis and tumorigenesis. Cancer Res. 2005, 65, 203–209. [Google Scholar]
- Slane, B.G.; Aykin-Burns, N.; Smith, B.J.; Kalen, A.L.; Goswami, P.C.; Domann, F.E.; Spitz, D.R. Mutation of succinate dehydrogenase subunit C results in increased O2.-, oxidative stress, and genomic instability. Cancer Res. 2006, 66, 7615–7620. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.F.; Low, P.; Dyason, J.C.; Wang, X.F.; Prochazka, L.; Witting, P.K.; Freeman, R.; Swettenham, E.; Valis, K.; Liu, J.; et al. Alpha-tocopheryl succinate induces apoptosis by targeting ubiquinone-binding sites in mitochondrial respiratory complex II. Oncogene 2008, 27, 4324–4335. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.F.; Freeman, R.; Liu, J.; Zobalova, R.; Marin-Hernandez, A.; Stantic, M.; Rohlena, J.; Valis, K.; Rodriguez-Enriquez, S.; Butcher, B.; et al. Suppression of tumor growth in vivo by the mitocan alpha-tocopheryl succinate requires respiratory complex II. Clin. Cancer Res. 2009, 15, 1593–1600. [Google Scholar] [CrossRef] [Green Version]
- Owens, K.M.; Aykin-Burns, N.; Dayal, D.; Coleman, M.C.; Domann, F.E.; Spitz, D.R. Genomic instability induced by mutant succinate dehydrogenase subunit D (SDHD) is mediated by O2(-•) and H2O2. Free Radic. Biol. Med. 2012, 52, 160–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzy, R.D.; Sharma, B.; Bell, E.; Chandel, N.S.; Schumacker, P.T. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol. Cell Biol. 2008, 28, 718–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, Y.; Ishii, K.A.; Aita, Y.; Ikeda, T.; Kawakami, Y.; Shimano, H.; Hara, H.; Takekoshi, K. Loss of SDHB Elevates Catecholamine Synthesis and Secretion Depending on ROS Production and HIF Stabilization. Neurochem. Res. 2016, 41, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Pang, Y.; Zhu, B.; Uher, O.; Caisova, V.; Huynh, T.T.; Taieb, D.; Hadrava Vanova, K.; Ghayee, H.K.; Neuzil, J.; et al. Therapeutic Targeting of SDHB-Mutated Pheochromocytoma/Paraganglioma with Pharmacologic Ascorbic Acid. Clin. Cancer Res. 2020, 26, 3868–3880. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Pang, Y.; Caisova, V.; Ding, J.; Yu, D.; Zhou, Y.; Huynh, T.T.; Ghayee, H.; Pacak, K.; Yang, C. Targeting NRF2-Governed Glutathione Synthesis for SDHB-Mutated Pheochromocytoma and Paraganglioma. Cancers 2020, 12. [Google Scholar] [CrossRef] [Green Version]
- Goncalves, J.; Moog, S.; Morin, A.; Gentric, G.; Müller, S.; Morrell, A.P.; Kluckova, K.; Stewart, T.J.; Andoniadou, C.L.; Lussey-Lepoutre, C.; et al. Loss of SDHB Promotes Dysregulated Iron Homeostasis, Oxidative Stress, and Sensitivity to Ascorbate. Cancer Res. 2021, 81, 3480–3494. [Google Scholar] [CrossRef]
- Florio, R.; De Lellis, L.; Veschi, S.; Verginelli, F.; Di Giacomo, V.; Gallorini, M.; Perconti, S.; Sanna, M.; Mariani-Costantini, R.; Natale, A.; et al. Effects of dichloroacetate as single agent or in combination with GW6471 and metformin in paraganglioma cells. Sci. Rep. 2018, 8, 13610. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Lu, Y.; Caisova, V.; Liu, Y.; Bullova, P.; Huynh, T.T.; Zhou, Y.; Yu, D.; Frysak, Z.; Hartmann, I.; et al. Targeting NAD(+)/PARP DNA Repair Pathway as a Novel Therapeutic Approach to SDHB-Mutated Cluster I Pheochromocytoma and Paraganglioma. Clin. Cancer Res. 2018, 24, 3423–3432. [Google Scholar] [CrossRef] [Green Version]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef]
- Hou, D.; Liu, Z.; Xu, X.; Liu, Q.; Zhang, X.; Kong, B.; Wei, J.-J.; Gong, Y.; Shao, C. Increased oxidative stress mediates the antitumor effect of PARP inhibition in ovarian cancer. Redox Biol. 2018, 17, 99–111. [Google Scholar] [CrossRef]
- Okon, I.S.; Zou, M.-H. Mitochondrial ROS and cancer drug resistance: Implications for therapy. Pharm. Res. 2015, 100, 170–174. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, V.; He, J. Reactive Oxygen Species, Metabolic Plasticity, and Drug Resistance in Cancer. Int. J. Mol. Sci. 2020, 21, 3412. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Naga Prasad, S.V.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martiniova, L.; Lai, E.W.; Elkahloun, A.G.; Abu-Asab, M.; Wickremasinghe, A.; Solis, D.C.; Perera, S.M.; Huynh, T.T.; Lubensky, I.A.; Tischler, A.S.; et al. Characterization of an animal model of aggressive metastatic pheochromocytoma linked to a specific gene signature. Clin. Exp. Metastasis 2009, 26, 239–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fliedner, S.M.J.; Kaludercic, N.; Jiang, X.-S.; Hansikova, H.; Hajkova, Z.; Sladkova, J.; Limpuangthip, A.; Backlund, P.S.; Wesley, R.; Martiniova, L.; et al. Warburg Effect’s Manifestation in Aggressive Pheochromocytomas and Paragangliomas: Insights from a Mouse Cell Model Applied to Human Tumor Tissue. PLoS ONE 2012, 7, e40949. [Google Scholar] [CrossRef]
- Pang, Y.; Yang, C.; Schovanek, J.; Wang, H.; Bullova, P.; Caisova, V.; Gupta, G.; Wolf, K.I.; Semenza, G.L.; Zhuang, Z.; et al. Anthracyclines suppress pheochromocytoma cell characteristics, including metastasis, through inhibition of the hypoxia signaling pathway. Oncotarget 2017, 8, 22313–22324. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Paech, F.; Bouitbir, J.; Krähenbühl, S. Hepatocellular Toxicity Associated with Tyrosine Kinase Inhibitors: Mitochondrial Damage and Inhibition of Glycolysis. Front. Pharm. 2017, 8, 367. [Google Scholar] [CrossRef] [Green Version]
- Paech, F.; Mingard, C.; Grünig, D.; Abegg, V.F.; Bouitbir, J.; Krähenbühl, S. Mechanisms of mitochondrial toxicity of the kinase inhibitors ponatinib, regorafenib and sorafenib in human hepatic HepG2 cells. Toxicology 2018, 395, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Park, Y.J.; Shin, J.H.; Kim, E.S.; Hwang, J.J.; Jin, D.H.; Kim, J.C.; Cho, D.H. A receptor tyrosine kinase inhibitor, Tyrphostin A9 induces cancer cell death through Drp1 dependent mitochondria fragmentation. Biochem. Biophys. Res. Commun. 2011, 408, 465–470. [Google Scholar] [CrossRef]
- Will, Y.; Dykens, J.A.; Nadanaciva, S.; Hirakawa, B.; Jamieson, J.; Marroquin, L.D.; Hynes, J.; Patyna, S.; Jessen, B.A. Effect of the Multitargeted Tyrosine Kinase Inhibitors Imatinib, Dasatinib, Sunitinib, and Sorafenib on Mitochondrial Function in Isolated Rat Heart Mitochondria and H9c2 Cells. Toxicol. Sci. 2008, 106, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Hernández, M.A.; De la Cruz-Ojeda, P.; López-Grueso, M.J.; Navarro-Villarán, E.; Requejo-Aguilar, R.; Castejón-Vega, B.; Negrete, M.; Gallego, P.; Vega-Ochoa, Á.; Victor, V.M.; et al. Integrated molecular signaling involving mitochondrial dysfunction and alteration of cell metabolism induced by tyrosine kinase inhibitors in cancer. Redox Biol. 2020, 36, 101510. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.Q.; Bezerra-Neto, J.E.; Mendonça, B.B.; Latronico, A.C.; Fragoso, M.C.B.V. Primary malignant tumors of the adrenal glands. Clinics 2018, 73, e756s. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Ramirez, M.; Chougnet, C.N.; Habra, M.A.; Palmer, J.L.; Leboulleux, S.; Cabanillas, M.E.; Caramella, C.; Anderson, P.; Al Ghuzlan, A.; Waguespack, S.G.; et al. Treatment with sunitinib for patients with progressive metastatic pheochromocytomas and sympathetic paragangliomas. J. Clin. Endocrinol. Metab. 2012, 97, 4040–4050. [Google Scholar] [CrossRef] [PubMed]
- Warkad, M.S.; Kim, C.H.; Kang, B.G.; Park, S.H.; Jung, J.S.; Feng, J.H.; Inci, G.; Kim, S.C.; Suh, H.W.; Lim, S.S.; et al. Metformin-induced ROS upregulation as amplified by apigenin causes profound anticancer activity while sparing normal cells. Sci. Rep. 2021, 11, 14002. [Google Scholar] [CrossRef]
- Li, B.; Zhou, P.; Xu, K.; Chen, T.; Jiao, J.; Wei, H.; Yang, X.; Xu, W.; Wan, W.; Xiao, J. Metformin induces cell cycle arrest, apoptosis and autophagy through ROS/JNK signaling pathway in human osteosarcoma. Int. J. Biol. Sci. 2020, 16, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Duan, Q.; Wang, T.; Ahmed, M.; Zhang, N.; Li, Y.; Li, L.; Yao, X. Mitochondrial Respiratory Chain Inhibitors Involved in ROS Production Induced by Acute High Concentrations of Iodide and the Effects of SOD as a Protective Factor. Oxid. Med. Cell Longev. 2015, 2015, 217670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogavero, A.; Maiorana, M.V.; Zanutto, S.; Varinelli, L.; Bozzi, F.; Belfiore, A.; Volpi, C.C.; Gloghini, A.; Pierotti, M.A.; Gariboldi, M. Metformin transiently inhibits colorectal cancer cell proliferation as a result of either AMPK activation or increased ROS production. Sci. Rep. 2017, 7, 15992. [Google Scholar] [CrossRef] [Green Version]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Park, W.H.; Han, Y.W.; Kim, S.H.; Kim, S.Z. An ROS generator, antimycin A, inhibits the growth of HeLa cells via apoptosis. J. Cell Biochem. 2007, 102, 98–109. [Google Scholar] [CrossRef]
- Park, D. Metformin Induces Oxidative Stress-Mediated Apoptosis without the Blockade of Glycolysis in H4IIE Hepatocellular Carcinoma Cells. Biol. Pharm. Bull. 2019, 42, 2002–2008. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Jiang, X.; Su, T.; Jiang, L.; Zhou, W.; Wang, W. Metformin Suppresses Proliferation and Viability of Rat Pheochromocytoma Cells. Med. Sci. Monit. 2017, 23, 3253–3260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, S.; Daley, B.; Klubo-Gwiezdzinska, J. The role of an anti-diabetic drug metformin in the treatment of endocrine tumors. J. Mol. Endocrinol. 2019, 63, R17–R35. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Liu, C.; Liu, W.; Zhang, H.; Zhang, R.; Liu, J.; Zhang, J.; Xu, C.; Liu, L.; Huang, S.; et al. Rotenone Induction of Hydrogen Peroxide Inhibits mTOR-mediated S6K1 and 4E-BP1/eIF4E Pathways, Leading to Neuronal Apoptosis. Toxicol. Sci. 2015, 143, 81–96. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lang, F.; Yang, C. NRF2 in human neoplasm: Cancer biology and potential therapeutic target. Pharm. Ther. 2021, 217, 107664. [Google Scholar] [CrossRef]
- Hempel, N.; Carrico, P.M.; Melendez, J.A. Manganese superoxide dismutase (Sod2) and redox-control of signaling events that drive metastasis. Anticancer. Agents Med. Chem. 2011, 11, 191–201. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hadrava Vanova, K.; Yang, C.; Meuter, L.; Neuzil, J.; Pacak, K. Reactive Oxygen Species: A Promising Therapeutic Target for SDHx-Mutated Pheochromocytoma and Paraganglioma. Cancers 2021, 13, 3769. https://doi.org/10.3390/cancers13153769
Hadrava Vanova K, Yang C, Meuter L, Neuzil J, Pacak K. Reactive Oxygen Species: A Promising Therapeutic Target for SDHx-Mutated Pheochromocytoma and Paraganglioma. Cancers. 2021; 13(15):3769. https://doi.org/10.3390/cancers13153769
Chicago/Turabian StyleHadrava Vanova, Katerina, Chunzhang Yang, Leah Meuter, Jiri Neuzil, and Karel Pacak. 2021. "Reactive Oxygen Species: A Promising Therapeutic Target for SDHx-Mutated Pheochromocytoma and Paraganglioma" Cancers 13, no. 15: 3769. https://doi.org/10.3390/cancers13153769
APA StyleHadrava Vanova, K., Yang, C., Meuter, L., Neuzil, J., & Pacak, K. (2021). Reactive Oxygen Species: A Promising Therapeutic Target for SDHx-Mutated Pheochromocytoma and Paraganglioma. Cancers, 13(15), 3769. https://doi.org/10.3390/cancers13153769