
Epigenetic Reprogramming of Tumor-Associated Fibroblasts in Lung Cancer: Therapeutic Opportunities

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Evidence That Tumor-Associated Fibroblasts (TAFs) Are Epigenetically Reprogrammed
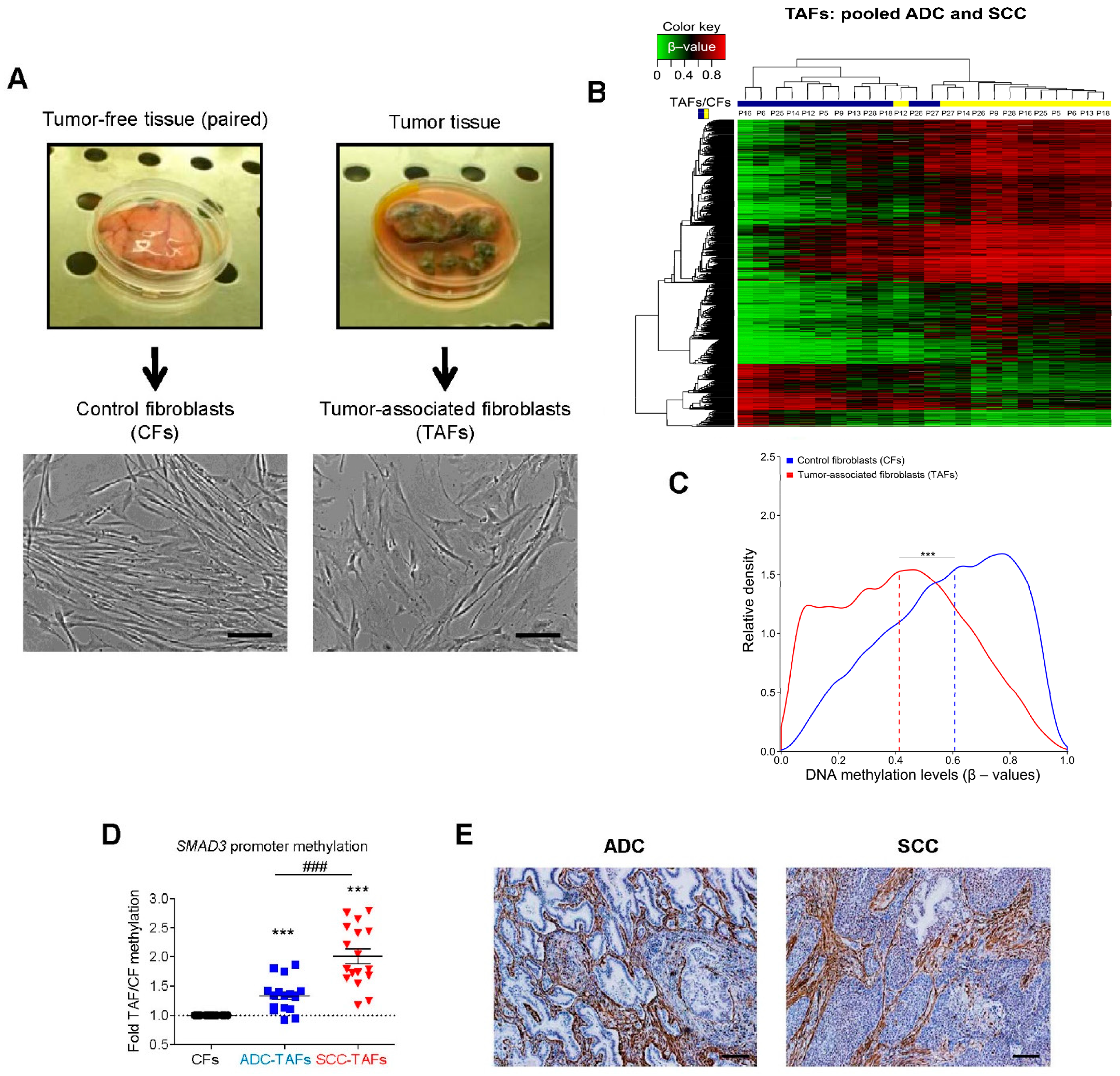
2.1. DNA Methylation Changes in TAFs
2.2. Histone Core Modifications in TAFs
2.3. Non-Coding RNA Alterations in TAFs
3. Epigenetics of TAF Activation: Cause or Consequence?
3.1. Global Hypomethylation and Selective Hypermethylation in Fibroblast Activation
3.2. Histone Core Modifications and DNMTs in Fibroblast Activation
3.3. MiRNAs in Fibroblast Activation
4. Emerging Mechanisms Underlying the Epigenetic Reprogramming of Lung TAFs
4.1. Crosstalk with Cancer Cells
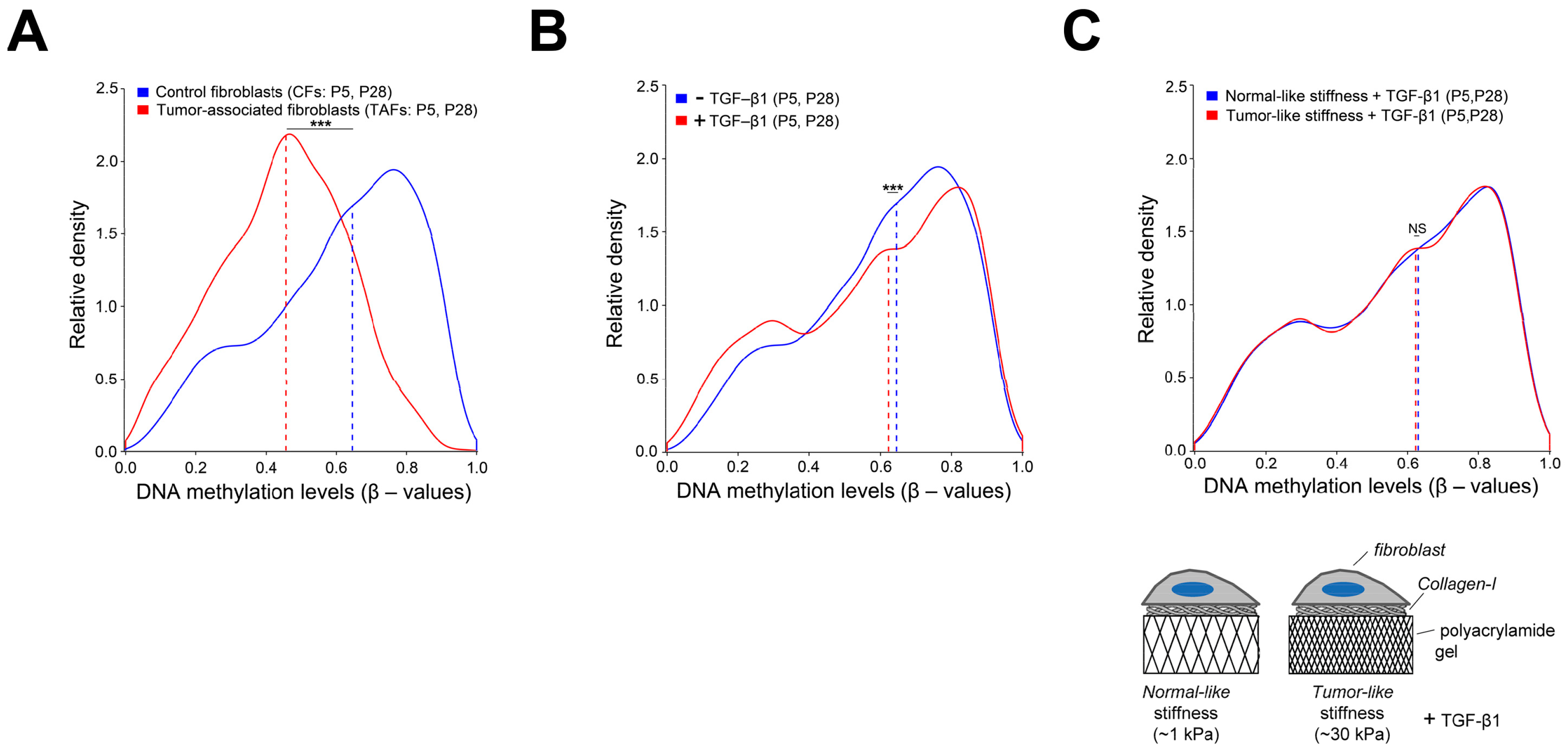
4.2. Extracellular and Intracellular Mechanical Cues
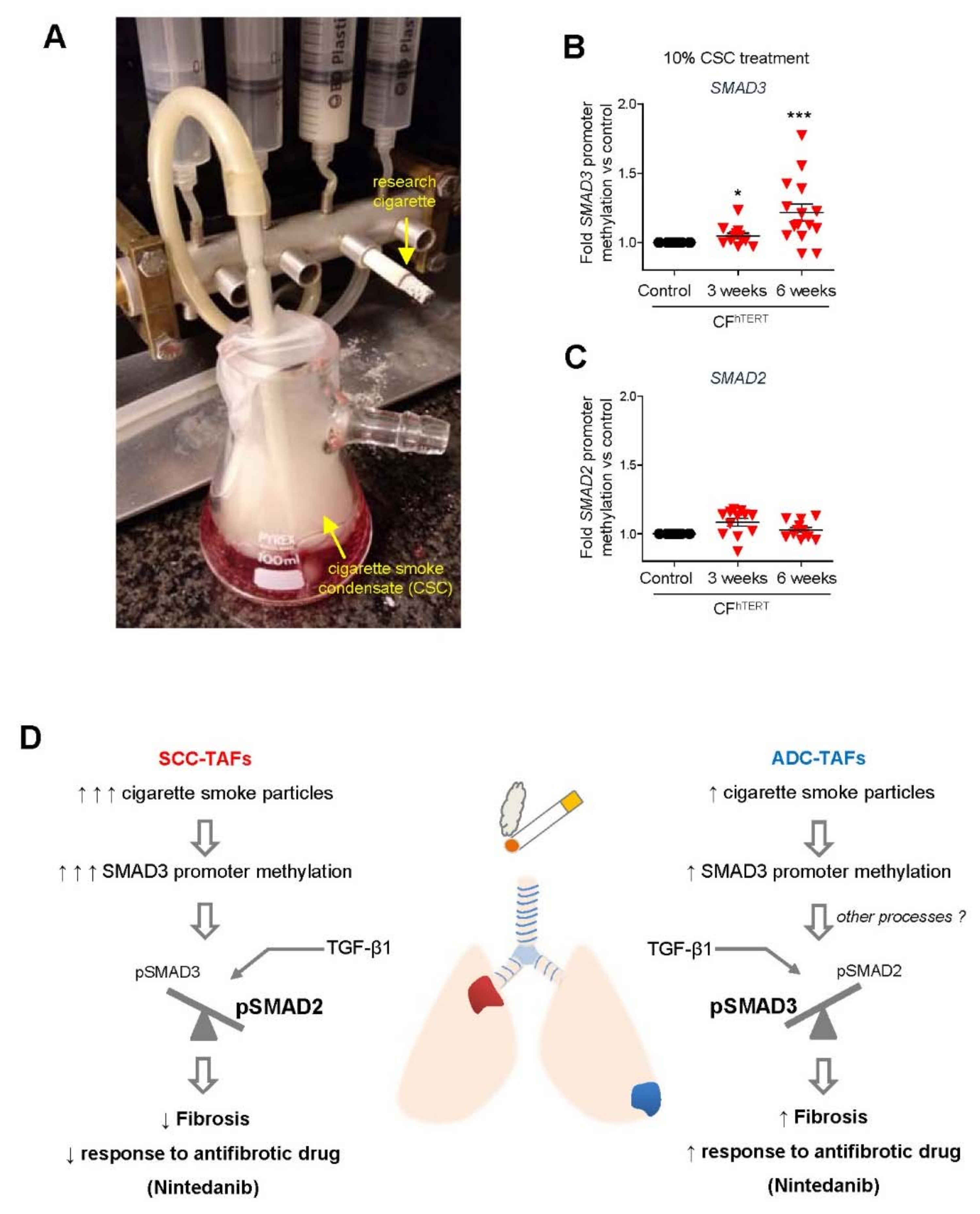
4.3. Smoking and Other Environmental Factors
4.4. Hypoxia
5. Potential Therapeutic Implications
5.1. Limitations of Antifibrotic Drugs in Lung Cancer: Unexpected Epigenetic Influence of Smoking
5.2. Drugs That Alter Histone Marks in Fibroblasts
5.3. Drugs That Modify DNA Methylation Marks in Fibroblasts and Mesenchymal Cells
5.4. Drugs of Nuclear Vitamin D Receptor
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.-K. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA A Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal Fibroblasts Present in Invasive Human Breast Carcinomas Promote Tumor Growth and Angiogenesis through Elevated SDF-1/CXCL12 Secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Ito, Y.; Mezawa, Y.; Sulidan, K.; Daigo, Y.; Hiraga, T.; Mogushi, K.; Wali, N.; Suzuki, H.; Itoh, T.; et al. Stromal fibroblasts induce metastatic tumor cell clusters via epithelial–mesenchymal plasticity. Life Sci. Alliance 2019, 2, e201900425. [Google Scholar] [CrossRef]
- Zhang, C.; Fu, L.; Fu, J.; Hu, L.; Yang, H.; Rong, T.-H.; Li, Y.; Liu, H.; Fu, S.-B.; Zeng, Y.-X.; et al. Fibroblast Growth Factor Receptor 2–Positive Fibroblasts Provide a Suitable Microenvironment for Tumor Development and Progression in Esophageal Carcinoma. Clin. Cancer Res. 2009, 15, 4017–4027. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Ronnov-Jessen, L.; Petersen, O.W.; Bissell, M.J. Cellular changes involved in conversion of normal to malignant breast: Importance of the stromal reaction. Physiol. Rev. 1996, 76, 69–125. [Google Scholar] [CrossRef]
- Öhlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef]
- Kim, D.J.; Dunleavey, J.; Xiao, L.; Ollila, D.W.; Troester, M.A.; Otey, C.A.; Li, W.; Barker, T.H.; Dudley, A.C. Suppression of TGFβ-mediated conversion of endothelial cells and fibroblasts into cancer associated (myo)fibroblasts via HDAC inhibition. Br. J. Cancer 2018, 118, 1359–1368. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K.; Martin, J.D.; Stylianopoulos, T. The Role of Mechanical Forces in Tumor Growth and Therapy. Annu. Rev. Biomed. Eng. 2014, 16, 321–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, M.; Luo, Y.; Mao, N.; Huang, G.; Teng, C.; Wang, H.; Wu, J.; Liao, X.; Yang, J. Cancer-Associated Fibroblasts Accelerate Malignant Progression of Non-Small Cell Lung Cancer via Connexin 43-Formed Unidirectional Gap Junctional Intercellular Communication. Cell. Physiol. Biochem. 2018, 51, 315–336. [Google Scholar] [CrossRef] [PubMed]
- Shintani, Y.; Abulaiti, A.; Kimura, T.; Funaki, S.; Nakagiri, T.; Inoue, M.; Sawabata, N.; Minami, M.; Morii, E.; Okumura, M. Pulmonary Fibroblasts Induce Epithelial Mesenchymal Transition and Some Characteristics of Stem Cells in Non-Small Cell Lung Cancer. Ann. Thorac. Surg. 2013, 96, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Wu, M.; Arcand, S.L.; Lavallée, S.; Hébert, J.; Tonin, P.N.; Basik, M. Breast Carcinoma–Associated Fibroblasts Rarely Contain p53 Mutations or Chromosomal Aberrations. Cancer Res. 2010, 70, 5770–5777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francastel, C.; Schübeler, D.; Martin, D.I.K.; Groudine, M. Nuclear compartmentalization and gene activity. Nat. Rev. Mol. Cell Biol. 2000, 1, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, D.; Wauters, E.; Boeckx, B.; Aibar, S.; Nittner, D.; Burton, O.; Bassez, A.; Decaluwé, H.; Pircher, A.; Eynde, K.V.D.; et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 2018, 24, 1277–1289. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef] [Green Version]
- Gabasa, M.; Radisky, E.S.; Ikemori, R.; Bertolini, G.; Arshakyan, M.; Hockla, A.; Duch, P.; Rondinone, O.; Llorente, A.; Maqueda, M.; et al. MMP1 drives tumor progression in large cell carcinoma of the lung through fibroblast senescence. Cancer Lett. 2021, 507, 1–12. [Google Scholar] [CrossRef]
- Ikemori, R.; Gabasa, M.; Duch, P.; Vizoso, M.; Bragado, P.; Arshakyan, M.; Luis, I.-C.; Marín, A.; Morán, S.; Castro, M.; et al. Epigenetic SMAD3 Repression in Tumor-Associated Fibroblasts Impairs Fibrosis and Response to the Antifibrotic Drug Nintedanib in Lung Squamous Cell Carcinoma. Cancer Res. 2019, 80, 276–290. [Google Scholar] [CrossRef] [Green Version]
- Puig, M.; Lugo, R.; Gabasa, M.; Giménez, A.; Velásquez, A.; Galgoczy, R.; Ramírez, J.; Gómez-Caro, A.; Busnadiego, Ó.; Rodriguez-Pascual, F.; et al. Matrix Stiffening and β1 Integrin Drive Subtype-Specific Fibroblast Accumulation in Lung Cancer. Mol. Cancer Res. 2014, 13, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Gabasa, M.; Ikemori, R.; Hilberg, F.; Reguart, N.; Alcaraz, J. Nintedanib selectively inhibits the activation and tumor-promoting effects of fibroblasts from lung adenocarcinoma patients. Br. J. Cancer 2017, 117, 1128–1138. [Google Scholar] [CrossRef] [Green Version]
- Reck, M.; Kaiser, R.; Mellemgaard, A.; Douillard, J.-Y.; Orlov, S.; Krzakowski, M.; von Pawel, J.; Gottfried, M.; Bondarenko, I.; Liao, M.; et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): A phase 3, double-blind, randomised controlled trial. Lancet Oncol. 2014, 15, 143–155. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, M. DNA methylation in cancer: Too much, but also too little. Oncogene 2002, 21, 5400–5413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Vlodrop, I.J.H.; Niessen, H.; Derks, S.; Baldewijns, M.M.L.L.; Van Criekinge, W.; Herman, J.G.; Van Engeland, M. Analysis of Promoter CpG Island Hypermethylation in Cancer: Location, Location, Location! Clin. Cancer Res. 2011, 17, 4225–4231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.-X.; He, C. Balance of DNA methylation and demethylation in cancer development. Genome Biol. 2012, 13, 173. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.E.; Davies, T.J.; Mc Auley, M.T. The role of DNA methylation in ageing and cancer. Proc. Nutr. Soc. 2018, 77, 412–422. [Google Scholar] [CrossRef]
- Jiang, L.; Gonda, T.A.; Gamble, M.V.; Salas, M.; Seshan, V.; Tu, S.; Twaddell, W.S.; Hegyi, P.; Lazar, G.; Steele, I.; et al. Global Hypomethylation of Genomic DNA in Cancer-Associated Myofibroblasts. Cancer Res. 2008, 68, 9900–9908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakya, R.; Gonda, T.; Quante, M.; Salas, M.; Kim, S.; Brooks, J.; Hirsch, S.; Davies, J.; Cullo, A.; Olive, K.; et al. Hypomethylating Therapy in an Aggressive Stroma-Rich Model of Pancreatic Carcinoma. Cancer Res. 2012, 73, 885–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.; Yao, J.; Cai, L.; Bachman, K.E.; Brûle, F.V.D.; Velculescu, V.; Polyak, K. Distinct epigenetic changes in the stromal cells of breast cancers. Nat. Genet. 2005, 37, 899–905. [Google Scholar] [CrossRef]
- Navab, R.; Strumpf, D.; Bandarchi, B.; Zhu, C.; Pintilie, M.; Ramnarine, V.R.; Ibrahimov, E.; Radulovich, N.; Leung, L.; Barczyk, M.; et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 7160–7165. [Google Scholar] [CrossRef] [Green Version]
- Vizoso, M.; Puig, M.; Carmona, F.; Maqueda, M.; Velásquez, A.; Gomez, A.; Labernadie, A.; Lugo, R.; Gabasa, M.; Rigat-Brugarolas, L.G.; et al. Aberrant DNA methylation in non-small cell lung cancer-associated fibroblasts. Carcinogenesis 2015, 36, 1453–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horie, M.; Miyashita, N.; Mikami, Y.; Noguchi, S.; Yamauchi, Y.; Suzukawa, M.; Fukami, T.; Ohta, K.; Asano, Y.; Satoshi, N.; et al. TBX4 is involved in the super-enhancer-driven transcriptional programs underlying features specific to lung fibroblasts. Am. J. Physiol. Cell. Mol. Physiol. 2018, 314, L177–L191. [Google Scholar] [CrossRef] [PubMed]
- Heintzman, N.D.; Stuart, R.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-wide Mapping of HATs and HDACs Reveals Distinct Functions in Active and Inactive Genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Eckert, M.; Coscia, F.; Chryplewicz, A.; Chang, J.W.; Hernandez, K.M.; Pan, S.; Tienda, S.M.; Nahotko, D.A.; Li, G.; Blaženović, I.; et al. Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nat. Cell Biol. 2019, 569, 723–728. [Google Scholar] [CrossRef]
- Wu, M.; Hu, W.; Wang, G.; Yao, Y.; Yu, X.-F. Nicotinamide N-Methyltransferase Is a Prognostic Biomarker and Correlated with Immune Infiltrates in Gastric Cancer. Front. Genet. 2020, 11, 580299. [Google Scholar] [CrossRef]
- Zhang, L.; Song, M.; Zhang, F.; Yuan, H.; Chang, W.; Yu, G.; Niu, Y. Accumulation of Nicotinamide N-Methyltransferase (NNMT) in Cancer-associated Fibroblasts: A Potential Prognostic and Predictive Biomarker for Gastric Carcinoma. J. Histochem. Cytochem. 2020, 69, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Sartini, D.; Morganti, S.; Guidi, E.; Rubini, C.; Zizzi, A.; Giuliante, R.; Pozzi, V.; Emanuelli, M. Nicotinamide N-methyltransferase in Non-small Cell Lung Cancer: Promising Results for Targeted Anti-cancer Therapy. Cell Biophys. 2013, 67, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Sartini, D.; Seta, R.; Pozzi, V.; Morganti, S.; Rubini, C.; Zizzi, A.; Tomasetti, M.; Santarelli, L.; Emanuelli, M. Role of nicotinamide N-methyltransferase in non-small cell lung cancer: In vitro effect of shRNA-mediated gene silencing on tumourigenicity. Biol. Chem. 2015, 396, 225–234. [Google Scholar] [CrossRef]
- Yasuda, T.; Koiwa, M.; Yonemura, A.; Miyake, K.; Kariya, R.; Kubota, S.; Yokomizo-Nakano, T.; Yasuda-Yoshihara, N.; Uchihara, T.; Itoyama, R.; et al. Inflammation-driven senescence-associated secretory phenotype in cancer-associated fibroblasts enhances peritoneal dissemination. Cell Rep. 2021, 34, 108779. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Huang, M.; Li, Q. Cancer-Associated Fibroblasts Promote Angiogenesis of Hepatocellular Carcinoma by VEGF-Mediated EZH2/VASH1 Pathway. Technol. Cancer Res. Treat. 2019, 18, 1533033819879905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Schoepp, M.; Ströse, A.J.; Haier, J. Dysregulation of miRNA Expression in Cancer Associated Fibroblasts (CAFs) and Its Consequences on the Tumor Microenvironment. Cancers 2017, 9, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Liu, J.; Liu, Y.; Wu, W.; Li, X.; Wu, Y.; Chen, H.; Zhang, K.; Gu, L. miR-101 represses lung cancer by inhibiting interaction of fibroblasts and cancer cells by down-regulating CXCL12. Biomed. Pharmacother. 2015, 74, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Yu, X.; Yang, F.; Zhang, Z.; Shen, J.; Sun, J.; Choksi, S.; Jitkaew, S.; Shu, Y. Reprogramming of Normal Fibroblasts into Cancer-Associated Fibroblasts by miRNAs-Mediated CCL2/VEGFA Signaling. PLoS Genet. 2016, 12, e1006244. [Google Scholar] [CrossRef]
- Kunita, A.; Morita, S.; Irisa, T.U.; Goto, A.; Niki, T.; Takai, D.; Nakajima, J.; Fukayama, M. MicroRNA-21 in cancer-associated fibroblasts supports lung adenocarcinoma progression. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Scotton, C.; Chambers, R.C. Molecular Targets in Pulmonary Fibrosis. Chest 2007, 132, 1311–1321. [Google Scholar] [CrossRef]
- Liu, F.; Mih, J.D.; Shea, B.S.; Kho, A.T.; Sharif, A.S.; Tager, A.M.; Tschumperlin, D.J. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J. Cell Biol. 2010, 190, 693–706. [Google Scholar] [CrossRef] [Green Version]
- Giménez, A.; Duch, P.; Puig, M.; Gabasa, M.; Xaubet, A.; Alcaraz, J. Dysregulated Collagen Homeostasis by Matrix Stiffening and TGF-β1 in Fibroblasts from Idiopathic Pulmonary Fibrosis Patients: Role of FAK/Akt. Int. J. Mol. Sci. 2017, 18, 2431. [Google Scholar] [CrossRef] [Green Version]
- Bierie, B.; Moses, H.L. TGFβ: The molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 2006, 6, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Dalal, B.I.; Keown, P.A.; Greenberg, A.H. Immunocytochemical localization of secreted transforming growth factor-beta 1 to the advancing edges of primary tumors and to lymph node metastases of human mammary carcinoma. Am. J. Pathol. 1993, 143, 381–389. [Google Scholar]
- Sun, X.; He, Y.; Huang, C.; Ma, T.-T.; Li, J. The epigenetic feedback loop between DNA methylation and microRNAs in fibrotic disease with an emphasis on DNA methyltransferases. Cell. Signal. 2013, 25, 1870–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, A.; Paoli, P.; Salvador, E.M.; White, S.; French, J.; Mann, J. Hepatic stellate cell transdifferentiation involves genome-wide remodeling of the DNA methylation landscape. J. Hepatol. 2016, 64, 661–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karouzakis, E.; Gay, R.E.; Michel, B.A.; Gay, S.; Neidhart, M. DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2009, 60, 3613–3622. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Gharaee-Kermani, M.; Wu, Z.; Phan, S. Epigenetic Regulation of Myofibroblast Differentiation by DNA Methylation. Am. J. Pathol. 2010, 177, 21–28. [Google Scholar] [CrossRef]
- Huang, S.K.; Scruggs, A.M.; Donaghy, J.; McEachin, R.C.; Fisher, A.S.; Richardson, B.C.; Peters-Golden, M. Prostaglandin E 2 increases fibroblast gene-specific and global DNA methylation via increased DNA methyltransferase expression. FASEB J. 2012, 26, 3703–3714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagood, J.S.; Prabhakaran, P.; Kumbla, P.; Salazar, L.; MacEwen, M.W.; Barker, T.H.; Ortiz, L.A.; Schoeb, T.; Siegal, G.P.; Alexander, C.B.; et al. Loss of Fibroblast Thy-1 Expression Correlates with Lung Fibrogenesis. Am. J. Pathol. 2005, 167, 365–379. [Google Scholar] [CrossRef] [Green Version]
- Hannan, R.T.; Peirce, S.; Barker, T.H. Fibroblasts: Diverse Cells Critical to Biomaterials Integration. ACS Biomater. Sci. Eng. 2017, 4, 1223–1232. [Google Scholar] [CrossRef]
- Hu, B.; Gharaee-Kermani, M.; Wu, Z.; Phan, S.H. Essential Role of MeCP2 in the Regulation of Myofibroblast Differentiation during Pulmonary Fibrosis. Am. J. Pathol. 2011, 178, 1500–1508. [Google Scholar] [CrossRef]
- Xiang, Z.; Zhou, Q.; Hu, M.; Sanders, Y.Y. MeCP2 epigenetically regulates alpha-smooth muscle actin in human lung fibroblasts. J. Cell. Biochem. 2020, 121, 3616–3625. [Google Scholar] [CrossRef]
- Tschumperlin, D.J.; Ligresti, G.; Hilscher, M.B.; Shah, V.H. Mechanosensing and fibrosis. J. Clin. Investig. 2018, 128, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Yu-Wai-Man, C.; Spencer-Dene, B.; Lee, R.M.H.; Hutchings, K.; Lisabeth, E.M.; Treisman, R.; Bailly, M.; Larsen, S.D.; Neubig, R.R.; Khaw, P.T. Local delivery of novel MRTF/SRF inhibitors prevents scar tissue formation in a preclinical model of fibrosis. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sala, L.; Valls, H.F.; Stanisavljevic, J.; Curto, J.; Vergés, J.; Peña, R.; Duch, P.; Alcaraz, J.; De Herreros, A.G.; Baulida, J. Abrogation of myofibroblast activities in metastasis and fibrosis by methyltransferase inhibition. Int. J. Cancer 2019, 145, 3064–3077. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Phan, S.; Thannickal, V.J.; Prunotto, M.; Desmoulière, A.; Varga, J.; De Wever, O.; Mareel, M.M.; Gabbiani, G. Recent Developments in Myofibroblast Biology: Paradigms for Connective Tissue Remodeling. Am. J. Pathol. 2012, 180, 1340–1355. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Shan, B.; Klingsberg, R.C.; Qin, X.; Lasky, J.A. Abrogation of TGF-β1-induced fibroblast-myofibroblast differentiation by histone deacetylase inhibition. Am. J. Physiol. Cell. Mol. Physiol. 2009, 297, L864–L870. [Google Scholar] [CrossRef] [Green Version]
- Bechtel-Walz, W.; McGoohan, S.; Zeisberg, E.M.; Müller, G.A.; Kalbacher, H.; Salant, D.; Müller, C.A.; Kalluri, R.; Zeisberg, M. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat. Med. 2010, 16, 544–550. [Google Scholar] [CrossRef] [Green Version]
- Docherty, N.G.; O’Sullivan, O.E.; Healy, D.A.; Murphy, M.; O’Neill, A.J.; Fitzpatrick, J.M.; Watson, R.W.G. TGF-β1-induced EMT can occur independently of its proapoptotic effects and is aided by EGF receptor activation. Am. J. Physiol. Physiol. 2006, 290, F1202–F1212. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhang, J.; Sun, X.; Su, Q.; You, C. Down-regulation of miR-29b in carcinoma associated fibroblasts promotes cell growth and metastasis of breast cancer. Oncotarget 2017, 8, 39559–39570. [Google Scholar] [CrossRef] [Green Version]
- Cushing, L.; Kuang, P.P.; Qian, J.; Shao, F.; Wu, J.; Little, F.; Thannickal, V.J.; Cardoso, W.V.; Lü, J. miR-29 Is a Major Regulator of Genes Associated with Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2011, 45, 287–294. [Google Scholar] [CrossRef]
- Mann, J.; Chu, D.C.; Maxwell, A.; Oakley, F.; Zhu, N.; Tsukamoto, H.; Mann, D.A. MeCP2 Controls an Epigenetic Pathway That Promotes Myofibroblast Transdifferentiation and Fibrosis. Gastroenterol. 2010, 138, 705–714.e4. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Sun, Y.; Hou, Y.; Peng, Q.; Wang, L.; Luo, H.; Tang, X.; Zeng, Z.; Liu, M. MiRNA expression analysis of cancer-associated fibroblasts and normal fibroblasts in breast cancer. Int. J. Biochem. Cell Biol. 2012, 44, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Sakamoto, N.; Oue, N.; Yashiro, M.; Sentani, K.; Yanagihara, K.; Hirakawa, K.; Yasui, W. Micro RNA-143 regulates collagen type III expression in stromal fibroblasts of scirrhous type gastric cancer. Cancer Sci. 2013, 105, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Arce, L.; Wang, M.; Putta, S.; Lanting, L.; Natarajan, R. A microRNA circuit mediates transforming growth factor-β1 autoregulation in renal glomerular mesangial cells. Kidney Int. 2011, 80, 358–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Serrano, C.H.; Benamar, S.; Croce, O.; et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 2015, 6, 10204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, T.; Lv, H.; Lv, G.; Li, T.; Wang, C.; Han, Q.; Yu, L.; Su, B.; Guo, L.; Huang, S.; et al. Tumor-derived exosomal miR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Balestrini, J.L.; Chaudhry, S.; Sarrazy, V.; Koehler, A.; Hinz, B. The mechanical memory of lung myofibroblasts. Integr. Biol. 2012, 4, 410–421. [Google Scholar] [CrossRef]
- Li, C.X.; Talele, N.P.; Boo, S.; Koehler, A.; Knee-Walden, E.; Balestrini, J.L.; Speight, P.; Kapus, A.; Hinz, B. MicroRNA-21 preserves the fibrotic mechanical memory of mesenchymal stem cells. Nat. Mater. 2016, 16, 379–389. [Google Scholar] [CrossRef]
- Liu, G.; Friggeri, A.; Yang, Y.; Milosevic, J.; Ding, Q.; Thannickal, V.J.; Kaminski, N.; Abraham, E.H. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J. Exp. Med. 2010, 207, 1589–1597. [Google Scholar] [CrossRef]
- Roca-Cusachs, P.; Alcaraz, J.; Sunyer, R.; Samitier, J.; Farre, R.; Navajas, D. Micropatterning of Single Endothelial Cell Shape Reveals a Tight Coupling between Nuclear Volume in G1 and Proliferation. Biophys. J. 2008, 94, 4984–4995. [Google Scholar] [CrossRef] [Green Version]
- Le Beyec, J.; Xu, R.; Lee, S.-Y.; Nelson, C.M.; Rizki, A.; Alcaraz, J.; Bissell, M.J. Cell shape regulates global histone acetylation in human mammary epithelial cells. Exp. Cell Res. 2007, 313, 3066–3075. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela-Fernández, A.; Cabrero, J.R.; Serrador, J.M.; Sánchez-Madrid, F. HDAC6: A key regulator of cytoskeleton, cell migration and cell–cell interactions. Trends Cell Biol. 2008, 18, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.N.; Nordheim, A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 2010, 11, 353–365. [Google Scholar] [CrossRef] [Green Version]
- Breitling, L.P.; Yang, R.; Korn, B.; Burwinkel, B.; Brenner, H. Tobacco-Smoking-Related Differential DNA Methylation: 27K Discovery and Replication. Am. J. Hum. Genet. 2011, 88, 450–457. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.; Hänninen, I.; Raitoharju, E.; Marttila, S.; Mishra, B.H.; Mononen, N.; Kähönen, M.; Hurme, M.; Raitakari, O.; Törönen, P.; et al. Epigenome-450K-wide methylation signatures of active cigarette smoking: The Young Finns Study. Biosci. Rep. 2020, 40, BSR20200596. [Google Scholar] [CrossRef] [PubMed]
- Zal, F.; Yarahmadi, A.; Totonchi, H.; Barazesh, M.; Sarabi, M.M. Nicotine attenuates global genomic DNA methylation by influencing DNMTs gene expression in human endometrial stromal cells. Genes Environ. 2020, 42, 6–8. [Google Scholar] [CrossRef]
- Kwon, Y.-M.; Park, J.-H.; Kim, H.; Shim, Y.M.; Kim, J.; Han, J.; Park, J.; Kim, D.-H. Different susceptibility of increased DNMT1 expression by exposure to tobacco smoke according to histology in primary non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2006, 133, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Mortusewicz, O.; Schermelleh, L.; Walter, J.; Cardoso, M.C.; Leonhardt, H. Recruitment of DNA methyltransferase I to DNA repair sites. Proc. Natl. Acad. Sci. USA 2005, 102, 8905–8909. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.-K.; Hsieh, Y.-S.; Lin, P.; Hsu, H.-S.; Chen, C.-Y.; Tang, Y.-A.; Lee, C.-F.; Wang, Y.-C. The tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. J. Clin. Investig. 2010, 120, 521–532. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Lin, I.G.; Hsieh, C.-L. Protein Binding Protects Sites on Stable Episomes and in the Chromosome from De Novo Methylation. Mol. Cell. Biol. 2001, 21, 3416–3424. [Google Scholar] [CrossRef] [Green Version]
- Samanta, D.; Gonzalez, A.L.; Nagathihalli, N.; Ye, F.; Carbone, D.P.; Datta, P.K. Smoking Attenuates Transforming Growth Factor-β–Mediated Tumor Suppression Function through Downregulation of Smad3 in Lung Cancer. Cancer Prev. Res. 2012, 5, 453–463. [Google Scholar] [CrossRef] [Green Version]
- Kong, F.-M.; Wang, S. Nondosimetric Risk Factors for Radiation-Induced Lung Toxicity. Semin. Radiat. Oncol. 2015, 25, 100–109. [Google Scholar] [CrossRef] [Green Version]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Shahrzad, S.; Bertrand, K.; Minhas, K.; Coomber, B. Induction of DNA Hypomethylation by Tumor Hypoxia. Epigenetics 2007, 2, 119–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Liu, L.; Zhao, Y.; Zhang, J.; Wang, D.; Chen, J.; He, Y.; Wu, J.; Zhang, Z.; Liu, Z. Hypoxia Induces Genomic DNA Demethylation through the Activation of HIF-1α and Transcriptional Upregulation of MAT2A in Hepatoma Cells. Mol. Cancer Ther. 2011, 10, 1113–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, C.M.; Neary, R.; Levendale, A.; Watson, C.J.; Baugh, J.A. Hypoxia-induced DNA hypermethylation in human pulmonary fibroblasts is associated with Thy-1 promoter methylation and the development of a pro-fibrotic phenotype. Respir. Res. 2012, 13, 74. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nat. Cell Biol. 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Barcellos-Hoff, M.H.; Lyden, D.; Wang, T.C. The evolution of the cancer niche during multistage carcinogenesis. Nat. Rev. Cancer 2013, 13, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Noel, A.; Foidart, J.-M. The role of stroma in breast carcinoma growth in vivo. J. Mammary Gland. Biol. Neoplasia 1998, 3, 215–225. [Google Scholar] [CrossRef]
- Tschumperlin, D.J.; Lagares, D. Mechano-therapeutics: Targeting Mechanical Signaling in Fibrosis and Tumor Stroma. Pharmacol. Ther. 2020, 212, 107575. [Google Scholar] [CrossRef]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.E.; Paget, J.T.E.; Khan, A.; Harrington, K. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef]
- D’Aniello, C.; Cermola, F.; Palamidessi, A.; Wanderlingh, L.G.; Gagliardi, M.; Migliaccio, A.; Varrone, F.; Casalino, L.; Matarazzo, M.R.; De Cesare, D.; et al. Collagen Prolyl Hydroxylation-Dependent Metabolic Perturbation Governs Epigenetic Remodeling and Mesenchymal Transition in Pluripotent and Cancer Cells. Cancer Res. 2019, 79, 3235–3250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D Receptor-Mediated Stromal Reprogramming Suppresses Pancreatitis and Enhances Pancreatic Cancer Therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorchs, L.; Ahmed, S.; Mayer, C.; Knauf, A.; Moro, C.F.; Svensson, M.; Heuchel, R.; Rangelova, E.; Bergman, P.; Kaipe, H. The vitamin D analogue calcipotriol promotes an anti-tumorigenic phenotype of human pancreatic CAFs but reduces T cell mediated immunity. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Ramnath, N.; Daignault-Newton, S.; Dy, G.K.; Muindi, J.R.; Adjei, A.; Elingrod, V.L.; Kalemkerian, G.P.; Cease, K.B.; Stella, P.J.; Brenner, D.E.; et al. A phase I/II pharmacokinetic and pharmacogenomic study of calcitriol in combination with cisplatin and docetaxel in advanced non-small-cell lung cancer. Cancer Chemother. Pharmacol. 2013, 71, 1173–1182. [Google Scholar] [CrossRef] [Green Version]
- Gray, J.E.; Saltos, A.; Tanvetyanon, T.; Haura, E.B.; Creelan, B.; Antonia, S.J.; Shafique, M.; Zheng, H.; Dai, W.; Saller, J.J.; et al. Phase I/Ib Study of Pembrolizumab Plus Vorinostat in Advanced/Metastatic Non–Small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 6623–6632. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Maitland, M.L.; Frankel, P.; Argiris, A.E.; Koczywas, M.; Gitlitz, B.; Thomas, S.; Espinoza-Delgado, I.; Vokes, E.E.; Gandara, D.R.; et al. Carboplatin and Paclitaxel in Combination with Either Vorinostat or Placebo for First-Line Therapy of Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2010, 28, 56–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, A.M.; Van Der Mijn, J.C.; Christos, P.; Wright, J.; Thomas, C.; Dutcher, J.P.; Nanus, D.M.; Tagawa, S.T.; Gudas, L.J. NCI 6896: A phase I trial of vorinostat (SAHA) and isotretinoin (13-cis retinoic acid) in the treatment of patients with advanced renal cell carcinoma. Investig. New Drugs 2020, 38, 1383–1389. [Google Scholar] [CrossRef]
- Brown, S.; Pawlyn, C.; Tillotson, A.-L.; Sherratt, D.; Flanagan, L.; Low, E.; Morgan, G.J.; Williams, C.; Kaiser, M.; Davies, F.E.; et al. Bortezomib, Vorinostat, and Dexamethasone Combination Therapy in Relapsed Myeloma: Results of the Phase 2 MUK four Trial. Clin. Lymphoma Myeloma Leuk. 2021, 21, 154–161.e3. [Google Scholar] [CrossRef]
- Su, G.; Sohn, T.; Ryu, B.; Kern, S.E. A novel histone deacetylase inhibitor identified by high-throughput transcriptional screening of a compound library. Cancer Res. 2000, 60, 3137–3142. [Google Scholar]
- Heerboth, S.; Lapinska, K.; Snyder, N.; Leary, M.; Rollinson, S.; Sarkar, S. Use of Epigenetic Drugs in Disease: An Overview. Genet. Epigenetics 2014, 6, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, H.; Hock, T.; Thannickal, V.J.; Sanders, Y.Y. Histone Deacetylase Inhibition Downregulates Collagen 3A1 in Fibrotic Lung Fibroblasts. Int. J. Mol. Sci. 2013, 14, 19605–19617. [Google Scholar] [CrossRef]
- Gjaltema, R.; Bank, R.A. Molecular insights into prolyl and lysyl hydroxylation of fibrillar collagens in health and disease. Crit. Rev. Biochem. Mol. Biol. 2016, 52, 74–95. [Google Scholar] [CrossRef] [Green Version]
- Musah, S.; Chen, J.; Schlueter, C.; Humphrey, D.M.; Stocke, K.; Hoyle, M.I.; Hoyle, G.W. Inhibition of chlorine-induced airway fibrosis by budesonide. Toxicol. Appl. Pharmacol. 2019, 363, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Veronesi, G.; Szabo, E.; DeCensi, A.; Gonzaga, A.G.; Bellomi, M.; Radice, D.; Ferretti, S.; Pelosi, G.; Lazzeroni, M.; Serrano, D.; et al. Randomized Phase II Trial of Inhaled Budesonide versus Placebo in High-Risk Individuals with CT Screen–Detected Lung Nodules. Cancer Prev. Res. 2010, 4, 34–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, J.; Oakley, F.; Akiboye, F.; Elsharkawy, A.; Thorne, A.W.; Mann, D.A. Regulation of myofibroblast transdifferentiation by DNA methylation and MeCP2: Implications for wound healing and fibrogenesis. Cell Death Differ. 2006, 14, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Mukai, Y.; Yamada, D.; Eguchi, H.; Iwagami, Y.; Asaoka, T.; Noda, T.; Kawamoto, K.; Gotoh, K.; Kobayashi, S.; Takeda, Y.; et al. Vitamin D Supplementation is a Promising Therapy for Pancreatic Ductal Adenocarcinoma in Conjunction with Current Chemoradiation Therapy. Ann. Surg. Oncol. 2018, 25, 1868–1879. [Google Scholar] [CrossRef] [PubMed]
- Fetahu, I.S.; Höbaus, J.; Kállay, E. Vitamin D and the epigenome. Front. Physiol. 2014, 5, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer-Mayorga, G.; Larriba, M.J.; Crespo, P.; Muñoz, A. Mechanisms of action of vitamin D in colon cancer. J. Steroid Biochem. Mol. Biol. 2019, 185, 1–6. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Major Known Targets in Fibroblasts | Drug | Cancer Type | Clinical Status | Reference |
|---|---|---|---|---|
| protein arginine methyltransferases (PRMTs) | arginine methyltransferase inhibitor 1 (AMI-1), sinefungin | Breast | in vivo preclinical study | [65] |
| Collagen prolyl hydroxylation | Budesonide | Breast | Clinically approved to treat asthma | [105] |
| Nuclear Vitamin D receptor | Calcipotriol, paricalcitol | Pancreas, colorectal cancer, NSCLC | Preclinical or Phase I/II | [106,107,108] |
| VEGFRs, PDGFRs, FGFRs, TGF-β1/SMAD3 | Nintedanib | NSCLC (ADC) | Clinically approved to treat advanced ADC | [19,21,22] |
| Histone deacetylases (HDACs) | Vorinostat | NSCLC, kidney, myeloma | Clinically approved for T-cell lymphoma | [109,110,111,112] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alcaraz, J.; Ikemori, R.; Llorente, A.; Díaz-Valdivia, N.; Reguart, N.; Vizoso, M. Epigenetic Reprogramming of Tumor-Associated Fibroblasts in Lung Cancer: Therapeutic Opportunities. Cancers 2021, 13, 3782. https://doi.org/10.3390/cancers13153782
Alcaraz J, Ikemori R, Llorente A, Díaz-Valdivia N, Reguart N, Vizoso M. Epigenetic Reprogramming of Tumor-Associated Fibroblasts in Lung Cancer: Therapeutic Opportunities. Cancers. 2021; 13(15):3782. https://doi.org/10.3390/cancers13153782
Chicago/Turabian StyleAlcaraz, Jordi, Rafael Ikemori, Alejandro Llorente, Natalia Díaz-Valdivia, Noemí Reguart, and Miguel Vizoso. 2021. "Epigenetic Reprogramming of Tumor-Associated Fibroblasts in Lung Cancer: Therapeutic Opportunities" Cancers 13, no. 15: 3782. https://doi.org/10.3390/cancers13153782
APA StyleAlcaraz, J., Ikemori, R., Llorente, A., Díaz-Valdivia, N., Reguart, N., & Vizoso, M. (2021). Epigenetic Reprogramming of Tumor-Associated Fibroblasts in Lung Cancer: Therapeutic Opportunities. Cancers, 13(15), 3782. https://doi.org/10.3390/cancers13153782