
Potential of Immunotherapies in Treating Hematological Cancer-Infection Comorbidities—A Mathematical Modelling Approach


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
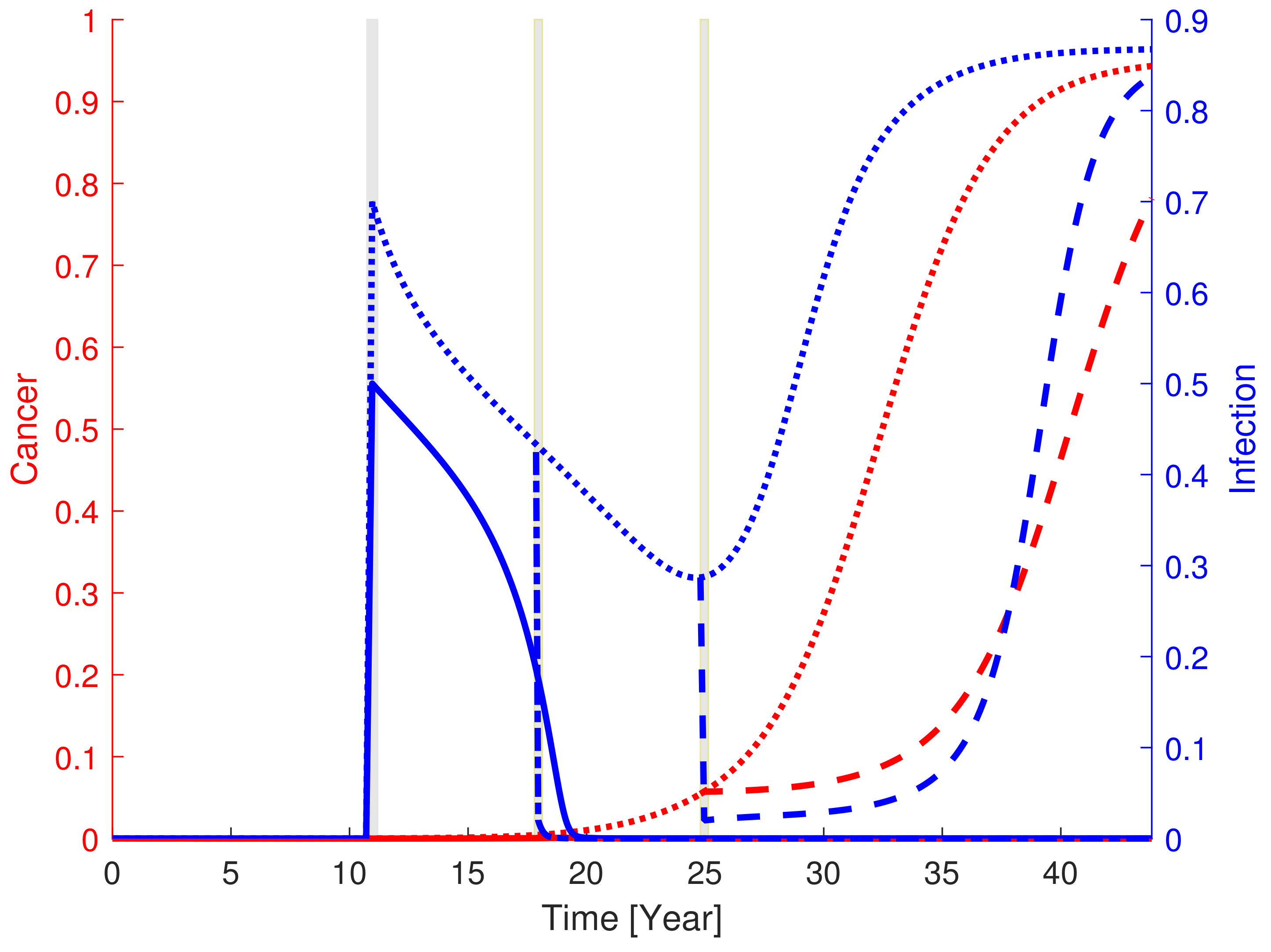
3.1. General In Silico Dynamics and Disease Progression
3.2. The Three E’s of Immunoediting Is a Consequence of the Cancer-Infection-Immune Response
3.3. Infection Trigger Cancer Escape from Immune Surveillance
3.4. Exhaustion of Cancer Specific Cytotoxic T-Cells Causes Cancer Escape
3.5. Early Treatment of Infection Prevents Cancer Progression
3.6. CAR T-Cell Immunotherapy Shows Good Effect but Is Improved in Combination with Antibiotics
4. Discussion
- Ongoing mutations may either be eradicated by the immunoediting, kept in low numbers in a dormant state or a malignant clone may escape the immunoediting and expand which results in diagnosable cancer that will progress toward full-blown cancer if left untreated. The dormant state may be thought of as a potentially pre-cancerous state, since malignant cells at low burden are rarely symptomatic. In hematopoietic cancers, such dormant states are referred to as clonal hematopoiesis of indeterminate potential (CHIP) and it requires an activation of the immune system by the malignant cells in order to control the cancer;
- The model illustrates how tumor immunoediting explains the transitions between health and disease depending on the inflammatory load caused by non-cancerous infectious factors. Such inflammation could include chronic inflammation, e.g., caused by inflammatory bowel diseases and severe virus infections such as COVID-19 or even obesity, aging and smoking;
- The model explains how an infection may compromise the immunosurveillance controlling the cancer as they are sharing pathways of the immune system. In particular, a severe infection or T-cell exhaustion may result in cancer escape;
- The model explains which pathophysiology, e.g., which disturbances of the common integrated system, results in cancer progression and which of these are ‘easily’ reversed or are harder to reverse by immunotherapies;
- In accordance with evidence-based knowledge, most patients show relapse after treatment is paused, e.g., in JAK2V617F-positive MPNs, while the treatment result may last in CALR-positive MPNs;
- In silico investigation of CAR T-cell therapy implies that strong and sufficient persistent immunotherapy may last;
- Sufficient early and strong treatment with CAR T-cell therapy shows good response for the virtual patient while postponed treatment may fail;
- Combining CAR T-cell therapy with immune-modulating antibiotic improves the effect of the treatment significantly and, in some cases, renders unsuccessful treatments successful;
- The model confirms the evidence-based experiences described by the “three E’s of immunoediting”, elimination, equilibrium and escape.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix A.1. Parameter Values
Appendix A.2. Sensitivity Analysis


Appendix A.3. Quasi-Steady State Approximation
References
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Desterke, C.; Martinaud, C.; Ruzehaji, N.; Le Bousse-Kerdiles, M. Inflammation as a keystone of bone marrow stroma alterations in primary myelofibrosis. Mediat. Inflamm. 2015, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palucka, A.K.; Coussens, L.M. The Basis of Oncoimmunology. Cell 2016, 164, 1233–1247. [Google Scholar] [CrossRef] [Green Version]
- Dunn, G.; Old, L.; Schreiber, R. The Three Es of Cancer Immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Eiró, N.; Vizoso, F. Inflammation and cancer. World J. Gastrointest Surg. 2012, 4, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Pietras, E.M.; Lakshminarasimhan, R.; Techner, J.M.; Fong, S.; Flach, J.; Binnewies, M.; Passegué, E. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J. Exp. Med. 2014, 221, 245–262. [Google Scholar] [CrossRef]
- Bald, T.; Smyth, M.J. Innate Cancer Immunoediting. J. Investig. Dermatol. 2020, 140, 745–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galon, J.; Angell, H.K.; Bedognetti, D.; Marincola, F.M. The Continuum of Cancer Immunosurveillance: Prognostic, Predictive, and Mechanistic Signatures. Immunity 2013, 39, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonardi, G.C.; Accardi, G.; Monastero, R.; Nicoletti, F.; Libra, M. Ageing: From inflammation to cancer. Immun. Ageing 2018, 15, 1–7. [Google Scholar] [CrossRef] [PubMed]
- DeGregori, J. Aging, inflammation and HSC. Blood 2020, 136, 153–154. [Google Scholar] [CrossRef]
- Markert, K.E.; Vazquez, A. Mathematical models of cancer metabolism. Cancer Metab. 2015, 3, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Fleischman, A.G.; Aichberger, K.J.; Luty, S.B.; Bumm, T.G.; Petersen, C.L.; Doratotaj, S.; Vasudevan, K.B.; Latocha, D.H.; Yang, F.; Press, R.D.; et al. TNFalpha facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood 2011, 118, 6392–6398. [Google Scholar] [CrossRef]
- Fleischman, A.G. Inflammation as a Driver of Clonal Evolution in Myeloproliferative Neoplasm. Mediat. Inflamm. 2015, 2015, 606819. [Google Scholar] [CrossRef] [Green Version]
- Hasselbalch, H.C.; Bjørn, M.E. MPNs as Inflammatory Diseases: The Evidence, Consequences, and Perspectives. Mediat. Inflamm. 2015, 2015, 102476. [Google Scholar] [CrossRef] [Green Version]
- Andersen, M.; Sajid, Z.; Pedersen, R.K.; Gudmand-Hoeyer, J.; Ellervik, C.; Skov, V.; Kjaer, L.; Pallisgaard, N.; Kruse, T.A.; Thomassen, M.; et al. Mathematical modelling as a proof of concept for MPNs as a human inflammation model for cancer development. PLoS ONE 2017, 12, e0183620. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, R.K.; Andersen, M.; Knudsen, T.A.; Sajid, Z.; Gudmand-Hoeyer, J.; Dam, M.J.; Skov, V.; Kjaer, L.; Ellervik, C.; Larsen, S.T.; et al. Data-Driven Analysis of Jak2V617F Kinetics During Interferon-Alpha2 Treatment of Patients with Polycythemia Vera and Related Neoplasms. Cancer Med. 2020, 3, 1–13. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Cytokine Profiling as a Novel Complementary Tool to Predict Prognosis in MPNs? HemaSphere 2020, 4, e482. [Google Scholar] [CrossRef] [PubMed]
- Øbro, N.F.; Grinfeld, J.; Belmonte, M.; Irvine, M.; Shepherd, M.S.; Rao, T.N.; Karow, A.; Riedel, L.M.; Harris, O.B.; Baxter, E.J.; et al. Longitudinal Cytokine Profiling Identifies GRO-alpha and EGF as Potential Biomarkers of Disease Progression in Essential Thrombocythemia. HemaSphere 2020, 4, 1–9. [Google Scholar] [CrossRef]
- Holmström, M.O.; Hasselbalch, H.C.; Andersen, M.H. Cancer immune therapy for Philadelphia chromosome-negative chronic myeloproliferative neoplasms. Cancers 2020, 12, 1763. [Google Scholar] [CrossRef]
- Osawa, M.; Hanada, K.-I.; Hamada, H.; Nakauchi, H. Long-Term Lymphohematopoietic Reconstitution by a Single CD34-Low/Negative Hematopoietic Stem Cell. Sci. Am. Assoc. Adv. Sci. 1996, 273, 242–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edelstein-Keshet, L. Mathematical Models in Biology; Classics in Applied Mathematics; Society for Industrial and Applied Mathematics: New York, NY, USA, 1988. [Google Scholar]
- Wodarz, D.; Komarova, N.L. Dynamics of Cancer: Mathematical Foundations of Oncology; World Scientific Publishing Co. Pte. Ltd.: Singapore, 2014. [Google Scholar]
- Allen, L.J.S. An Introduction to Mathematical Biology; Pearson/Prentice Hall: Upper Saddle River, NJ, USA, 2007. [Google Scholar]
- Kuznetsov, V.A.; Makalkin, I.A.; Taylor, M.A.; Perelson, A.S. Nonlinear dynamics of immunogenic tumors: Parameter estimation and global bifurcation analysis. Bull. Math. Biol. 1994, 56, 295–321. [Google Scholar] [CrossRef]
- Kuznetsov, V.A.; Knott, G.D. Modeling Tumor Regrowth and Immunotherapy. Math. Comput. Model. 2001, 33, 1275–1287. [Google Scholar] [CrossRef] [Green Version]
- Eftimie, R.; Bramson, J.L.; Earn, D.J. Interactions between the immune system and cancer: A brief review of non-spatial mathematical models. Bull. Math. Biol. 2011, 73, 2–32. [Google Scholar] [CrossRef]
- Eladdadi, A.; Kim, P.; Editors, D.M.E. Mathematical Models of Tumor-Immune System Dynamics; Springer: New York, NY, USA, 2014. [Google Scholar]
- Wilkie, K.P.; Aktar, F. Mathematically modelling inflammation as a promoter of tumour growth. Math. Med. Biol. 2020, 37, 491–514. [Google Scholar] [CrossRef]
- Wilkie, K.P.; Hahnfeldt, P. Modeling the Dichotomy of the Immune Response to Cancer: Cytotoxic Effects and Tumor-Promoting Inflammation. Bull. Math. Biol. 2017, 79, 1426–1448. [Google Scholar] [CrossRef]
- De Pillis, L.G.; Radunskaya, A.E.; Wiseman, C.L. A validated mathematical model of cell-mediated immune response to tumor growth. Cancer Res. 2005, 65, 7950–7958. [Google Scholar] [CrossRef] [Green Version]
- Kaech, S.; Cui, W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 2012, 12, 749–761. [Google Scholar] [CrossRef]
- Chaudhury, A.; Zhu, X.; Chu, L.; Goliaei, A.; June, C.H.; Kearns, J.D.; Stein, A.M. Chimeric Antigen Receptor T Cell Therapies: A Review of Cellular Kinetic-Pharmacodynamic Modeling Approaches. J. Clin. Pharmacol. 2020, 60, S147–S159. [Google Scholar] [CrossRef]
- León-Triana, O.; Pérez-Martínez, A.; Ramírez-Orellana, M.; Pérez-García, V.M. Dual-target CAR-Ts with on-and off-tumour activity may override immune suppression in solid cancers: A mathematical proof of concept. Cancers 2021, 13, 703. [Google Scholar] [CrossRef] [PubMed]
- Box, G.E. Science and statistics. J. Am. Stat. Assoc. 1976, 71, 791–799. [Google Scholar] [CrossRef]
- Sessions, R. Section Arts & Leisure. New York Times, 8 January 1950; 89. [Google Scholar]
- Krammer, P.; Arnold, R.; Lavrik, I.N. Life and death in peripheral T cells. Nat. Rev. Immunol. 2007, 7, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Carrington, E.M.; Zhang, Y.; Heinzel, S.; Lew, A.M. Life and Death of Activated T Cells: How Are They Different from Naïve T Cells? Front. Immunol. 2017, 8, 1809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrams, S.; Brahmi, Z. Mechanism of K562-induced human natural killer cell inactivation using highly enriched effector cells isolated via a new single-step heep erythrocyte rosette assay. Ann. Inst. Pasteur Immunol. 1988, 139, 361–381. [Google Scholar] [CrossRef]
- Callewaert, D.; Meyers, J.; Radcliff, G. Kinetics of cellular cytotoxicity mediated by cloned cytotoxic T lymphocytes. Immonobiology 1988, 178, 203–214. [Google Scholar] [CrossRef]
- Russ, B.; Olshansky, M.; Smallwood, H.; Li, J.; Denton, A.; Prier, J.; Stock, A.; Croom, H.; Cullen, J.; Nguyen, M.; et al. Distinct Epigenetic Signatures Delineate Transcriptional Programs during Virus-Specific CD8+ T Cell Differentiation. Immun. Resour. 2014, 41, 853–865. [Google Scholar] [CrossRef] [Green Version]
- Whitman, E.; Barber, A. NKG2D receptor activation of NF-κB enhances inflammatory cytokineproduction in murine effector CD8+T cells. Mol. Immunol. 2015, 63, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Katz, G.; Voss, K.; Yan, T.; Kim, Y.; Kortum, R.; Scott, D.; Snow, A. FOXP3 renders activated human regulatory T cells resistant to restimulation induced cell death by suppressing SAP expression. Cell. Immunol. 2018, 327, 54–61. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, M.; Nie, H.; Yuan, Y. PD-1 and PD-L1 in cancer immunotherapy: Clinical implications and future considerations. Hum. Vaccine Immunother. 2019, 15, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Liang, K. Mathematical model of infection kinetics and its analysis for COVID-19, SARS and MERS. Infect. Genet. Evol. 2020, 82, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Remmerswaal, E.; Havenith, S.; Idumm, M.; Van Leeuwen, E.; Van Donselaar, K.A.; Ten Brinke, A.; Van Der Bom-Baylon, N.; Bemelman, F.J.; Van Lier, R.A.; Ten Berge, I.J. Human virus-specific effector-type T cells accumulate in blood but not in lymph nodes. Blood 2012, 119, 1702–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, L.M.; Zeiser, R. Immunotherapy in Myeloproliferative Diseases. Cells 2020, 9, 1559. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Tian, T.; Zhang, X. A mathematical model of cell-mediated immune response to tumor. Math. Biosci. Eng. 2021, 18, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Bowen, J.R.; Acrivos, A.; Oppenheim, A.K. Singular perturbation refinement to quasi-steady state approximation in chemical kinetics. Chem. Eng. Sci. 1963, 18, 177–188. [Google Scholar] [CrossRef]
- Hek, G. Geometric singular perturbation theory in biological practice. J. Math. Biol. 2010, 36, 334–372. [Google Scholar] [CrossRef] [Green Version]
- Andersen, M.; Hasselbalch, H.C.; Kjaer, L.; Skov, V.; Ottesen, J.T. Global dynamics of healthy and cancer cells competing in the hematopoietic system. Math. Biosci. 2020, 326, 1–15. [Google Scholar] [CrossRef]
- Ottesen, J.T.; Pedersen, R.K.; Sajid, Z.; Gudmand-Hoeyer, J.; Bangsgaard, K.O.; Skov, V.; Kjær, L.; Knudsen, T.A.; Pallisgaard, N.; Hasselbalch, H.C.; et al. Bridging blood cancers and inflammation: The reduced Cancitis model. J. Theor. Biol. 2019, 465, 90–108. [Google Scholar] [CrossRef] [Green Version]
- Ottesen, J.T.; Stiehl, T.; Andersen, M. Blood Cancer and Immune Surveillance. In Systems Medicine: Integrative, Qualitative and Computational Approaches; Wolkenhauer, O., Ed.; Elsevier: Oxford, UK, 2021; Volume 3, pp. 261–268. ISBN 978-0-12-816078-7. [Google Scholar] [CrossRef]
- Pan, J.; Yang, J.F.; Deng, B.P.; Zhao, X.J.; Zhang, X.; Lin, Y.H.; Wu, Y.N.; Deng, Z.L.; Zhang, Y.L.; Liu, S.H.; et al. High efficacy and safety of low-dose CD19-directed CAR-T cell therapy in 51 refractory or relapsed B acute lymphoblastic leukemia patients. Leukemia 2017, 31, 2587–2593. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R. Cancer Immunoediting: From Surveillance to Escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Abbott, M.; Ustoyev, Y. Cancer and the Immune System: The History and Background of Immunotherapy. Semin. Oncol. Nurs. 2019, 35, 5. [Google Scholar] [CrossRef]
- Pérez-García, V.M.; León-Triana, O.; Rosa, M.; Pérez-Martínez, A. CAR T cells for T-cell leukemias: Insights from mathematical models. Commun. Nonlinear Sci. Numer. Simul. 2021, 96, e105684. [Google Scholar] [CrossRef]
- Ottesen, J.; Pedersen, R.; Dam, M.; Knudsen, T.; Skov, V.; Kjær, L.; Andersen, M. Mathematical Modeling of MPNs Offers Understanding and Decision Support for Personalized Treatment. Cancers 2020, 12, e2119. [Google Scholar] [CrossRef] [PubMed]
- Allahverdy, A.; Moghaddam, A.K.; Rahbar, S.; Shafiekhani, S.; Mirzaie, H.R.; Amanpour, S.; Etemadi, Y.; Hadjati, J.; Jafari, A.H. An agent-based model for investigating the effect of myeloid-derived suppressor cells and its depletion on tumor immune surveillance. J. Med. Signals Sens. 2019, 9, 15–23. [Google Scholar] [CrossRef]
- Carvalho, T.; Rodrigues, D.S. A Mathematical Model on the Immune System Role in Achieving Better Outcomes of Cancer Chemotherapy. Tendencias Mat. Apl. Comput. 2019, 20, 343–357. [Google Scholar] [CrossRef]
- Makhlouf, A.M.; El-Shennawy, L.; Elkaranshawy, H.A. Mathematical Modelling for the Role of CD4+T Cells in Tumor-Immune Interactions. Comput. Math. Methods Med. 2020, 7187602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unni, P.; Seshaiyer, P. Mathematical Modeling, Analysis, and Simulation of Tumor Dynamics with Drug Interventions. Comput. Math. Methods Med. 2019, 4079298. [Google Scholar] [CrossRef] [PubMed]
- Gurcan, F.; Kartal, S.; Ozturk, I.; Bozkurt, F. Stability and bifurcation analysis of a mathematical model for tumor-immune interaction with piecewise constant arguments of delay. Chaos Solitons Fractals 2014, 68, 169–179. [Google Scholar] [CrossRef]
- Matzavinos, A.; Chaplain, M.A.; Kuznetsov, V.A. Mathematical modelling of the spatio-temporal response of cytotoxic T-lymphocytes to a solid tumour. Math. Med. Biol. 2004, 21, 1–34. [Google Scholar] [CrossRef] [PubMed]
- De Pillis, L.; Radunskaya, A. The dynamics of an optimally controlled tumor model: A case study. Math. Comput. Model. 2003, 37, 1221–1244. [Google Scholar] [CrossRef]
- Gentry, S.; Jackson, T. A Mathematical Model of Cancer Stem Cell Driven Tumor Initiation: Implications of Niche Size and Loss of Homeostatic Regulatory Mechanisms. PLoS ONE 2013, 8, e71128. [Google Scholar] [CrossRef] [PubMed]
- Dingli, D.; Michor, F. Successful Therapy Must Eradicate Cancer Stem Cells. Stem Cells 2006, 24, 2603–2610. [Google Scholar] [CrossRef] [Green Version]
- Haeno, H.; Levine, R.L.; Gilliland, D.G.; Michor, F. A progenitor cell origin of myeloid malignancies. Proc. Natl. Acad. Sci. USA 2009, 106, 16616–16621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slavin, S.; Strober, S. Spontaneous murine B-cell leukaemia. Nature 1978, 272, 624–626. [Google Scholar] [CrossRef] [PubMed]
- Khajanchi, S.; Banerjee, S. Stability and bifurcation analysis of delay induced tumor immune interaction model. Appl. Math. Comput. 2014, 248, 652–671. [Google Scholar] [CrossRef]
- Siu, H.; Vitetta, E.S.; May, R.D.; Uhr, J.W. Tumor dormancy. I. Regression of BCL1 tumor and induction of a dormant tumor state in mice chimeric at the major histocompatibility complex. J. Immunol. 1986, 137, 1376–1382. [Google Scholar]
- Nanda, S.; Moore, H.; Lenhart, S. Optimal control of treatment in a mathematical model of chronic myelogenous leukemia. Math. Biosci. 2007, 210, 143–156. [Google Scholar] [CrossRef]
- Essunger, P.; Perelson, A. Modeling HIV infection of CD4þ T-cell subpopulations. J. Theor. Biol. 1994, 170, 367–391. [Google Scholar] [CrossRef]
- Janeway, C.; Travers, P.; Walport, M.; Shlomchik, M. Immunobiology: The Immune System in Health and Disease; Garland Publishing: New York, NY, USA, 2001. [Google Scholar]
- Mohri, H.; Perelson, A.; Tung, K.; Ribeiro, R.; Ramratnam, B.; Markowitz, M.; Kost, R.; Hurley, A.; Weinberger, L.; Cesar, D.; et al. Increased turnover of T lymphocytes in HIV-1 infection and its reduction by antiretroviral therapy. J. Exp. Med. 2001, 194, 1277–1287. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, R.; Mohri, H.; Ho, D.; Perelson, A. In vivo dynamics of T cell activation, proliferation, and death in HIV-1 infection: Why are CD4+ but not CD8+ T cells depleted? Proc. Natl. Acad. Sci. USA 2002, 99, 15572–15577. [Google Scholar] [CrossRef] [Green Version]
- Martin, G. Sepsis, severe sepsis and septic shock: Changes in incidence, pathogens and outcomes. Expert Rev Anti-Infect. Ther. 2012, 10, 701–706. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, R.M.; Qin, L.; Chavez, L.L.; Li, D.; Self, S.G.; Perelson, A.S. Estimation of the Initial Viral Growth Rate and Basic Reproductive Number during Acute HIV-1 Infection. J. Virol. 2010, 84, 6096–6102. Available online: https://jvi.asm.org/content/84/12/6096.full.pdf (accessed on 31 March 2010). [CrossRef] [PubMed] [Green Version]
- Bradford, B.; Mabbott, N. Prion Disease and the Innate Immune System. Viruses 2018, 4, 3389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, R. Subacute sclerosing panencephalitis. Postgrad. Med. J. 2002, 78, 63–70. [Google Scholar] [CrossRef]
- Hemachudha, T.; Ugolini, G.; Wacharapluesadee, S.; Sungkarat, W.; Shuangshoti, S.; Laothamatas, J. Human rabies: Neuropathogenesis, diagnosis, and management. Lancet Neurol. 2013, 12, 498–513. [Google Scholar] [CrossRef]
- Almocera, A.E.S.; Quiroz, G.; Hernandez-Vargas, E.A. Stability analysis in COVID-19 within-host model with immune response. Commun. Nonlinear Sci. Numer. Simul. 2021, 95, 105584. [Google Scholar] [CrossRef]
- Ghosh, I. Within host dynamics of SARS-CoV-2 in humans: Modeling immune responses and antiviral treatments. arXiv 2020, arXiv:2006.02936. [Google Scholar]
- Kuehn, C. General Fenichel Theory. In Multiple Time Scale Dynamics; Applied Mathematical Sciences; Springer: Cham, Switzerland, 2015; Volume 191. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3.04 | 20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ottesen, J.T.; Andersen, M. Potential of Immunotherapies in Treating Hematological Cancer-Infection Comorbidities—A Mathematical Modelling Approach. Cancers 2021, 13, 3789. https://doi.org/10.3390/cancers13153789
Ottesen JT, Andersen M. Potential of Immunotherapies in Treating Hematological Cancer-Infection Comorbidities—A Mathematical Modelling Approach. Cancers. 2021; 13(15):3789. https://doi.org/10.3390/cancers13153789
Chicago/Turabian StyleOttesen, Johnny T., and Morten Andersen. 2021. "Potential of Immunotherapies in Treating Hematological Cancer-Infection Comorbidities—A Mathematical Modelling Approach" Cancers 13, no. 15: 3789. https://doi.org/10.3390/cancers13153789
APA StyleOttesen, J. T., & Andersen, M. (2021). Potential of Immunotherapies in Treating Hematological Cancer-Infection Comorbidities—A Mathematical Modelling Approach. Cancers, 13(15), 3789. https://doi.org/10.3390/cancers13153789